
Abstract
This paper documents an experimental study to compare the behavior of bis-(3-sodiumsulfopropyl) disulfide (SPS)–Cl–polyethylene glycol (PEG) and 3-mercaptopropanesulfonic acid sodium salt (MPSA)-Cl-PEG additive systems during copper electrodeposition. These systems are analogous to those used industrially to plate copper lines onto integrated circuits. Galvanostatic experiments show that in the absence of chloride ion, either SPS or MPSA added to the plating solution inhibited copper deposition. Upon the addition of  , a rapid transition from inhibition to acceleration was observed for both additives, illustrating the critical role played by the chloride ion in these systems. Potentiostatic experiments performed for the Cl–PEG–SPS and Cl–PEG–MPSA additive systems show that the potential dependency of the Cl–PEG–SPS system was much stronger than that of the MPSA–Cl–PEG system. Differences between the SPS and MPSA additive systems suggest that acceleration may occur through an MPSA pathway. Finally, results with chemical compounds similar to MPSA indicate that the thiol group is associated with inhibition and that a synergistic accelerating relationship exists between the sulfonate group and chloride, presumably due to complex formation.
, a rapid transition from inhibition to acceleration was observed for both additives, illustrating the critical role played by the chloride ion in these systems. Potentiostatic experiments performed for the Cl–PEG–SPS and Cl–PEG–MPSA additive systems show that the potential dependency of the Cl–PEG–SPS system was much stronger than that of the MPSA–Cl–PEG system. Differences between the SPS and MPSA additive systems suggest that acceleration may occur through an MPSA pathway. Finally, results with chemical compounds similar to MPSA indicate that the thiol group is associated with inhibition and that a synergistic accelerating relationship exists between the sulfonate group and chloride, presumably due to complex formation.
Export citation and abstract BibTeX RIS
Two model systems consisting of an acidic electroplating solution containing either (i) polyethylene glycol (PEG), chloride ions  , and bis-(3-sodiumsulfopropyl) disulfide (SPS) or (ii) polyethylene glycol (PEG), chloride ions
, and bis-(3-sodiumsulfopropyl) disulfide (SPS) or (ii) polyethylene glycol (PEG), chloride ions  , and 3-mercaptopropanesulfonic acid sodium salt (MPSA) have demonstrated bottom-up filling (superfilling) of trenches with copper, similar to the behavior observed for industrial baths used for fabrication of on-chip copper interconnects.1–3 For these two model additive systems, it has generally been regarded that the combination of
, and 3-mercaptopropanesulfonic acid sodium salt (MPSA) have demonstrated bottom-up filling (superfilling) of trenches with copper, similar to the behavior observed for industrial baths used for fabrication of on-chip copper interconnects.1–3 For these two model additive systems, it has generally been regarded that the combination of  and PEG results in a strong inhibiting effect on copper deposition, while SPS/MPSA creates an accelerating effect.
and PEG results in a strong inhibiting effect on copper deposition, while SPS/MPSA creates an accelerating effect.
The inhibition resulting from the presence of PEG and  during copper electrodeposition has been addressed in a number of studies.4–9 More recent studies have examined the influence of the accelerator (SPS or MPSA) and the role of the accelerator in superfilling.10–13 Dow et al. concluded that acceleration was due to
during copper electrodeposition has been addressed in a number of studies.4–9 More recent studies have examined the influence of the accelerator (SPS or MPSA) and the role of the accelerator in superfilling.10–13 Dow et al. concluded that acceleration was due to  complex formation that facilitated electron transport.10 Moffat et al. attributed acceleration to disruption of the inhibition layer by the sulfonate end group.11 Yet another explanation was offered by Vereecken et al. , who stated that acceleration was due to the formation of a stable cuprous complex.12 The work of Schultz et al. indicated that a complex with sulfonate, cuprous ion, and chloride may be important in the acceleration process.13 They also concluded from analysis of the surface that a dense, strongly absorbed surface layer was not involved in acceleration. The present experimental study, which was performed in parallel with these recent studies, presents results that are complementary to those previously published to provide additional insight into the behavior of the accelerator in both the SPS and MPSA systems.14, 15
complex formation that facilitated electron transport.10 Moffat et al. attributed acceleration to disruption of the inhibition layer by the sulfonate end group.11 Yet another explanation was offered by Vereecken et al. , who stated that acceleration was due to the formation of a stable cuprous complex.12 The work of Schultz et al. indicated that a complex with sulfonate, cuprous ion, and chloride may be important in the acceleration process.13 They also concluded from analysis of the surface that a dense, strongly absorbed surface layer was not involved in acceleration. The present experimental study, which was performed in parallel with these recent studies, presents results that are complementary to those previously published to provide additional insight into the behavior of the accelerator in both the SPS and MPSA systems.14, 15
Experimental
Copper electrodeposition experiments were performed with use of a copper-plated platinum rotating disk ( diam) working electrode and a copper counter electrode (Cu 99.99%). All potentials are relative to a saturated mercurous sulfate reference. The composition of the basic electrolyte (BE) was
diam) working electrode and a copper counter electrode (Cu 99.99%). All potentials are relative to a saturated mercurous sulfate reference. The composition of the basic electrolyte (BE) was 
 . and
. and 
 , to which one or more of the three additives PEG (
, to which one or more of the three additives PEG ( , Aldrich),
, Aldrich),  (added as NaCl, reagent, Mallinckrodt), and/or either SPS or MPSA (Rasching, Germany) were added. The amount of PEG and
(added as NaCl, reagent, Mallinckrodt), and/or either SPS or MPSA (Rasching, Germany) were added. The amount of PEG and  added was 300 and
added was 300 and  , respectively. At the beginning of each experiment a copper layer was deposited on a platinum disk in the basic electrolyte without additives at
, respectively. At the beginning of each experiment a copper layer was deposited on a platinum disk in the basic electrolyte without additives at  for
for  . The electrolyte temperature was kept constant at
. The electrolyte temperature was kept constant at  . The rotation rate was
. The rotation rate was  for all experiments.
for all experiments.
Galvanostatic deposition experiments were performed at a current density of  . Deposition experiments were also performed under potentiostatic conditions at
. Deposition experiments were also performed under potentiostatic conditions at  or
or  vs a saturated sulfate electrode (SSE). All MPSA experiments were performed in a
vs a saturated sulfate electrode (SSE). All MPSA experiments were performed in a  environment to avoid additive degradation by oxidation. Additives were added sequentially to the basic electrolyte in order to elucidate additive behavior and interactions.
environment to avoid additive degradation by oxidation. Additives were added sequentially to the basic electrolyte in order to elucidate additive behavior and interactions.
An additional set of experiments was performed with MPSA, 1, 3-propanedithiol (PDT), and 1, 3-propanedisulfonic acid disodium salt (PDSA) (Sigma-Aldrich) to explore the role of the sulfonate and thiol end groups. MPSA has one thiol and one sulfonate end group, PDT has two thiol end groups, and PDSA has two sulfonate end groups. Experiments similar to the MPSA experiments described above were performed where additives were added sequentially. These experiments were performed in deaerated electrolyte in a cell where a Nafion membrane was used to separate the cathode and anode compartments.
Results and Discussion
Interaction between  and SPS
and SPS
In order to understand the role that SPS plays in the Cl–SPS–PEG additive system, the influence of SPS and a combination of SPS/Cl on copper deposition was investigated. First, experiments were performed to examine the effect of SPS on copper deposition. Figure 1 shows that SPS alone (prior to  addition) inhibited copper deposition as evidenced by a drop in potential observed during galvanostatic deposition. Similar observations have been recently reported in the literature10, 16, 17 and are important in understanding the role of specific additives in the deposition process. The inhibition shown in Fig. 1 was significantly more pronounced at higher SPS concentrations (50 vs
addition) inhibited copper deposition as evidenced by a drop in potential observed during galvanostatic deposition. Similar observations have been recently reported in the literature10, 16, 17 and are important in understanding the role of specific additives in the deposition process. The inhibition shown in Fig. 1 was significantly more pronounced at higher SPS concentrations (50 vs  ). In both cases, the curves reached a plateau that was dependent on concentration, perhaps due to a steady-state balance between adsorption and desorption/consumption. Several minutes were required to reach steady state. The dependency of this inhibition on potential was also studied by applying potentials in the range of normal copper electrodeposition (from
). In both cases, the curves reached a plateau that was dependent on concentration, perhaps due to a steady-state balance between adsorption and desorption/consumption. Several minutes were required to reach steady state. The dependency of this inhibition on potential was also studied by applying potentials in the range of normal copper electrodeposition (from  vs SSE) and inhibition of copper deposition was observed for all applied potentials.
vs SSE) and inhibition of copper deposition was observed for all applied potentials.
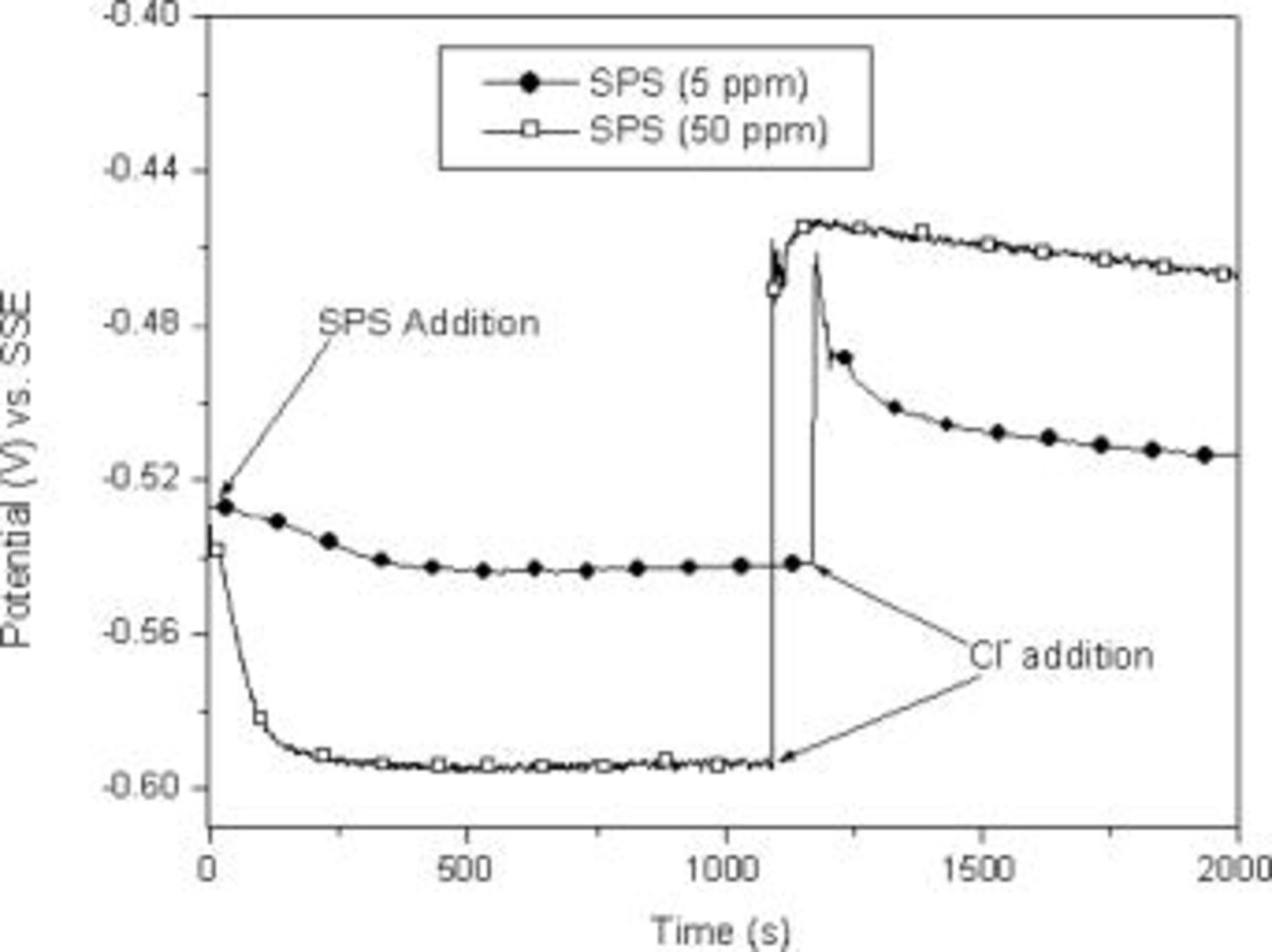
Figure 1. Sequential addition of SPS and 
 (galvanostatic electrodeposition at
(galvanostatic electrodeposition at  ).
).
It was hypothesized that the inhibition was associated with an adsorbed surface layer and an experiment was performed to qualitatively examine that layer. Copper was deposited in  SPS for about
SPS for about  , after which the working electrode was removed from the electrolyte, rinsed in deionized (DI) water, and returned to the same solution where galvanostatic plating was resumed. Figure 2 shows that the potential observed after water rinsing was almost the same as that before the sample was taken out of the solution. This observation is consistent with the presence of a strongly adsorbed surface layer that remained intact in spite of the water rinsing. Thus, copper electrodeposition experiments with a single additive, SPS, showed concentration-dependent inhibition involving a strongly adsorbed surface layer.
, after which the working electrode was removed from the electrolyte, rinsed in deionized (DI) water, and returned to the same solution where galvanostatic plating was resumed. Figure 2 shows that the potential observed after water rinsing was almost the same as that before the sample was taken out of the solution. This observation is consistent with the presence of a strongly adsorbed surface layer that remained intact in spite of the water rinsing. Thus, copper electrodeposition experiments with a single additive, SPS, showed concentration-dependent inhibition involving a strongly adsorbed surface layer.
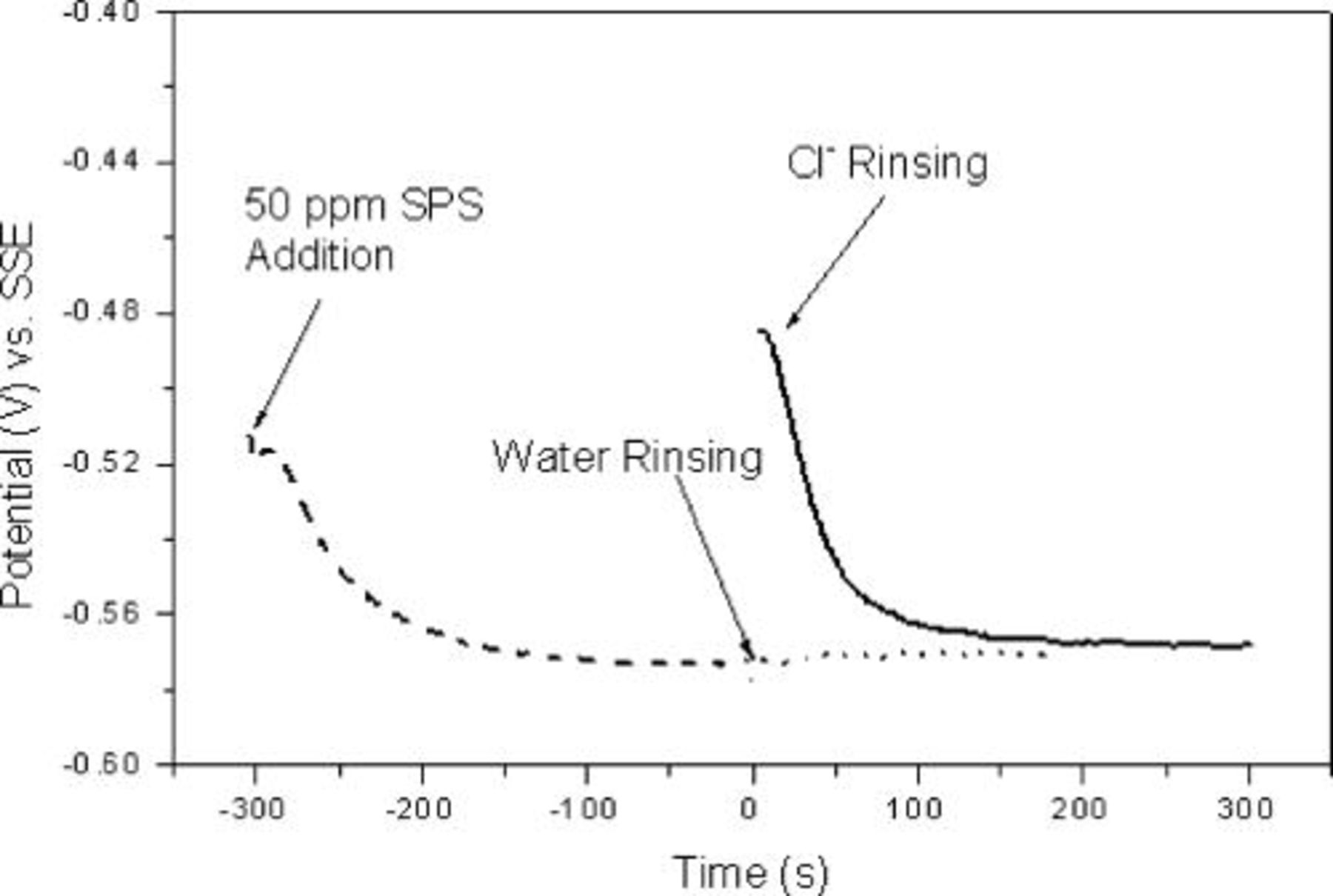
Figure 2. Water and  rinsing experiments (galvanostatic electrodeposition at
rinsing experiments (galvanostatic electrodeposition at  current density).
current density).
The interaction of  and SPS was investigated by the sequential addition of
and SPS was investigated by the sequential addition of 
 to the SPS-containing electrolyte. The results of similar experiments were recently reported by Dow et al.10 who added the SPS and chloride in the opposite order. Our results, also in Fig. 1, show a rapid jump in potential upon the addition of the chloride. With both
to the SPS-containing electrolyte. The results of similar experiments were recently reported by Dow et al.10 who added the SPS and chloride in the opposite order. Our results, also in Fig. 1, show a rapid jump in potential upon the addition of the chloride. With both  and SPS in the solution, significant acceleration of copper deposition was observed. As shown by Dow et al. , this acceleration was greater than that observed for electrodeposition from basic electrolyte containing
and SPS in the solution, significant acceleration of copper deposition was observed. As shown by Dow et al. , this acceleration was greater than that observed for electrodeposition from basic electrolyte containing  only. An important difference between our results and those of Dow et al. is that we observed a very rapid transition from inhibition to acceleration, whereas a slow transition was observed when the additives were added in the reverse order. Thus, the process responsible for the slow transition occurs in the absence of chloride and, therefore, must not involve chemical interactions between SPS and
only. An important difference between our results and those of Dow et al. is that we observed a very rapid transition from inhibition to acceleration, whereas a slow transition was observed when the additives were added in the reverse order. Thus, the process responsible for the slow transition occurs in the absence of chloride and, therefore, must not involve chemical interactions between SPS and  . Acceleration was dependent upon SPS concentration, with a higher SPS concentration resulting in increased acceleration of copper deposition in the presence of chloride.
. Acceleration was dependent upon SPS concentration, with a higher SPS concentration resulting in increased acceleration of copper deposition in the presence of chloride.
A possible explanation for transition from inhibition to acceleration observed upon the addition of chloride ions is that  was instrumental in removing or modifying the adsorbed inhibition layer. To test this hypothesis, the rinsing experiment was repeated with the addition of a
was instrumental in removing or modifying the adsorbed inhibition layer. To test this hypothesis, the rinsing experiment was repeated with the addition of a  -containing rinse solution. Specifically, the sample was rinsed in a solution containing
-containing rinse solution. Specifically, the sample was rinsed in a solution containing 
 followed by rinsing in DI water prior to placing it back into the electrolyte. The results (Fig. 2) show that the chloride ions effectively removed the inhibition, as evidenced by the high starting potential (in contrast to the low starting potential observed for the water-only rinse). Figure 2 shows that the behavior of the rinsed sample was similar to that of the fresh copper sample but with a higher starting potential. A strong inhibition on copper deposition was observed in both cases after the system reached steady state. Thus, the acceleration related to Cl–SPS diminished rapidly in the absence of
followed by rinsing in DI water prior to placing it back into the electrolyte. The results (Fig. 2) show that the chloride ions effectively removed the inhibition, as evidenced by the high starting potential (in contrast to the low starting potential observed for the water-only rinse). Figure 2 shows that the behavior of the rinsed sample was similar to that of the fresh copper sample but with a higher starting potential. A strong inhibition on copper deposition was observed in both cases after the system reached steady state. Thus, the acceleration related to Cl–SPS diminished rapidly in the absence of  . It is clear from the above results that any mechanism that describes acceleration under the conditions considered above must include the role of
. It is clear from the above results that any mechanism that describes acceleration under the conditions considered above must include the role of  as a key component and account for the synergistic behavior of SPS and
as a key component and account for the synergistic behavior of SPS and  .
.
Interaction between and MPSA
Experiments similar to those reported in Fig. 1 for SPS were also performed with MPSA (Fig. 3). The behavior of the MPSA was essentially the same as that observed for SPS, except that the behavior was observed at a much lower concentration. In fact, it was possible to adjust the MPSA concentration so that the observed behavior matched that of SPS as shown in Fig. 3 ( SPS and
SPS and  MPSA). The fact that the same concentration matched the behavior both with and without chloride (both acceleration and inhibition) seems to indicate that the surface chemistry was similar in both cases.
MPSA). The fact that the same concentration matched the behavior both with and without chloride (both acceleration and inhibition) seems to indicate that the surface chemistry was similar in both cases.
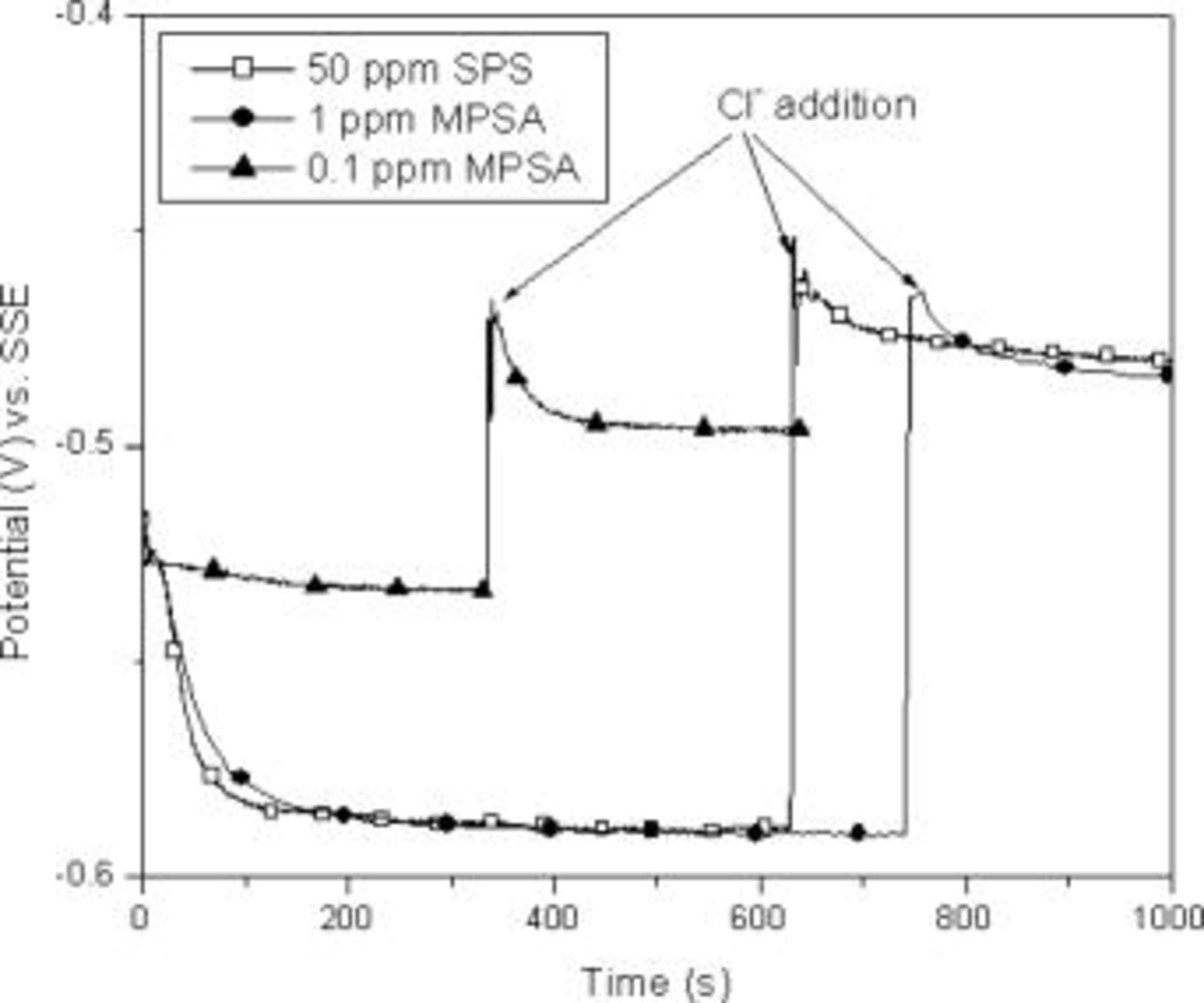
Figure 3. Comparison of the behavior of binary additive systems for sequential addition of SPS–Cl or MSP–Cl (at  current density).
current density).
The similar behavior observed for SPS and MPSA in our experiments is in contrast to the different behavior observed for these same systems when the order of additive addition was reversed.10 Specifically, Dow et al. observed a much more gradual transition when SPS was added to a solution containing chloride.10 When both sets of experiments are viewed together, the following appear to be true about the process responsible for the slow transition observed by Dow: (i) it had already occurred during the inhibition portion of the experiment when chloride was added last (our experiments) and (ii) it does not involve  . The experiments also demonstrate that the interaction with chloride occurs rapidly.
. The experiments also demonstrate that the interaction with chloride occurs rapidly.
The above observations are consistent with a mechanism that involves the conversion of SPS to MPSA and acceleration through the same MPSA pathway. The large difference in the required concentration is the result of the conversion of only a fraction of the SPS to MPSA. This mechanism is consistent with statements in the literature that the disulfide bond is reduced before adsorption can take place.12, 13 Also, studies on surfaces derivatized in either SPS or MPSA demonstrated the same acceleration behavior.11
Potential-dependent behavior
The potential-dependent behavior of the Cl–PEG–SPS and Cl–PEG–MPSA systems was examined in order to gain insight into the differences between the two systems. The three additives (Cl–PEG–SPS or Cl–PEG–MPSA) were added to the plating solution at time zero, and the change in current was recorded as a function of time at constant potential. Figure 4 illustrates that when a high overpotential was applied  , both
, both  SPS and
SPS and  MPSA behaved similarly, showing significant acceleration relative to the Cl–PEG-only electrolyte and demonstrating once again that a much lower concentration of MPSA is needed to produce the same acceleration behavior. When a low overpotential was applied
MPSA behaved similarly, showing significant acceleration relative to the Cl–PEG-only electrolyte and demonstrating once again that a much lower concentration of MPSA is needed to produce the same acceleration behavior. When a low overpotential was applied  , the acceleration effect of SPS
, the acceleration effect of SPS  –Cl–PEG was quite small. An increase in the SPS concentration to
–Cl–PEG was quite small. An increase in the SPS concentration to  at the same potential led to increased acceleration, although the acceleration current was still relatively small. In contrast, MPSA
at the same potential led to increased acceleration, although the acceleration current was still relatively small. In contrast, MPSA  –Cl–PEG showed significant acceleration at
–Cl–PEG showed significant acceleration at  . The results demonstrate that the MPSA system is much less sensitive to changes in potential than is the SPS system. In addition, the time constant associated with the transient behavior was a strong function of potential for the SPS system but remained relatively constant with the change in potential for MPSA (see Fig. 4). In similar experiments, Moffatt et al. also showed a strong potential dependence for both the time constant and steady-state current for SPS,11 although no comparison to MPSA was made.
. The results demonstrate that the MPSA system is much less sensitive to changes in potential than is the SPS system. In addition, the time constant associated with the transient behavior was a strong function of potential for the SPS system but remained relatively constant with the change in potential for MPSA (see Fig. 4). In similar experiments, Moffatt et al. also showed a strong potential dependence for both the time constant and steady-state current for SPS,11 although no comparison to MPSA was made.
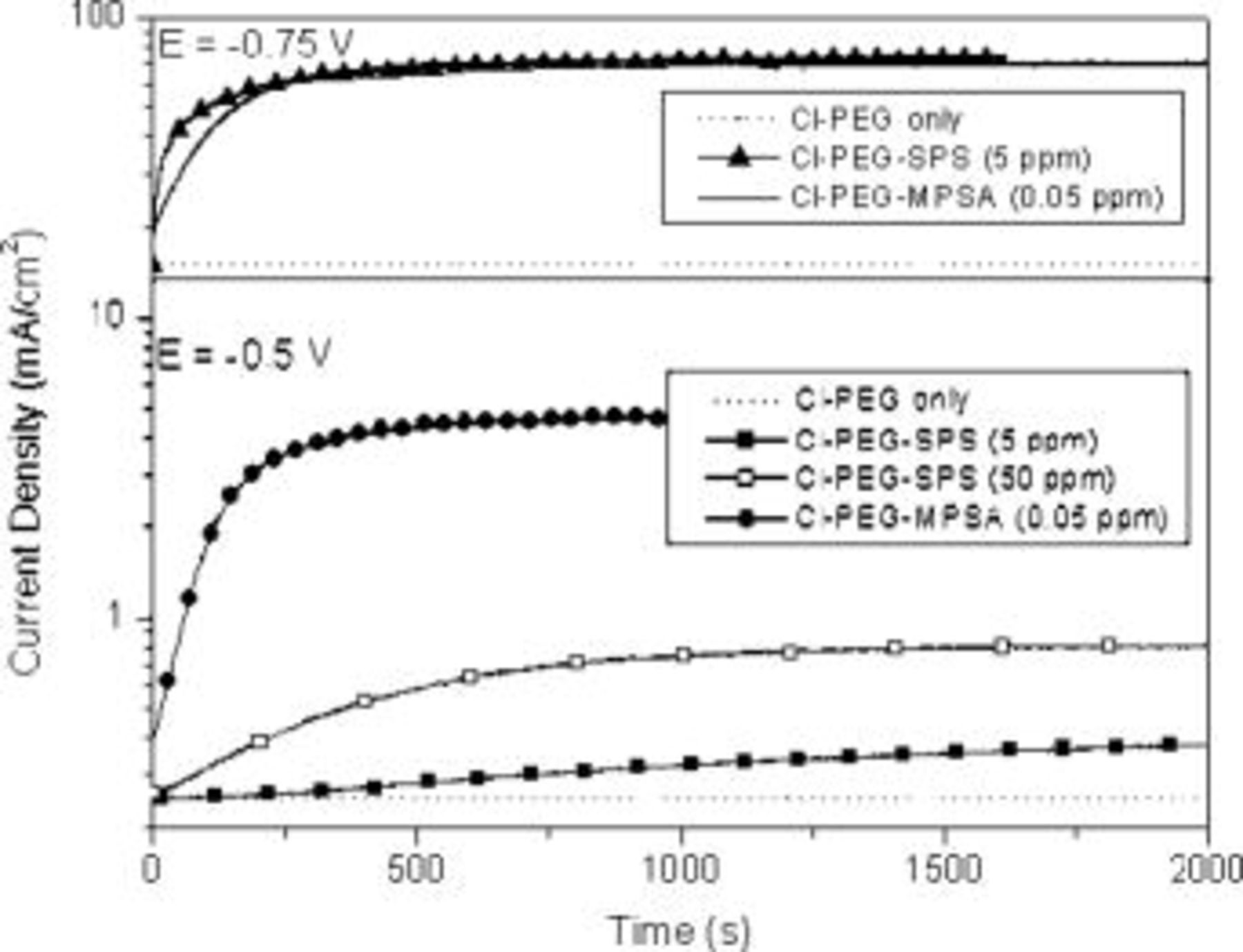
Figure 4. Potentiostatic behavior of Cl–PEG–SPS and Cl–PEG–MPSA additive systems (BE with 
 ,
,  PEG, and
PEG, and 
 MPSA).
MPSA).
A possible explanation for the difference in behavior between the MPSA and SPS systems is that acceleration occurs through an MPSA pathway, and that a fraction of the SPS in the Cl–PEG–SPS additive system is converted to MPSA through a potential-dependent reaction. This explanation is consistent with both the observed concentration difference needed to impact deposition and the potential dependency of the SPS system. Electroreduction of SPS to MPSA may occur according to the reaction
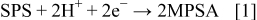
and is likely associated with the observed strong potential dependency of the SPS system. Cathodic reduction of SPS to MPSA was reported by Zhukauskaite and Malinauskas.18, 19 Healy et al. also reported cathodic current in the potential range of interest to copper electrodeposition for a sulfuric acid solution containing SPS; they saw no evidence of cathodic reactions at the copper electrode for experiments performed under similar conditions with MPSA instead of SPS.20 Dow et al. recently included Reaction 1 as part of their mechanistic description of copper superfilling.10 In contrast, Vereecken et al. did not see evidence of SPS reduction in their rotating disk electrode experiments.12 Although the results presented here are not sufficient to establish the role of Reaction 1, it is clear that the behavior of SPS is much more strongly dependent on potential than that of MPSA. Taken in the context of the results presented in the previous section, which indicate that the acceleration chemistry is similar for both SPS and MPSA, the hypothesis that SPS is converted to MPSA via a potential-dependent reaction is consistent with the observed behavior.
Role of chemical end groups
Additional experiments were performed in order to investigate the role of the thiol and sulfonate end groups. These experiments were performed with two analogues of MPSA, which has one thiol end group and one sulfonate end group separated by a propane backbone. The first analogue is PDT, which has two thiol end groups with a propane backbone. The second is PDSA, which has two sulfonate end groups with a propane backbone. The results are shown in Fig. 5, where a simple illustration of each molecule has been included to emphasize the role of the different end groups. In each case, the additive concentration (MPSA, PDT, or PDSA) was  . The addition of PDT to the basic electrolyte led to significant inhibition, much greater than that observed for MPSA. Inhibition that increased gradually with time was also observed for MPSA, consistent with the results presented earlier. In contrast, the addition of PDSA resulted in a slight acceleration that increased very gradually with time. It seems clear from these results that the thiol group in MPSA is responsible for the inhibition behavior observed with no chloride in the solution.
. The addition of PDT to the basic electrolyte led to significant inhibition, much greater than that observed for MPSA. Inhibition that increased gradually with time was also observed for MPSA, consistent with the results presented earlier. In contrast, the addition of PDSA resulted in a slight acceleration that increased very gradually with time. It seems clear from these results that the thiol group in MPSA is responsible for the inhibition behavior observed with no chloride in the solution.
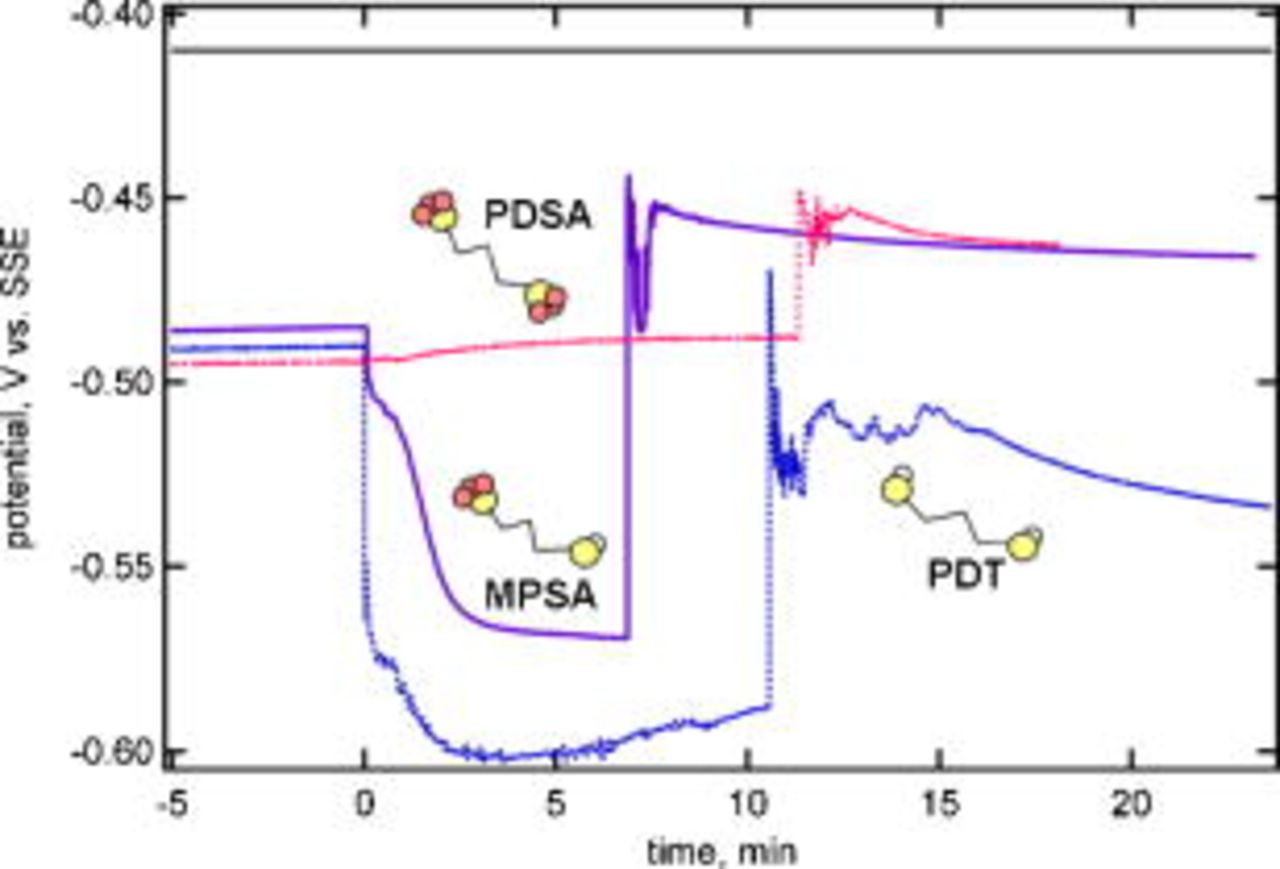
Figure 5. (Color online) Behavior of MPSA, PDT, and PDSA at  when added to the basic electrolyte. The upward step change in the potential corresponds to the sequential addition of chloride.
when added to the basic electrolyte. The upward step change in the potential corresponds to the sequential addition of chloride.
Figure 5 also shows that for all three additives, the addition of chloride resulted in a sharp increase in the potential (decrease in the overpotential), followed by oscillations and then a steady gradual decline in the potential. For PDT, the solution remained inhibiting; even though the potential increased significantly, it was below the value observed for the basic electrolyte without PDT or chloride. It is well known that the addition of small amounts of chloride accelerates deposition.21 Therefore, it seems likely that chloride added to the PDT solution only partially displaces the PDT because the solution remains inhibiting. It seems probable that the chloride displacement occurs preferentially at particular sites (e.g., step sites or kink sites). The change in behavior upon chloride addition may also reflect acceleration of deposition by chloride on areas of the surface where PDT was not adsorbed.
The replacement of one of the thiol groups with a sulfonate group (MPSA) leads to a switch in behavior from inhibiting to accelerating upon the addition of chloride. The increase may be due to partial removal of the inhibiting surface layer by displacement with chloride. However, this is not the entire story as seen from the results for PDSA, which does not contain thiol end groups. The PDSA, which was slightly accelerating prior to chloride addition, led to significantly more acceleration in the presence of chloride. Thus, there appears to be a synergistic effect between the sulfonate and chloride that accelerates deposition. Given the behavior for PDSA and PDT, it is logical to conclude that the sharp increase in potential observed for MPSA is due in part to thiol displacement and in part to synergistic acceleration by chloride and the sulfonate groups. Note that the final potential reached for PDSA and MPSA was the same, indicating that acceleration depended more on the number of accelerator molecules containing sulfonate than on the absolute number of sulfonate groups present.
Dow et al. attributed acceleration to the formation of inner-sphere complexes in the presence of chloride ion, which facilitated electron transfer.10 While this mechanism is believed to be qualitatively correct, the conclusion of these authors that the ligand complexes were based on coordination with sulfide is, at best, incomplete. The results presented above clearly establish the critical role of the sulfonate end group in the acceleration process. Evidence for the preferential formation of cuprous complexes with the sulfonate end of the MPSA molecule in the presence of chloride was recently reported.13 One possibility consistent with the above is that acceleration is due to a complex between cupric ion, chloride, and the sulfonate end group that facilitates reduction of the cupric to cuprous, which reduction is believed to be the rate-limiting step at the overpotentials considered. This explanation is also consistent with the observation of an increased cuprous concentration at the surface at high overpotentials in the presence of the accelerator.12
Finally, although similar levels of acceleration were observed for PDSA and MPSA in the presence of chloride on the surface of a rotating disk electrode, this does not necessarily mean that PDSA is an effective catalyst for bottom-up filling. It is possible that the thiol group is needed to tether the accelerator to the surface as suggested by Moffat et al.11 In stating this, it is noted that no evidence was found for any kind of ordered array on the surface for MPSA and SPS in the presence of chloride, although low coverage adsorption was not precluded.13 Also, studies on derivatized surfaces11 seem to indicate that the catalyst "floats," or that it remains located very near the growing surface without being bound to the surface. Thus, although it seems likely that a sulfonate complex plays a critical role in the acceleration process, there are a number of issues related to superfilling that remain to be resolved.
Conclusions
Electrodeposition from acidic copper electrolytes containing either SPS or MPSA was inhibited due to a strongly adsorbed surface layer that was dependent on the concentration of SPS/MPSA. The inhibition was readily eliminated upon the addition of chloride and resulted in an overall acceleration that was also dependent on the SPS/MPSA concentration. Both SPS and MPSA showed a rapid transition from inhibiting to accelerating upon the addition of chloride, in contrast to the previous observation of a much slower transition for SPS when the order of additive addition was reversed.10 It follows that the process responsible for the slow transition (likely disulfide cleavage) had already occurred during the inhibition portion of the experiment when chloride was added last (our experiments) and does not involve  . The experiments also demonstrate that the interaction with chloride occurs rapidly. Although the observed acceleration behavior was similar for MPSA and SPS when the concentrations were "matched," the behavior of SPS was much more strongly dependent on potential than that of MPSA. The hypothesis that SPS is converted to MPSA via a potential-dependent reaction appears to be consistent with the observed behavior.
. The experiments also demonstrate that the interaction with chloride occurs rapidly. Although the observed acceleration behavior was similar for MPSA and SPS when the concentrations were "matched," the behavior of SPS was much more strongly dependent on potential than that of MPSA. The hypothesis that SPS is converted to MPSA via a potential-dependent reaction appears to be consistent with the observed behavior.
Finally, experiments with MPSA, PDT, and PDSA clearly demonstrate that the sulfonate group is responsible for acceleration in the presence of chloride. This acceleration is believed to occur through complex formation that facilitates electron transfer.
Acknowledgment
The authors gratefully acknowledge the National Science Foundation (CTS-0215786), the American chemical Society (PRF42461–G5), and Brigham Young University for their support of this work.