
Two reproducible new phases of 2D boron sheets have been found on Ag(1 1 1). One of them shares the identical atomic structure of the previously reported S1 phase (β12 sheet) but has a different rotational relationship with the substrate, and thus exhibits very different features in scanning tunneling microscopy (STM) images. The other new phase has a hexagonal symmetry and is proposed to be the long-expected α-sheet. Both of these two boron sheets are confirmed to be metallic by scanning tunneling spectroscopy.
Highlights of 2017

Welcome to the Journal of Physics: Condensed Matter highlights of 2017, our annual selection of the best papers published in the previous year, which represent the breadth and excellence of the work published in the journal.
We would like to thank all of the journal's authors, reviewers, readers and Editorial Board, for their invaluable dedication and support over the last year.
We hope that you enjoy reading these papers and that you will consider publishing your next paper with Journal of Physics: Condensed Matter.
Tom Sharp
Executive Editor
Journal of Physics : Condensed Matter
Surfaces and interfaces
Show article list
Assessing the amorphousness and periodicity of common domain boundaries in silica bilayers on Ru(0 0 0 1)
View article, Assessing the amorphousness and periodicity of common domain boundaries in silica bilayers on Ru(0 0 0 1)
PDF, Assessing the amorphousness and periodicity of common domain boundaries in silica bilayers on Ru(0 0 0 1)
Domain boundaries are hypothesized to play a role in the crystalline to amorphous transition. Here we examine domain boundary structures in comparison to crystalline and amorphous structures in bilayer silica grown on Ru(0 0 0 1). Atomically resolved scanning probe microscopy data of boundaries in crystalline bilayer films are analyzed to determine structural motifs. A rich variety of boundary structures including rotational, closed-loop, antiphase, and complex boundaries are identified. Repeating units with ring sizes of 558 and 57 form the two most common domain boundary types. Quantitative metrics are utilized to assess the structural composition and degree of order for the chemically equivalent crystalline, domain boundary, and amorphous structures. It is found that domain boundaries in the crystalline phase show similarities to the amorphous phase in their ring statistics and, in some cases, in terms of the observed ring neighborhoods. However, by assessing order and periodicity, domain boundaries are shown to be distinct from the glassy state. The role of the Ru(0 0 0 1) substrate in influencing grain boundary structure is also discussed.
Open access
Fermi surface map of large-scale single-orientation graphene on SiO2
View article, Fermi surface map of large-scale single-orientation graphene on SiO2
PDF, Fermi surface map of large-scale single-orientation graphene on SiO2
Large scale tetraoctylammonium-assisted electrochemical transfer of graphene grown on single-crystalline Ir(1 1 1) films by chemical vapour deposition is reported. The transferred samples are characterized in air with optical microscopy, Raman spectroscopy and four point transport measurements, providing the sheet resistance and the Hall carrier concentration. In vacuum we apply low energy electron diffraction and photoelectron spectroscopy that indicate transferred large-scale single orientation graphene. Angular resolved photoemission reveals a Fermi surface and a Dirac point energy which are consistent with charge neutral graphene.
Blocking of injected holes at the charge extracting interface
View article, Blocking of injected holes at the charge extracting interface
PDF, Blocking of injected holes at the charge extracting interface
For an organic crystal with four metal electrodes, we have investigated the charge transfer between the injecting electrode 1 and the extracting electrodes 2–4. Each of the measured I−V curves was decomposed into three components, characterizing the injecting and extracting interfaces, and the squaraine (SQ) crystal. We considered a case, where the resistance of the organic crystal was significantly smaller than that of the interfaces. The different values of the contact resistances measured at different electrodes are related to differing values of the barrier height  and its width d, which were determined using the Simmons approximation. When the extracting interface has a larger contact resistance compared to that of the SQ crystal and that of the injecting interface, part of the injected holes remains located within the SQ crystal. The voltage V0, at which the injected hole density begins to exceed the extracted hole density within the SQ crystal, depends on the ratio
and its width d, which were determined using the Simmons approximation. When the extracting interface has a larger contact resistance compared to that of the SQ crystal and that of the injecting interface, part of the injected holes remains located within the SQ crystal. The voltage V0, at which the injected hole density begins to exceed the extracted hole density within the SQ crystal, depends on the ratio  (indices ex and in describe the extracting and injecting electrodes, respectively). The smaller δ is, the larger is voltage V0.
(indices ex and in describe the extracting and injecting electrodes, respectively). The smaller δ is, the larger is voltage V0.
Thermoelectric properties of SnSe2 monolayer
View article, Thermoelectric properties of SnSe2 monolayer
PDF, Thermoelectric properties of SnSe2 monolayer
The 2H (MoS2-type) phase of 2D transition metal dichalcogenides (TMDCs) has been extensively studied and exhibits excellent electronic and optoelectronic properties, but the high phonon thermal conductivity is detrimental to the thermoelectric performances. Here, we use first-principles methods combined with Boltzmann transport theory to calculate the electronic and phononic transport properties of 1T (CdI2-type) SnSe2 monolayer, a recently realized 2D metal dichalcogenide semiconductor. The calculated band gap is 0.85 eV, which is a little larger than the bulk value. Lower phonon thermal conductivity and higher power factor are obtained in 1T-SnSe2 monolayer compared to 2H-TMDCs monolayers. The low phonon thermal conductivity (3.27 W mK−1 at room temperature) is mainly due to the low phonon frequency of acoustic modes and the coupling of acoustic modes with optical modes. We also find that the p-type has better thermoelectric performance than the n-type, and the figure of merit within p-type can reach 0.94 at 600 K for 1T-SnSe2 monolayer, which is higher than those of most 2H-TMDCs monolayers, making 1T-SnSe2 monolayer a promising candidate for thermoelectric applications.
Interface electronic structure at the topological insulator–ferrimagnetic insulator junction
View article, Interface electronic structure at the topological insulator–ferrimagnetic insulator junction
PDF, Interface electronic structure at the topological insulator–ferrimagnetic insulator junction
An interface electron state at the junction between a three-dimensional topological insulator film, Bi2Se3, and a ferrimagnetic insulator film, Y3Fe5O12 (YIG), was investigated by measurements of angle-resolved photoelectron spectroscopy and x-ray absorption magnetic circular dichroism. The surface state of the Bi2Se3 film was directly observed and localized 3d spin states of the Fe3+ in the YIG film were confirmed. The proximity effect is likely described in terms of the exchange interaction between the localized Fe 3d electrons in the YIG film and delocalized electrons of the surface and bulk states in the Bi2Se3 film.
Surface plasmon resonance in gold nanoparticles: a review
View article, Surface plasmon resonance in gold nanoparticles: a review
PDF, Surface plasmon resonance in gold nanoparticles: a review
In the last two decades, plasmon resonance in gold nanoparticles (Au NPs) has been the subject of intense research efforts. Plasmon physics is intriguing and its precise modelling proved to be challenging. In fact, plasmons are highly responsive to a multitude of factors, either intrinsic to the Au NPs or from the environment, and recently the need emerged for the correction of standard electromagnetic approaches with quantum effects. Applications related to plasmon absorption and scattering in Au NPs are impressively numerous, ranging from sensing to photothermal effects to cell imaging. Also, plasmon-enhanced phenomena are highly interesting for multiple purposes, including, for instance, Raman spectroscopy of nearby analytes, catalysis, or sunlight energy conversion. In addition, plasmon excitation is involved in a series of advanced physical processes such as non-linear optics, optical trapping, magneto-plasmonics, and optical activity. Here, we provide the general overview of the field and the background for appropriate modelling of the physical phenomena. Then, we report on the current state of the art and most recent applications of plasmon resonance in Au NPs.
Open access
Intrusion and extrusion of a liquid on nanostructured surfaces
View article, Intrusion and extrusion of a liquid on nanostructured surfaces
PDF, Intrusion and extrusion of a liquid on nanostructured surfaces
Superhydrophobicity is connected to the presence of gas pockets within surface asperities. Upon increasing the pressure this 'suspended' state may collapse, causing the complete wetting of the rough surface. In order to quantitatively characterize this process on nanostructured surfaces, we perform rare-event atomistic simulations at different pressures and for several texture geometries. Such an approach allows us to identify for each pressure the stable and metastable states and the free energy barriers separating them. Results show that, by starting from the superhydrophobic state and increasing the pressure, the suspended state abruptly collapses at a critical intrusion pressure. If the pressure is subsequently decreased, the system remains trapped in the metastable state corresponding to the wet surface. The liquid can be extruded from the nanostructures only at very negative pressures, by reaching the critical extrusion pressure (spinodal for the confined liquid). The intrusion and extrusion curves form a hysteresis cycle determined by the large free energy barriers separating the suspended and wet states. These barriers, which grow very quickly for pressures departing from the intrusion/extrusion pressure, are shown to strongly depend on the texture geometry.
Soft matter, biophysics and liquids
Show article list
Smectic phases in ionic liquid crystals
Ionic liquid crystals (ILCs) are anisotropic mesogenic molecules which carry charges and therefore combine properties of liquid crystals, e.g. the formation of mesophases, and of ionic liquids, such as low melting temperatures and tiny triple-point pressures. Previous density functional calculations have revealed that the phase behavior of ILCs is strongly affected by their molecular properties, i.e. their aspect ratio, the loci of the charges, and their interaction strengths. Here, we report new findings concerning the phase behavior of ILCs as obtained by density functional theory and Monte Carlo simulations. The most important result is the occurrence of a novel, wide smectic-A phase  , at low temperature, the layer spacing of which is larger than that of the ordinary high-temperature smectic-A phase
, at low temperature, the layer spacing of which is larger than that of the ordinary high-temperature smectic-A phase  . Unlike the ordinary smectic SA phase, the structure of the
. Unlike the ordinary smectic SA phase, the structure of the  phase consists of alternating layers of particles oriented parallel to the layer normal and oriented perpendicular to it.
phase consists of alternating layers of particles oriented parallel to the layer normal and oriented perpendicular to it.
Open access
Self-assembly of two-dimensional binary quasicrystals: a possible route to a DNA quasicrystal
View article, Self-assembly of two-dimensional binary quasicrystals: a possible route to a DNA quasicrystal
PDF, Self-assembly of two-dimensional binary quasicrystals: a possible route to a DNA quasicrystal
We use Monte Carlo simulations and free-energy techniques to show that binary solutions of penta- and hexavalent two-dimensional patchy particles can form thermodynamically stable quasicrystals even at very narrow patch widths, provided their patch interactions are chosen in an appropriate way. Such patchy particles can be thought of as a coarse-grained representation of DNA multi-arm 'star' motifs, which can be chosen to bond with one another very specifically by tuning the DNA sequences of the protruding arms. We explore several possible design strategies and conclude that DNA star tiles that are designed to interact with one another in a specific but not overly constrained way could potentially be used to construct soft quasicrystals in experiment. We verify that such star tiles can form stable dodecagonal motifs using oxDNA, a realistic coarse-grained model of DNA.
Open access
The tilt-dependent potential of mean force of a pair of DNA oligomers from all-atom molecular dynamics simulations
View article, The tilt-dependent potential of mean force of a pair of DNA oligomers from all-atom molecular dynamics simulations
PDF, The tilt-dependent potential of mean force of a pair of DNA oligomers from all-atom molecular dynamics simulations
Electrostatic interactions between DNA molecules have been extensively studied experimentally and theoretically, but several aspects (e.g. its role in determining the pitch of the cholesteric DNA phase) still remain unclear. Here, we performed large-scale all-atom molecular dynamics simulations in explicit water and 150 mM sodium chloride, to reconstruct the potential of mean force (PMF) of two DNA oligomers 24 base pairs long as a function of their interaxial angle and intermolecular distance. We find that the potential of mean force is dominated by total DNA charge, and not by the helical geometry of its charged groups. The theory of homogeneously charged cylinders fits well all our simulation data, and the fit yields the optimal value of the total compensated charge on DNA to ≈65% of its total fixed charge (arising from the phosphorous atoms), close to the value expected from Manning's theory of ion condensation. The PMF calculated from our simulations does not show a significant dependence on the handedness of the angle between the two DNA molecules, or its size is on the order of  . Thermal noise for molecules of the studied length seems to mask the effect of detailed helical charge patterns of DNA. The fact that in monovalent salt the effective interaction between two DNA molecules is independent on the handedness of the tilt may suggest that alternative mechanisms are required to understand the cholesteric phase of DNA.
. Thermal noise for molecules of the studied length seems to mask the effect of detailed helical charge patterns of DNA. The fact that in monovalent salt the effective interaction between two DNA molecules is independent on the handedness of the tilt may suggest that alternative mechanisms are required to understand the cholesteric phase of DNA.
Open access
Liquid crystals in micron-scale droplets, shells and fibers
View article, Liquid crystals in micron-scale droplets, shells and fibers
PDF, Liquid crystals in micron-scale droplets, shells and fibers
The extraordinary responsiveness and large diversity of self-assembled structures of liquid crystals are well documented and they have been extensively used in devices like displays. For long, this application route strongly influenced academic research, which frequently focused on the performance of liquid crystals in display-like geometries, typically between flat, rigid substrates of glass or similar solids. Today a new trend is clearly visible, where liquid crystals confined within curved, often soft and flexible, interfaces are in focus. Innovation in microfluidic technology has opened for high-throughput production of liquid crystal droplets or shells with exquisite monodispersity, and modern characterization methods allow detailed analysis of complex director arrangements. The introduction of electrospinning in liquid crystal research has enabled encapsulation in optically transparent polymeric cylinders with very small radius, allowing studies of confinement effects that were not easily accessible before. It also opened the prospect of functionalizing textile fibers with liquid crystals in the core, triggering activities that target wearable devices with true textile form factor for seamless integration in clothing. Together, these developments have brought issues center stage that might previously have been considered esoteric, like the interaction of topological defects on spherical surfaces, saddle-splay curvature-induced spontaneous chiral symmetry breaking, or the non-trivial shape changes of curved liquid crystal elastomers with non-uniform director fields that undergo a phase transition to an isotropic state. The new research thrusts are motivated equally by the intriguing soft matter physics showcased by liquid crystals in these unconventional geometries, and by the many novel application opportunities that arise when we can reproducibly manufacture these systems on a commercial scale. This review attempts to summarize the current understanding of liquid crystals in spherical and cylindrical geometry, the state of the art of producing such samples, as well as the perspectives for innovative applications that have been put forward.
Proofreading of DNA polymerase: a new kinetic model with higher-order terminal effects
View article, Proofreading of DNA polymerase: a new kinetic model with higher-order terminal effects
PDF, Proofreading of DNA polymerase: a new kinetic model with higher-order terminal effects
The fidelity of DNA replication by DNA polymerase (DNAP) has long been an important issue in biology. While numerous experiments have revealed details of the molecular structure and working mechanism of DNAP which consists of both a polymerase site and an exonuclease (proofreading) site, there were quite a few theoretical studies on the fidelity issue. The first model which explicitly considered both sites was proposed in the 1970s and the basic idea was widely accepted by later models. However, all these models did not systematically investigate the dominant factor on DNAP fidelity, i.e. the higher-order terminal effects through which the polymerization pathway and the proofreading pathway coordinate to achieve high fidelity. In this paper, we propose a new and comprehensive kinetic model of DNAP based on some recent experimental observations, which includes previous models as special cases. We present a rigorous and unified treatment of the corresponding steady-state kinetic equations of any-order terminal effects, and derive analytical expressions for fidelity in terms of kinetic parameters under bio-relevant conditions. These expressions offer new insights on how the higher-order terminal effects contribute substantially to the fidelity in an order-by-order way, and also show that the polymerization-and-proofreading mechanism is dominated only by very few key parameters. We then apply these results to calculate the fidelity of some real DNAPs, which are in good agreements with previous intuitive estimates given by experimentalists.
Insight into the electrical properties and chain conformation of spherical polyelectrolyte brushes by dielectric spectroscopy
View article, Insight into the electrical properties and chain conformation of spherical polyelectrolyte brushes by dielectric spectroscopy
PDF, Insight into the electrical properties and chain conformation of spherical polyelectrolyte brushes by dielectric spectroscopy
We report here a dielectric study on three kinds of anionic spherical polyelectrolyte brush (SPBs, consisting of a polystyrene (PS) core and three different poly (acrylic acid) chains grafted onto the core) suspensions over a frequency ranging from 40 Hz to 110 MHz. The relaxation behavior of the SPB suspensions shows significant changes in the brush-layer properties when the mass fraction of SPBs and the pH of the suspensions change. Two definite relaxations related to the interfacial polarization are observed around 100 kHz and 10 MHz. A single-layer spherical-shell model is applied to describe the SPB suspensions wherein the suspended SPB is modeled as a spherical-shell composite particle in which an insulated PS sphere is surrounded by a conducting ion-permeable shell (the polyelectrolyte chain layer). We developed the curve-fitting procedure to analyze the dielectric spectrum in order to obtain the dielectric properties of the components of the SPBs, especially the properties of the polyelectrolyte brush. Based on this method and model, the permittivity and conductivity of the brush layer, ζ potential, etc are calculated. The ordered orientation of the water molecules in the layer leads to an additional electrical dipole moment; increasing pH causes the brush layer to swell. In addition, the repulsive force between the SPB particles are evaluated using the brush-layer thickness, which is obtained by fitting dielectric spectra, combined with relative theoretical formulas. Increasing PH values or SPB concentration would improve the stability of the SPBs dispersion.
Modeling shear-induced particle ordering and deformation in a dense soft particle suspension
View article, Modeling shear-induced particle ordering and deformation in a dense soft particle suspension
PDF, Modeling shear-induced particle ordering and deformation in a dense soft particle suspension
We apply the lattice Boltzmann method and the bead-spring network model of deformable particles (DPs) to study shear-induced particle ordering and deformation and the corresponding rheological behavior for dense DP suspensions confined in a narrow gap under steady external shear. The particle configuration is characterized with small-angle scattering intensity, the real-space 2D local order parameter, and the particle shape factors including deformation, stretching and tilt angles. We investigate how particle ordering and deformation vary with the particle volume fraction ϕ (=0.45–0.65) and the external shear rate characterized with the capillary number Ca (=0.003–0.191). The degree of particle deformation increases mildly with ϕ but significantly with Ca. Under moderate shear rate (Ca = 0.105), the inter-particle structure evolves from string-like ordering to layered hexagonal close packing (HCP) as ϕ increases. A long wavelength particle slithering motion emerges for sufficiently large ϕ. For ϕ = 0.61, the structure maintains layered HCP for Ca = 0.031–0.143 but gradually becomes disordered for larger and smaller Ca. The correlation in particle zigzag movements depends sensitively on ϕ and particle ordering. Layer-by-layer analysis reveals how the non-slippery hard walls affect particle ordering and deformation. The shear-induced reconfiguration of DPs observed in the simulation agrees qualitatively with experimental results of sheared uniform emulsions. The apparent suspension viscosity increases with ϕ but exhibits much weaker dependence compared to hard-sphere suspensions, indicating that particle deformation and unjamming under shear can significantly reduce the viscous stress. Furthermore, the suspension shear-thins, corresponding to increased inter-DP ordering and particle deformation with Ca. This work provides useful insights into the microstructure-rheology relationship of concentrated deformable particle suspensions.
Distinguishing advective and powered motion in self-propelled colloids
View article, Distinguishing advective and powered motion in self-propelled colloids
PDF, Distinguishing advective and powered motion in self-propelled colloids
Self-powered motion in catalytic colloidal particles provides a compelling example of active matter, i.e. systems that engage in single-particle and collective behavior far from equilibrium. The long-time, long-distance behavior of such systems is of particular interest, since it connects their individual micro-scale behavior to macro-scale phenomena. In such analyses, it is important to distinguish motion due to subtle advective effects—which also has long time scales and length scales—from long-timescale phenomena that derive from intrinsically powered motion. Here, we develop a methodology to analyze the statistical properties of the translational and rotational motions of powered colloids to distinguish, for example, active chemotaxis from passive advection by bulk flow.
Physics of chemical processes
Show article list
Statistical analysis of the reduction process of graphene oxide probed by Raman spectroscopy mapping
View article, Statistical analysis of the reduction process of graphene oxide probed by Raman spectroscopy mapping
PDF, Statistical analysis of the reduction process of graphene oxide probed by Raman spectroscopy mapping
We propose a method for monitoring the large-scale homogeneity of the reduction process of graphene oxide. For this purpose, a Raman mapping technique is employed to probe the evolution of the phonon properties of two different graphene oxide (GO) thin films upon controllable thermal reduction. The reduction of GO is reflected by the upshift of the statistical distribution of the relative intensity ratio of the G and D peaks (ID/IG) of the Raman spectra and is consistent with the ratio obtained for chemically reduced GO. In addition, the shifts of the position distributions of the main Raman modes ( ,
,  ) and their cross-correlation with the ID/IG ratio provides evidence of a change of the doping level, demonstrating the influence of reduction processes on GO films.
) and their cross-correlation with the ID/IG ratio provides evidence of a change of the doping level, demonstrating the influence of reduction processes on GO films.
A full understanding of oxygen reduction reaction mechanism on Au(1 1 1) surface
View article, A full understanding of oxygen reduction reaction mechanism on Au(1 1 1) surface
PDF, A full understanding of oxygen reduction reaction mechanism on Au(1 1 1) surface
Oxygen reduction and hydrogen peroxide reduction are technologically important reactions in energy-conversion devices. In this work, a full understanding of oxygen reduction reaction (ORR) mechanism on Au(1 1 1) surface is investigated by density functional theory (DFT) calculations, including the reaction mechanisms of O2 dissociation, OOH dissociation, and H2O2 dissociation. Among these ORR mechanisms on Au(1 1 1), the activation energy of  hydrogenation reaction is much lower than that of
hydrogenation reaction is much lower than that of  dissociation, indicating that
dissociation, indicating that  hydrogenation reaction is more appropriate at the first step than
hydrogenation reaction is more appropriate at the first step than  dissociation. In the following, H2O2 can be formed with the lower activation energy compared with the OOH dissociation reaction, and finally H2O2 could be generated as a detectable product due to the high activation energy of H2O2 dissociation reaction. Furthermore, the potential dependent free energy study suggests that the H2O2 formation is thermodynamically favorable up to 0.4 V on Au(1 1 1), reducing the overpotential for 2e− ORR process. And the elementary step of first H2O formation becomes non-spontaneous at 0.4 V, indicating the difficulty of 4e− reduction pathway. Our DFT calculations show that H2O2 can be generated on Au(1 1 1) and the first electron transfer is the rate determining step. Our results show that gold surface could be used as a good catalyst for small-scale manufacture and on-site production of H2O2.
dissociation. In the following, H2O2 can be formed with the lower activation energy compared with the OOH dissociation reaction, and finally H2O2 could be generated as a detectable product due to the high activation energy of H2O2 dissociation reaction. Furthermore, the potential dependent free energy study suggests that the H2O2 formation is thermodynamically favorable up to 0.4 V on Au(1 1 1), reducing the overpotential for 2e− ORR process. And the elementary step of first H2O formation becomes non-spontaneous at 0.4 V, indicating the difficulty of 4e− reduction pathway. Our DFT calculations show that H2O2 can be generated on Au(1 1 1) and the first electron transfer is the rate determining step. Our results show that gold surface could be used as a good catalyst for small-scale manufacture and on-site production of H2O2.
MoS2-supported gold nanoparticle for CO hydrogenation
View article, MoS2-supported gold nanoparticle for CO hydrogenation
PDF, MoS2-supported gold nanoparticle for CO hydrogenation
Employing dispersion-corrected density functional theory, we examine the geometry, electronic structure, and reactivity of 13-atom Au nanoparticle supported on defect-laden single-layer MoS2. The planar structure of Au13 favored in isolated phase, transforms into the three-dimensional structure when supported on MoS2. We find that charge is transferred from MoS2 to Au13, and that the electron density is also distributed away from the Au13/MoS2 interfacial region—making Au sites away from the interface catalytically active. Owing to effect of the support, the Au d states become narrower, and the frontier states appear close to the Fermi level. Consequently, in contrast to the reactivity of Au13/TiO2 toward methanol decomposition, Au13/MoS2 offers excellent activity toward methanol synthesis, as demonstrated here, via CO hydrogenation.
Nanostructures and nanoelectronics
Show article list
Open access
Two-probe STM experiments at the atomic level
View article, Two-probe STM experiments at the atomic level
PDF, Two-probe STM experiments at the atomic level
Direct characterization of planar atomic or molecular scale devices and circuits on a supporting surface by multi-probe measurements requires unprecedented stability of single atom contacts and manipulation of scanning probes over large, nanometer scale area with atomic precision. In this work, we describe the full methodology behind atomically defined two-probe scanning tunneling microscopy (STM) experiments performed on a model system: dangling bond dimer wire supported on a hydrogenated germanium (0 0 1) surface. We show that 70 nm long atomic wire can be simultaneously approached by two independent STM scanners with exact probe to probe distance reaching down to 30 nm. This allows direct wire characterization by two-probe I–V characteristics at distances below 50 nm. Our technical results presented in this work open a new area for multi-probe research, which can be now performed with precision so far accessible only by single-probe scanning probe microscopy (SPM) experiments.
Edge states of hydrogen terminated monolayer materials: silicene, germanene and stanene ribbons
View article, Edge states of hydrogen terminated monolayer materials: silicene, germanene and stanene ribbons
PDF, Edge states of hydrogen terminated monolayer materials: silicene, germanene and stanene ribbons
We investigate the energy dispersion of the edge states in zigzag silicene, germanene and stanene nanoribbons with and without hydrogen termination based on a multi-orbital tight-binding model. Since the low buckled structures are crucial for these materials, both the π and σ orbitals have a strong influence on the edge states, different from the case for graphene nanoribbons. The obtained dispersion of helical edge states is nonlinear, similar to that obtained by first-principles calculations. On the other hand, the dispersion derived from the single-orbital tight-binding model is always linear. Therefore, we find that the non-linearity comes from the multi-orbital effects, and accurate results cannot be obtained by the single-orbital model but can be obtained by the multi-orbital tight-binding model. We show that the multi-orbital model is essential for correctly understanding the dispersion of the edge states in tetragen nanoribbons with a low buckled geometry.
Optical response of a line node semimetal
View article, Optical response of a line node semimetal
PDF, Optical response of a line node semimetal
We calculate the AC optical response of a line node semimetal with emphasis on characteristic behaviours which can be used to distinguish them from point node materials such as Dirac and Weyl semimetals. The interband optical background at zero temperature displays a flat region at small photon energies ( ) analogue to the universal background seen in graphene. However, in contrast to graphene, the height of the constant region is not universal but depends inversely on the Fermi velocity of the charge carriers and directly on the radius (b) in momentum space of the nodal circle. The parameter b is a defining energy scale and determines the range of photon energy over which the flat response persists. At high energies
) analogue to the universal background seen in graphene. However, in contrast to graphene, the height of the constant region is not universal but depends inversely on the Fermi velocity of the charge carriers and directly on the radius (b) in momentum space of the nodal circle. The parameter b is a defining energy scale and determines the range of photon energy over which the flat response persists. At high energies  , the interband response becomes linear in
, the interband response becomes linear in  in agreement with the case for 3D-Dirac fermions with point node. The optical spectral weight contained in the interband or Drude conductivity shows the same two distinct regimes. At low temperature (T) (chemical potential (μ)), it rises linearly with
in agreement with the case for 3D-Dirac fermions with point node. The optical spectral weight contained in the interband or Drude conductivity shows the same two distinct regimes. At low temperature (T) (chemical potential (μ)), it rises linearly with  and is proportional to b. At high temperature,
and is proportional to b. At high temperature,  , a
, a  law is obtained, which is independent of b. At T = 0, the Lorentz number takes on the conventional value
law is obtained, which is independent of b. At T = 0, the Lorentz number takes on the conventional value  for all values of μ. It increases with increasing temperature to reach a first plateau of 2.4Lo provided
for all values of μ. It increases with increasing temperature to reach a first plateau of 2.4Lo provided  but
but  . At high temperature, T > b, a second plateau of height 4.2Lo emerges. The first plateau is characteristic of 2D-Dirac while the second corresponds to 3D-Dirac. The thermopower as a function of temperature also shows an evolution from a 2D to 3D behaviour.
. At high temperature, T > b, a second plateau of height 4.2Lo emerges. The first plateau is characteristic of 2D-Dirac while the second corresponds to 3D-Dirac. The thermopower as a function of temperature also shows an evolution from a 2D to 3D behaviour.
Low-energy theory for strained graphene: an approach up to second-order in the strain tensor
View article, Low-energy theory for strained graphene: an approach up to second-order in the strain tensor
PDF, Low-energy theory for strained graphene: an approach up to second-order in the strain tensor
An analytical study of low-energy electronic excited states in uniformly strained graphene is carried out up to second-order in the strain tensor. We report a new effective Dirac Hamiltonian with an anisotropic Fermi velocity tensor, which reveals the graphene trigonal symmetry being absent in first-order low-energy theories. In particular, we demonstrate the dependence of the Dirac-cone elliptical deformation on the stretching direction with respect to graphene lattice orientation. We further analytically calculate the optical conductivity tensor of strained graphene and its transmittance for a linearly polarized light with normal incidence. Finally, the obtained analytical expression of the Dirac point shift allows a better determination and understanding of pseudomagnetic fields induced by nonuniform strains.
Hidden symmetries in N-layer dielectric stacks
View article, Hidden symmetries in N-layer dielectric stacks
PDF, Hidden symmetries in N-layer dielectric stacks
The optical properties of a multilayer system with arbitrary N layers of dielectric media are investigated. Each layer is one of two dielectric media, with a thickness one-quarter the wavelength of light in that medium, corresponding to a central frequency f0. Using the transfer matrix method, the transmittance T is calculated for all possible 2N sequences for small N. Unexpectedly, it is found that instead of 2N different values of T at f0 (T0), there are only  discrete values of T0, for even N, and (N + 1) for odd N. We explain this high degeneracy in T0 values by finding symmetry operations on the sequences that do not change T0. Analytical formulae were derived for the T0 values and their degeneracies as functions of N and an integer parameter for each sequence we call 'charge'. Additionally, the bandwidth at f0 and filter response of the transmission spectra are investigated, revealing asymptotic behavior at large N.
discrete values of T0, for even N, and (N + 1) for odd N. We explain this high degeneracy in T0 values by finding symmetry operations on the sequences that do not change T0. Analytical formulae were derived for the T0 values and their degeneracies as functions of N and an integer parameter for each sequence we call 'charge'. Additionally, the bandwidth at f0 and filter response of the transmission spectra are investigated, revealing asymptotic behavior at large N.
Quantum transport across van der Waals domain walls in bilayer graphene
View article, Quantum transport across van der Waals domain walls in bilayer graphene
PDF, Quantum transport across van der Waals domain walls in bilayer graphene
Bilayer graphene can exhibit deformations such that the two graphene sheets are locally detached from each other resulting in a structure consisting of domains with different van der Waals inter-layer coupling. Here we investigate how the presence of these domains affects the transport properties of bilayer graphene. We derive analytical expressions for the transmission probability, and the corresponding conductance, across walls separating different inter-layer coupling domains. We find that the transmission can exhibit a valley-dependent layer asymmetry and that the domain walls have a considerable effect on the chiral tunnelling properties of the charge carriers. We show that transport measurements allow one to obtain the strength with which the two layers are coupled. We perform numerical calculations for systems with two domain walls and find that the availability of multiple transport channels in bilayer graphene significantly modifies the conductance dependence on inter-layer potential asymmetry.
Structure, dynamics and phase transitions
Show article list
Open access
On the persistence of polar domains in ultrathin ferroelectric capacitors
View article, On the persistence of polar domains in ultrathin ferroelectric capacitors
PDF, On the persistence of polar domains in ultrathin ferroelectric capacitors
The instability of ferroelectric ordering in ultra-thin films is one of the most important fundamental issues pertaining realization of a number of electronic devices with enhanced functionality, such as ferroelectric and multiferroic tunnel junctions or ferroelectric field effect transistors. In this paper, we investigate the polarization state of archetypal ultrathin (several nanometres) ferroelectric heterostructures: epitaxial single-crystalline BaTiO3 films sandwiched between the most habitual perovskite electrodes, SrRuO3, on top of the most used perovskite substrate, SrTiO3. We use a combination of piezoresponse force microscopy, dielectric measurements and structural characterization to provide conclusive evidence for the ferroelectric nature of the relaxed polarization state in ultrathin BaTiO3 capacitors. We show that even the high screening efficiency of SrRuO3 electrodes is still insufficient to stabilize polarization in SrRuO3/BaTiO3/SrRuO3 heterostructures at room temperature. We identify the key role of domain wall motion in determining the macroscopic electrical properties of ultrathin capacitors and discuss their dielectric response in the light of the recent interest in negative capacitance behaviour.
Modeling hybrid perovskites by molecular dynamics
View article, Modeling hybrid perovskites by molecular dynamics
PDF, Modeling hybrid perovskites by molecular dynamics
The topical review describes the recent progress in the modeling of hybrid perovskites by molecular dynamics simulations. Hybrid perovskites and in particular methylammonium lead halide (MAPI) have a tremendous technological relevance representing the fastest-advancing solar material to date. They also represent the paradigm of an organic–inorganic crystalline material with some conceptual peculiarities: an inorganic semiconductor for what concerns the electronic and absorption properties with a hybrid and solution processable organic–inorganic body. After briefly explaining the basic concepts of ab initio and classical molecular dynamics, the model potential recently developed for hybrid perovskites is described together with its physical motivation as a simple ionic model able to reproduce the main dynamical properties of the material. Advantages and limits of the two strategies (either ab initio or classical) are discussed in comparison with the time and length scales (from pico to microsecond scale) necessary to comprehensively study the relevant properties of hybrid perovskites from molecular reorientations to electrocaloric effects. The state-of-the-art of the molecular dynamics modeling of hybrid perovskites is reviewed by focusing on a selection of showcase applications of methylammonium lead halide: molecular cations disorder; temperature evolution of vibrations; thermally activated defects diffusion; thermal transport. We finally discuss the perspectives in the modeling of hybrid perovskites by molecular dynamics.
On entropy change measurements around first order phase transitions in caloric materials
View article, On entropy change measurements around first order phase transitions in caloric materials
PDF, On entropy change measurements around first order phase transitions in caloric materials
In this work we discuss the measurement protocols for indirect determination of the isothermal entropy change associated with first order phase transitions in caloric materials. The magneto-structural phase transitions giving rise to giant magnetocaloric effects in Cu-doped MnAs and FeRh are used as case studies to exemplify how badly designed protocols may affect isothermal measurements and lead to incorrect entropy change estimations. Isothermal measurement protocols which allow correct assessment of the entropy change around first order phase transitions in both direct and inverse cases are presented.
High-pressure structural, elastic, and thermodynamic properties of zircon-type HoPO4 and TmPO4
View article, High-pressure structural, elastic, and thermodynamic properties of zircon-type HoPO4 and TmPO4
PDF, High-pressure structural, elastic, and thermodynamic properties of zircon-type HoPO4 and TmPO4
Zircon-type holmium phosphate (HoPO4) and thulium phosphate (TmPO4) have been studied by single-crystal x-ray diffraction and ab initio calculations. We report on the influence of pressure on the crystal structure, and on the elastic and thermodynamic properties. The equation of state for both compounds is accurately determined. We have also obtained information on the polyhedral compressibility which is used to explain the anisotropic axial compressibility and the bulk compressibility. Both compounds are ductile and more resistive to volume compression than to shear deformation at all pressures. Furthermore, the elastic anisotropy is enhanced upon compression. Finally, the calculations indicate that the possible causes that make the zircon structure unstable are mechanical instabilities and the softening of a silent B1u mode.
Pressure-dependent semiconductor to semimetal and Lifshitz transitions in 2H-MoTe2: Raman and first-principles studies
View article, Pressure-dependent semiconductor to semimetal and Lifshitz transitions in 2H-MoTe2: Raman and first-principles studies
PDF, Pressure-dependent semiconductor to semimetal and Lifshitz transitions in 2H-MoTe2: Raman and first-principles studies
High pressure Raman spectroscopy of bulk 2H-MoTe2 up to ∼29 GPa is shown to reveal two phase transitions (at ∼6 and 16.5 GPa), which are analyzed using first-principles density functional theoretical calculations. The transition at 6 GPa is marked by changes in the pressure coefficients of A1g and  Raman mode frequencies as well as in their relative intensity. Our calculations show that this is an isostructural semiconductor to a semimetal transition. The transition at ∼16.5 GPa is identified with the changes in linewidths of the Raman modes as well as in the pressure coefficients of their frequencies. Our theoretical analysis clearly shows that the structure remains the same up to 30 GPa. However, the topology of the Fermi-surface evolves as a function of pressure, and abrupt appearance of electron and hole pockets at
Raman mode frequencies as well as in their relative intensity. Our calculations show that this is an isostructural semiconductor to a semimetal transition. The transition at ∼16.5 GPa is identified with the changes in linewidths of the Raman modes as well as in the pressure coefficients of their frequencies. Our theoretical analysis clearly shows that the structure remains the same up to 30 GPa. However, the topology of the Fermi-surface evolves as a function of pressure, and abrupt appearance of electron and hole pockets at  GPa marks a Lifshitz transition.
GPa marks a Lifshitz transition.
Electronic structure
Show article list
Open access
Theoretical modeling of charge trapping in crystalline and amorphous Al2O3
View article, Theoretical modeling of charge trapping in crystalline and amorphous Al2O3
PDF, Theoretical modeling of charge trapping in crystalline and amorphous Al2O3
The characteristics of intrinsic electron and hole trapping in crystalline and amorphous Al2O3 have been studied using density functional theory (DFT). Special attention was paid to enforcing the piece-wise linearity of the total energy with respect to electron number through the use of a range separated, hybrid functional PBE0-TC-LRC (Guidon et al 2009 J. Chem. Theory Comput. 5 3010) in order to accurately model the behaviour of localized states. The tuned functional is shown to reproduce the geometric and electronic structures of the perfect crystal as well as the spectroscopic characteristics of MgAl hole centre in corundum α-Al2O3. An ensemble of ten amorphous Al2O3 structures was generated using classical molecular dynamics and a melt and quench method and their structural characteristics compared with the experimental data. The electronic structure of amorphous systems was characterized using the inverse participation ratio method. Electrons and holes were then introduced into both crystalline and amorphous alumina structures and their properties calculated. Holes are shown to trap spontaneously in both crystalline and amorphous alumina. In the crystalline phase they localize on single O ion with the trapping energy of 0.38 eV. In amorphous phase, holes localize on two nearest neighbour oxygen sites with an average trapping energy of 1.26 eV, with hole trapping sites separated on average by about 8.0 Å. No electron trapping is observed in the material. Our results suggest that trapping of positive charge can be much more severe and stable in amorphous alumina rather than in crystalline samples.
Terahertz plasmonic excitations in Bi2Se3 topological insulator
View article, Terahertz plasmonic excitations in Bi2Se3 topological insulator
PDF, Terahertz plasmonic excitations in Bi2Se3 topological insulator
After the discovery of Dirac electrons in condensed matter physics, more specifically in graphene and its derivatives, their potentialities in the fields of plasmonics and photonics have been readily recognized, leading to a plethora of applications in active and tunable optical devices. Massless Dirac carriers have been further found in three-dimensional topological insulators. These exotic quantum systems have an insulating gap in the bulk and intrinsic Dirac metallic states at any surface, sustaining not only single-particle excitations but also plasmonic collective modes. In this paper we will review the plasmon excitations in different microstructures patterned on Bi2Se3 topological insulator thin films as measured by terahertz spectroscopy. We discuss the dependence of the plasmon absorption versus the microstructure shape, wavevector, and magnetic field. Finally we will discuss the topological protection of both the Dirac single-particle and plasmon excitations.
Intrinsic valley polarization of magnetic VSe2 monolayers
View article, Intrinsic valley polarization of magnetic VSe2 monolayers
PDF, Intrinsic valley polarization of magnetic VSe2 monolayers
Intrinsic valley polarization can be obtained in VSe2 monolayers with broken inversion symmetry and time reversal symmetry. First-principles investigations reveal that the magnitude of the valley splitting in magnetic VSe2 induced by spin–orbit coupling reaches as high as 78.2 meV and can be linearly tuned by biaxial strain. Besides conventional polarized light, hole doping or illumination with light of proper frequency can offer effective routes to realize valley polarization. Moreover, spin–orbit coupling in monolayer VSe2 breaks not only the valley degeneracy but also the three-fold rotational symmetry in band structure. The intrinsic and tunable valley splitting and the breaking of optical isotropy bring additional benefits to valleytronic and optoelectronic applications.
Optical and fundamental band gaps disparity in transparent conducting oxides: new findings for the  and
and  systems
systems
and systems
View article, Optical and fundamental band gaps disparity in transparent conducting oxides: new findings for the and systems
PDF, Optical and fundamental band gaps disparity in transparent conducting oxides: new findings for the and systems
The optical band gap, extracted from absorption measurements, defines the figure of merit for transparent conducting oxides (TCOs). In many oxides, such as  or
or  , inversion symmetry introduces a selection rule that blocks transitions from the valence-band maximum to the conduction-band minimum. This raises the absorption threshold and enlarges the optical gap relative to the fundamental band gap. Here, we present density-functional computations identifying two optical gaps, either of which can be detected, depending on the optical light intensity. Under strong illumination, weak transitions from
, inversion symmetry introduces a selection rule that blocks transitions from the valence-band maximum to the conduction-band minimum. This raises the absorption threshold and enlarges the optical gap relative to the fundamental band gap. Here, we present density-functional computations identifying two optical gaps, either of which can be detected, depending on the optical light intensity. Under strong illumination, weak transitions from  -points near the valence-band maximum contribute significantly to the absorption spectrum and define an optical gap matching the fundamental gap. Low optical intensities by contrast give prominence to the large optical gap determined by the selection rule. While experimental conditions have favored observation of the former optical gap in
-points near the valence-band maximum contribute significantly to the absorption spectrum and define an optical gap matching the fundamental gap. Low optical intensities by contrast give prominence to the large optical gap determined by the selection rule. While experimental conditions have favored observation of the former optical gap in  , in contrast, absorption measurements in
, in contrast, absorption measurements in  have focused on the latter. Our findings explain the disparity between the optical and fundamental gaps in bixbyite
have focused on the latter. Our findings explain the disparity between the optical and fundamental gaps in bixbyite  and predict that, measured under low illumination, the optical gap for rutile
and predict that, measured under low illumination, the optical gap for rutile  will increase, from 3.60 eV to 4.34 eV.
will increase, from 3.60 eV to 4.34 eV.
Compressive strain induced enhancement in thermoelectric-power-factor in monolayer MoS2 nanosheet
View article, Compressive strain induced enhancement in thermoelectric-power-factor in monolayer MoS2 nanosheet
PDF, Compressive strain induced enhancement in thermoelectric-power-factor in monolayer MoS2 nanosheet
Strain and temperature induced tunability in the thermoelectric properties in monolayer MoS2 (ML-MoS2) has been demonstrated using density functional theory coupled to semi-classical Boltzmann transport theory. Compressive strain, in general and uniaxial compressive strain (along the zig-zag direction), in particular, is found to be most effective in enhancing the thermoelectric power factor, owing to the higher electronic mobility and its sensitivity to lattice compression along this direction. Variation in the Seebeck coefficient and electronic band gap with strain is found to follow the Goldsmid–Sharp relation. n-type doping is found to raise the relaxation time-scaled thermoelectric power factor higher than p-type doping and this divide widens with increasing temperature. The relaxation time-scaled thermoelectric power factor in optimally n-doped ML-MoS2 is found to undergo maximal enhancement under the application of 3% uniaxial compressive strain along the zig-zag direction, when both the (direct) electronic band gap and the Seebeck coefficient reach their maximum, while the electron mobility drops down drastically from 73.08 to 44.15 cm2 V−1 s−1. Such strain sensitive thermoelectric responses in ML-MoS2 could open doorways for a variety of applications in emerging areas in 2D-thermoelectrics, such as on-chip thermoelectric power generation and waste thermal energy harvesting.
Existence of semi-Dirac cones and symmetry of two-dimensional materials
View article, Existence of semi-Dirac cones and symmetry of two-dimensional materials
PDF, Existence of semi-Dirac cones and symmetry of two-dimensional materials
There have been growing efforts to find new two-dimensional (2D) materials with anisotropic properties due to their potential applications in electronics. Although in such a search, a symmetry based analysis can be useful, it has not been reported so far. Using group theory we have found sufficient conditions for the existence of a linear dispersion in one direction and quadratic one in perpendicular direction, in the vicinity of points of symmetry in the Brillouin zone (BZ) of any non-magnetic, 2D material with negligible spin–orbit coupling. We have formulated a set of symmetry conditions that lead to the semi-Dirac dispersion and analyzed all possible symmetries of 2D materials. In four, out of all eighty symmetry groups, combined time-reversal and crystal symmetry leads, at given points in the BZ, to such dispersion. The result is valid irrespectively of strength of electronic correlations in the system, model used to calculate the band structure, or the actual crystal structure that realizes given groups. We have illustrated our findings using a tight-binding example.
Open access
A new approach towards spintronics–spintronics with no magnets
View article, A new approach towards spintronics–spintronics with no magnets
PDF, A new approach towards spintronics–spintronics with no magnets
We review a recently discovered phenomenon, the chiral induced spin selectivity (CISS) effect, that can enable a new technology for the injection of spin polarized current without the need for a permanent magnetic layer. The effect occurs in chiral molecules and systems without parity symmetry, i.e. systems that do not have inversion symmetry. The theoretical foundations for the effect are presented first and then followed by several examples of spin-valves that are based on chiral systems. The CISS-based spin valves introduce the possibility to inject spin current without the use of a permanent magnet and to achieve relatively large magnetoresistance at room temperature.
Correlated electrons systems
Show article list
Topological phases of the kicked Harper–Kitaev model with ultracold atoms
View article, Topological phases of the kicked Harper–Kitaev model with ultracold atoms
PDF, Topological phases of the kicked Harper–Kitaev model with ultracold atoms
We propose using ultracold atoms trapped in a one-dimensional periodically driven optical lattice to realize the Harper–Kitaev model, where the on-site energies are periodically kicked. Such a system provides a natural platform to study both Chern insulators and Majorana fermions. Based on calculating the quasienergy spectra, we find that both Floquet Majorana modes and Hall chiral edge modes could appear at the sample boundary in the gaps between the quasienergy bands. We also study the competition of topological superconductor and Chern insulator states in the model. We calculate the 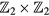 index and Floquet Chern number to characterize the above two different topological states, including the topological phase transitions in the kicked Harper–Kitaev model with the increase in the strength of the kick.
index and Floquet Chern number to characterize the above two different topological states, including the topological phase transitions in the kicked Harper–Kitaev model with the increase in the strength of the kick.
Open access
Symmetry analysis of transport properties in helical superconductor junctions
View article, Symmetry analysis of transport properties in helical superconductor junctions
PDF, Symmetry analysis of transport properties in helical superconductor junctions
We study the discrete symmetries satisfied by helical p-wave superconductors with the d-vectors  or
or  and the transformations brought by symmetry operations to ferromagnet and spin-singlet superconductors, which show intimate associations with the transport properties in heterojunctions, including helical superconductors. In particular, the partial symmetries of the Hamiltonian under spin-rotation and gauge-rotation operations are responsible for the novel invariances of the conductance in tunnel junctions and the new selection rules for the lowest current and peculiar phase diagrams in Josephson junctions, which were reported recently. The symmetries of constructed free energies for Josephson junctions are also analyzed, and are consistent with the results from the Hamiltonian.
and the transformations brought by symmetry operations to ferromagnet and spin-singlet superconductors, which show intimate associations with the transport properties in heterojunctions, including helical superconductors. In particular, the partial symmetries of the Hamiltonian under spin-rotation and gauge-rotation operations are responsible for the novel invariances of the conductance in tunnel junctions and the new selection rules for the lowest current and peculiar phase diagrams in Josephson junctions, which were reported recently. The symmetries of constructed free energies for Josephson junctions are also analyzed, and are consistent with the results from the Hamiltonian.
Mesoscopic quantum effects in a bad metal, hydrogen-doped vanadium dioxide
View article, Mesoscopic quantum effects in a bad metal, hydrogen-doped vanadium dioxide
PDF, Mesoscopic quantum effects in a bad metal, hydrogen-doped vanadium dioxide
The standard treatment of quantum corrections to semiclassical electronic conduction assumes that charge carriers propagate many wavelengths between scattering events, and succeeds in explaining multiple phenomena (weak localization magnetoresistance (WLMR), universal conductance fluctuations, Aharonov–Bohm oscillations) observed in polycrystalline metals and doped semiconductors in various dimensionalities. We report apparent WLMR and conductance fluctuations in HxVO2, a poor metal (in violation of the Mott–Ioffe–Regel limit) stabilized by the suppression of the VO2 metal-insulator transition through atomic hydrogen doping. Epitaxial thin films, single-crystal nanobeams, and nanosheets show similar phenomenology, though the details of the apparent WLMR seem to depend on the combined effects of the strain environment and presumed doping level. Self-consistent quantitative analysis of the WLMR is challenging given this and the high resistivity of the material, since the quantitative expressions for WLMR are derived assuming good metallicity. These observations raise the issue of how to assess and analyze mesoscopic quantum effects in poor metals.
On the performance of natural orbital functional approximations in the Hubbard model
View article, On the performance of natural orbital functional approximations in the Hubbard model
PDF, On the performance of natural orbital functional approximations in the Hubbard model
Strongly correlated materials are now under intense development, and natural orbital functional (NOF) methods seem to be able to capture the physics of these systems. We present a benchmark based on the Hubbard model for a class of commonly used NOF approximations (also known as reduced density matrix functional approximations). Our findings highlight the importance of imposing ensemble N-representability conditions in order to obtain consistent results in systems with either weak or strong electronic correlation, such as the Hubbard system with a varying two-particle interaction parameter. Based on the accuracy of the results obtained using PNOF7, which retrieves a large amount of the total strong nondynamic correlation, the Hubbard model points out that N-representability gives solid foundations for NOF development.
Anisotropic superconducting property studies of single crystal PbTaSe2
View article, Anisotropic superconducting property studies of single crystal PbTaSe2
PDF, Anisotropic superconducting property studies of single crystal PbTaSe2
The anisotropic superconducting properties of PbTaSe2 single crystal is reported. Superconductivity with Tc = 3.83 ± 0.02 K has been characterized fully with electrical resistivity ρ(T), magnetic susceptibility χ(T), and specific heat Cp(T) measurements using single crystal samples. The superconductivity is type-II with lower critical field Hc1 and upper critical field Hc2 of 65 and 450 Oe (H⊥ to the ab-plane), 140 and 1500 Oe (H|| to the ab-plane), respectively. These results indicate that the superconductivity of PbTaSe2 is anisotropic. The superconducting anisotropy, electron–phonon coupling λep, superconducting energy gap Δ0, and the specific heat jump ΔC/γTc at Tc confirms that PbTaSe2 can be categorized as a bulk superconductor.
Unconventional superconductivity in generalized Hubbard model: role of electron–hole symmetry breaking terms
View article, Unconventional superconductivity in generalized Hubbard model: role of electron–hole symmetry breaking terms
PDF, Unconventional superconductivity in generalized Hubbard model: role of electron–hole symmetry breaking terms
We investigate the effect of the electron–hole (e–h) symmetry breaking on d-wave superconductivity induced by non-local effects of correlations in the generalized Hubbard model. The symmetry breaking is introduced in a two-fold manner: by the next-to-nearest neighbor hopping of electrons and by the charge-bond interaction—the off-diagonal term of the Coulomb potential. Both terms lead to a pronounced asymmetry of the superconducting order parameter. The next-to-nearest neighbor hopping enhances superconductivity for h-doping, while diminishes it for e-doping. The charge-bond interaction alone leads to the opposite effect and, additionally, to the kinetic-energy gain upon condensation in the underdoped regime. With both terms included, with similar amplitudes, the height of the superconducting dome and the critical doping remain in favor of h-doping. The influence of the charge-bond interaction on deviations from  symmetry of the shape of the gap at the Fermi surface in the momentum space is briefly discussed.
symmetry of the shape of the gap at the Fermi surface in the momentum space is briefly discussed.
Physics of materials
Show article list
Open access
Superconducting properties of the s±-wave state: Fe-based superconductors
View article, Superconducting properties of the s±-wave state: Fe-based superconductors
PDF, Superconducting properties of the s±-wave state: Fe-based superconductors
Although the pairing mechanism of Fe-based superconductors (FeSCs) has not yet been settled with consensus with regard to the pairing symmetry and the superconducting (SC) gap function, the vast majority of experiments support the existence of spin-singlet sign-changing s-wave SC gaps on multi-bands ( -wave state). This multi-band
-wave state). This multi-band  -wave state is a very unique gap state per se and displays numerous unexpected novel SC properties, such as a strong reduction of the coherence peak, non-trivial impurity effects, nodal-gap-like nuclear magnetic resonance signals, various Volovik effects in the specific heat (SH) and thermal conductivity, and anomalous scaling behaviors with a SH jump and condensation energy versus Tc, etc. In particular, many of these non-trivial SC properties can easily be mistaken as evidence for a nodal-gap state such as a d-wave gap. In this review, we provide detailed explanations of the theoretical principles for the various non-trivial SC properties of the
-wave state is a very unique gap state per se and displays numerous unexpected novel SC properties, such as a strong reduction of the coherence peak, non-trivial impurity effects, nodal-gap-like nuclear magnetic resonance signals, various Volovik effects in the specific heat (SH) and thermal conductivity, and anomalous scaling behaviors with a SH jump and condensation energy versus Tc, etc. In particular, many of these non-trivial SC properties can easily be mistaken as evidence for a nodal-gap state such as a d-wave gap. In this review, we provide detailed explanations of the theoretical principles for the various non-trivial SC properties of the  -wave pairing state, and then critically compare the theoretical predictions with experiments on FeSCs. This will provide a pedagogical overview of to what extent we can coherently understand the wide range of different experiments on FeSCs within the
-wave pairing state, and then critically compare the theoretical predictions with experiments on FeSCs. This will provide a pedagogical overview of to what extent we can coherently understand the wide range of different experiments on FeSCs within the  -wave gap model.
-wave gap model.
Pressure-induced superconductivity in the giant Rashba system BiTeI
View article, Pressure-induced superconductivity in the giant Rashba system BiTeI
PDF, Pressure-induced superconductivity in the giant Rashba system BiTeI
At ambient pressure, BiTeI exhibits a giant Rashba splitting of the bulk electronic bands. At low pressures, BiTeI undergoes a transition from trivial insulator to topological insulator. At still higher pressures, two structural transitions are known to occur. We have carried out a series of electrical resistivity and AC magnetic susceptibility measurements on BiTeI at pressure up to ∼40 GPa in an effort to characterize the properties of the high-pressure phases. A previous calculation found that the high-pressure orthorhombic P4/nmm structure BiTeI is a metal. We find that this structure is superconducting with Tc values as high as 6 K. AC magnetic susceptibility measurements support the bulk nature of the superconductivity. Using electronic structure and phonon calculations, we compute Tc and find that our data is consistent with phonon-mediated superconductivity.
Quantum-trajectory analysis for charge transfer in solid materials induced by strong laser fields
View article, Quantum-trajectory analysis for charge transfer in solid materials induced by strong laser fields
PDF, Quantum-trajectory analysis for charge transfer in solid materials induced by strong laser fields
We investigate the dependence of charge transfer on the intensity of driving laser field when SiO2 crystal is irradiated by an 800 nm laser. It is surprising that the direction of charge transfer undergoes a sudden reversal when the driving laser intensity exceeds critical values with different carrier–envelope phases. By applying quantum-trajectory analysis, we find that the Bloch oscillation plays an important role in charge transfer in solids. Also, we study the interaction of a strong laser with gallium nitride (GaN), which is widely used in optoelectronics. A pump–probe scheme is applied to control the quantum trajectories of the electrons in the conduction band. The signal of charge transfer is controlled successfully by means of a theoretically proposed approach.
Thermal transport in monolayer InSe
Two-dimensional InSe, a recently synthesized semiconductor having a moderate band gap, has gained attention due to its ultra high mobility and high photo-responsivity. In this work, we calculate the lattice thermal conductivity (κ) of monolayer InSe by solving the phonon Boltzmann transport equation (BTE) with first-principles calculated inter atomic force constants. κ of monolayer InSe is isotropic and found to be around 27.6 W m  at room temperature along the in-plane direction. The size dependence of κ shows the size effect can persist up to 20 μm. Further, κ can be reduced to half by tuning the sample size to 300 nm. This low value suggests that κ might be a limiting factor for emerging nanoelectronic applications of monolayer InSe.
at room temperature along the in-plane direction. The size dependence of κ shows the size effect can persist up to 20 μm. Further, κ can be reduced to half by tuning the sample size to 300 nm. This low value suggests that κ might be a limiting factor for emerging nanoelectronic applications of monolayer InSe.
Topological phase transition and evolution of edge states in In-rich InGaN/GaN quantum wells under hydrostatic pressure
View article, Topological phase transition and evolution of edge states in In-rich InGaN/GaN quantum wells under hydrostatic pressure
PDF, Topological phase transition and evolution of edge states in In-rich InGaN/GaN quantum wells under hydrostatic pressure
Combining the k · p method with the third-order elasticity theory, we perform a theoretical study of the pressure-induced topological phase transition and the pressure evolution of topologically protected edge states in InN/GaN and In-rich InGaN/GaN quantum wells. We show that for a certain range of the quantum well parameters, thanks to a negative band gap pressure coefficient, it is possible to continuously drive the system from the normal insulator state through the topological insulator into the semimetal phase. The critical pressure for the topological phase transition depends not only on the quantum well thickness but also on the width of the Hall bar, which determines the coupling between the edge states localized at the opposite edges. We also find that in narrow Hall bar structures, near the topological phase transition, a significant Rashba-type spin splitting of the lower and upper branches of the edge state dispersion curve appears. This effect originates from the lack of the mirror symmetry of the quantum well potential caused by the built-in electric field, and can be suppressed by increasing the Hall bar width. When the pressure increases, the energy dispersion of the edge states becomes more parabolic-like and the spin splitting decreases. A further increase of pressure leads to the transition to a semimetal phase, which occurs due to the closure of the indirect 2D bulk band gap. The difference between the critical pressure at which the system becomes semimetallic, and the pressure for the topological phase transition, correlates with the variation of the pressure coefficient of the band gap in the normal insulator state.
Open access
Structure of rare-earth chalcogenide glasses by neutron and x-ray diffraction
View article, Structure of rare-earth chalcogenide glasses by neutron and x-ray diffraction
PDF, Structure of rare-earth chalcogenide glasses by neutron and x-ray diffraction
The method of neutron diffraction with isomorphic substitution was used to measure the structure of the rare-earth chalcogenide glasses  (Ga2X3)0.33(GeX2)0.60 with
(Ga2X3)0.33(GeX2)0.60 with  or Ce and
or Ce and  or Se. X-ray diffraction was also used to measure the structure of the sulphide glass. The results are consistent with networks that are built from GeX4 and GaX4 tetrahedra, and give R–S and R–Se coordination numbers of 8.0(2) and 8.5(4), respectively. The minimum nearest-neighbour R–R distance associated with rare-earth clustering is discussed.
or Se. X-ray diffraction was also used to measure the structure of the sulphide glass. The results are consistent with networks that are built from GeX4 and GaX4 tetrahedra, and give R–S and R–Se coordination numbers of 8.0(2) and 8.5(4), respectively. The minimum nearest-neighbour R–R distance associated with rare-earth clustering is discussed.
Open access
Diffusion and aggregation of oxygen vacancies in amorphous silica
View article, Diffusion and aggregation of oxygen vacancies in amorphous silica
PDF, Diffusion and aggregation of oxygen vacancies in amorphous silica
Using density functional theory (DFT) calculations, we investigated oxygen vacancy diffusion and aggregation in relation to dielectric breakdown in amorphous silicon dioxide (a-SiO2). Our calculations indicate the existence of favourable sites for the formation of vacancy dimers and trimers in the amorphous network with maximum binding energies of approximately 0.13 eV and 0.18 eV, respectively. However, an average energy barrier height for neutral vacancy diffusion is found to be about 4.6 eV, rendering this process unfeasible. At Fermi level positions above 6.4 eV with respect to the top of the valence band, oxygen vacancies can trap up to two extra electrons. Average barriers for the diffusion of negative and double negatively charged vacancies are found to be 2.7 eV and 2.0 eV, respectively. These barriers are higher than or comparable to thermal ionization energies of extra electrons from oxygen vacancies into the conduction band of a-SiO2. In addition, we discuss the competing pathways for electron trapping in oxygen deficient a-SiO2 caused by the existence of intrinsic electron traps and oxygen vacancies. These results provide new insights into the role of oxygen vacancies in degradation and dielectric breakdown in amorphous silicon oxides.
Magnetism
Show article list
Magnon Hall effect without Dzyaloshinskii–Moriya interaction
View article, Magnon Hall effect without Dzyaloshinskii–Moriya interaction
PDF, Magnon Hall effect without Dzyaloshinskii–Moriya interaction
Topological magnon bands and magnon Hall effect in insulating collinear ferromagnets are induced by the Dzyaloshinskii–Moriya interaction (DMI) even at zero magnetic field. In the geometrically frustrated star lattice, a coplanar/noncollinear  magnetic ordering may be present due to spin frustration. This magnetic structure, however, does not exhibit topological magnon effects even with DMI in contrast to collinear ferromagnets. We show that a magnetic field applied perpendicular to the star plane induces a non-coplanar spin configuration with nonzero spin scalar chirality, which provides topological effects without the need of DMI. The non-coplanar spin texture originates from the topology of the spin configurations and does not need the presence of DMI or magnetic ordering, which suggests that this phenomenon may be present in the chiral spin liquid phases of frustrated magnetic systems. We propose that these anomalous topological magnon effects can be accessible in polymeric iron (III) acetate—a star-lattice antiferromagnet with both spin frustration and long-range magnetic ordering.
magnetic ordering may be present due to spin frustration. This magnetic structure, however, does not exhibit topological magnon effects even with DMI in contrast to collinear ferromagnets. We show that a magnetic field applied perpendicular to the star plane induces a non-coplanar spin configuration with nonzero spin scalar chirality, which provides topological effects without the need of DMI. The non-coplanar spin texture originates from the topology of the spin configurations and does not need the presence of DMI or magnetic ordering, which suggests that this phenomenon may be present in the chiral spin liquid phases of frustrated magnetic systems. We propose that these anomalous topological magnon effects can be accessible in polymeric iron (III) acetate—a star-lattice antiferromagnet with both spin frustration and long-range magnetic ordering.
Open access
Vortices and vortex lattices in quantum ferrofluids
View article, Vortices and vortex lattices in quantum ferrofluids
PDF, Vortices and vortex lattices in quantum ferrofluids
The experimental realization of quantum-degenerate Bose gases made of atoms with sizeable magnetic dipole moments has created a new type of fluid, known as a quantum ferrofluid, which combines the extraordinary properties of superfluidity and ferrofluidity. A hallmark of superfluids is that they are constrained to rotate through vortices with quantized circulation. In quantum ferrofluids the long-range dipolar interactions add new ingredients by inducing magnetostriction and instabilities, and also affect the structural properties of vortices and vortex lattices. Here we give a review of the theory of vortices in dipolar Bose–Einstein condensates, exploring the interplay of magnetism with vorticity and contrasting this with the established behaviour in non-dipolar condensates. We cover single vortex solutions, including structure, energy and stability, vortex pairs, including interactions and dynamics, and also vortex lattices. Our discussion is founded on the mean-field theory provided by the dipolar Gross–Pitaevskii equation, ranging from analytic treatments based on the Thomas–Fermi (hydrodynamic) and variational approaches to full numerical simulations. Routes for generating vortices in dipolar condensates are discussed, with particular attention paid to rotating condensates, where surface instabilities drive the nucleation of vortices, and lead to the emergence of rich and varied vortex lattice structures. We also present an outlook, including potential extensions to degenerate Fermi gases, quantum Hall physics, toroidal systems and the Berezinskii–Kosterlitz–Thouless transition.
Open access
Ultrafast demagnetization in bulk versus thin films: an ab initio study
View article, Ultrafast demagnetization in bulk versus thin films: an ab initio study
PDF, Ultrafast demagnetization in bulk versus thin films: an ab initio study
We report ab initio simulations of the quantum dynamics of electronic charge and spins when subjected to intense laser pulses. By performing these purely electron-dynamics calculations for a thin film and for the bulk of Ni, we conclude that formation of surfaces has a dramatic influence of amplifying the laser induced demagnetization. The reason for this amplification is enhanced spin-currents on the surface of the thin films. We show that the underlying physics of demagnetization for bulk is dominated by spin-flips induced by spin–orbit coupling. In the case of thin films, the dominant cause of demagnetization is a combination of the flow of spin-currents and spin-flips. Furthermore, a comparison of our results with experimental data shows that below ∼120 fs processes of demagnetization are entirely dominated by purely electronic processes followed by which dissipative effects like the Elliott–Yafet mechanism start to contribute significantly.
Magnetic states of MnP: muon-spin rotation studies
View article, Magnetic states of MnP: muon-spin rotation studies
PDF, Magnetic states of MnP: muon-spin rotation studies
Muon-spin rotation data collected at ambient pressure (p) and at p = 2.42 GPa in MnP were analyzed to check their consistency with various low- and high-pressure magnetic structures reported in the literature. Our analysis confirms that in MnP the low-temperature and low-pressure helimagnetic phase is characterised by an increased value of the average magnetic moment compared to the high-temperature ferromagnetic phase. An elliptical double-helical structure with a propagation vector  , an a-axis moment elongated by approximately 18% and an additional tilt of the rotation plane towards c-direction by
, an a-axis moment elongated by approximately 18% and an additional tilt of the rotation plane towards c-direction by  –8° leads to a good agreement between the theory and the experiment. The analysis of the high-pressure μSR data reveals that the new magnetic order appearing for pressures exceeding 1.5 GPa can not be described by keeping the propagation vector
–8° leads to a good agreement between the theory and the experiment. The analysis of the high-pressure μSR data reveals that the new magnetic order appearing for pressures exceeding 1.5 GPa can not be described by keeping the propagation vector  . Even the extreme case—decoupling the double-helical structure into four individual helices—remains inconsistent with the experiment. It is shown that the high-pressure magnetic phase which is a precursor of superconductivity is an incommensurate helical state with
. Even the extreme case—decoupling the double-helical structure into four individual helices—remains inconsistent with the experiment. It is shown that the high-pressure magnetic phase which is a precursor of superconductivity is an incommensurate helical state with  .
.
Disordered ferromagnetism in Ho2NiMnO6 double perovskite
View article, Disordered ferromagnetism in Ho2NiMnO6 double perovskite
PDF, Disordered ferromagnetism in Ho2NiMnO6 double perovskite
Magnetic and dielectric properties of the double perovskite Ho2NiMnO6 are reported. The compound is synthesized by nitrate route and is found to crystallize in monoclinic P21/n space group. Lattice parameters obtained by refining powder x-ray diffraction data are; a = 5.218(2) Å, b = 5.543(2) Å, c = 7.480(3) Å and the monoclinic angle is  (4). A phase transition is observed at
(4). A phase transition is observed at  K in the temperature-dependent magnetization curve, M(T). The inverse magnetic susceptibility, (1/
K in the temperature-dependent magnetization curve, M(T). The inverse magnetic susceptibility, (1/ ) fits reasonably well with modified Curie–Weiss law by incorporating the paramagnetic response of Ho3+. 1/
) fits reasonably well with modified Curie–Weiss law by incorporating the paramagnetic response of Ho3+. 1/ manifests as an upward deviation from ideal Curie–Weiss behaviour well above the ferromagnetic transition. Signs of inherent Griffiths phase pertaining to the Ni/Mn subsystem are visible when one subtracts the Ho3+ paramagnetic contribution from total susceptibility and does the power-law analysis. The magnetic hysteresis at 2 K gives the maximum value of magnetization
manifests as an upward deviation from ideal Curie–Weiss behaviour well above the ferromagnetic transition. Signs of inherent Griffiths phase pertaining to the Ni/Mn subsystem are visible when one subtracts the Ho3+ paramagnetic contribution from total susceptibility and does the power-law analysis. The magnetic hysteresis at 2 K gives the maximum value of magnetization 
 /f.u. at 50 kOe. Field-derivative of magnetization at 2 K shows discontinuities which indicates the existence of metamagnetic transitions in this compound. This needs to be probed further. Out of the two dielectric relaxations observed, the one at low temperature may be attributed to phononic frequencies and that at higher temperature may be due to Maxwell–Wagner relaxation. A correlation between magnetic and lattice degrees of freedom is plausible since the anomaly in dielectric constant coincides with TC.
/f.u. at 50 kOe. Field-derivative of magnetization at 2 K shows discontinuities which indicates the existence of metamagnetic transitions in this compound. This needs to be probed further. Out of the two dielectric relaxations observed, the one at low temperature may be attributed to phononic frequencies and that at higher temperature may be due to Maxwell–Wagner relaxation. A correlation between magnetic and lattice degrees of freedom is plausible since the anomaly in dielectric constant coincides with TC.
Exchange anisotropy in the skyrmion host GaV4S8
View article, Exchange anisotropy in the skyrmion host GaV4S8
PDF, Exchange anisotropy in the skyrmion host GaV4S8
Using ferromagnetic resonance spectroscopy at 34 GHz we explored the magnetic anisotropy of single-crystalline GaV4S8 in the field-polarized magnetic state. We describe the data in terms of an easy-axis type uniaxial anisotropy with an anisotropy constant  erg cm−3 at 2 K, corresponding to a relative exchange anisotropy
erg cm−3 at 2 K, corresponding to a relative exchange anisotropy  %, and about
%, and about  erg cm−3 near 11 K, i.e. at temperatures where the skyrmion-lattice phase was recently discovered. The relatively large value of K1 explains the confinement of the skyrmion tubes to the
erg cm−3 near 11 K, i.e. at temperatures where the skyrmion-lattice phase was recently discovered. The relatively large value of K1 explains the confinement of the skyrmion tubes to the  easy axes. A distinct set of resonances in the spectra is attributed to the co-existence of different rhombohedral domains. Complementary broadband spectroscopy demonstrates that non-collinear spin states may sensitively be detected by electron spin resonance techniques.
easy axes. A distinct set of resonances in the spectra is attributed to the co-existence of different rhombohedral domains. Complementary broadband spectroscopy demonstrates that non-collinear spin states may sensitively be detected by electron spin resonance techniques.
Weak d0 ferromagnetism: Zn vacancy condensation in ZnS nanocrystals
View article, Weak d0 ferromagnetism: Zn vacancy condensation in ZnS nanocrystals
PDF, Weak d0 ferromagnetism: Zn vacancy condensation in ZnS nanocrystals
We provide the explanation of the large discrepancy of three orders of magnitude between the experimentally measured and theoretically calculated magnetic moments in ZnS nanocrystals. We assume that the condensation of Zn vacancies into a single droplet takes place. The energy calculations reveal that the droplet phase is more favorable than the uniformly distributed vacancy configuration. The other assumption made is that a small magnetic moment could arise at the interface between the ZnS crystal and vacancy cluster. The calculations however dismiss this hypothesis because the magnetization of the layered system also vanishes. Thus we suggest that the experimentally low magnetization values could be explained from one of the two following pictures: (a) there are two phases where the vacancy cluster with the zero magnetic moment coexists along with the other phase, in which there are uniformly distributed Zn vacancies with low concentrations or (b) there is only a single vacancy phase—a vacancy droplet being in the metastable state with a weak nonvanishing magnetic moment.
Magnetic ordering and dielectric relaxation in the double perovskite YBaCuFeO5
View article, Magnetic ordering and dielectric relaxation in the double perovskite YBaCuFeO5
PDF, Magnetic ordering and dielectric relaxation in the double perovskite YBaCuFeO5
Using magnetization, dielectric constant, and neutron diffraction measurements on a high quality single crystal of YBaCuFeO5 (YBCFO), we demonstrate that the crystal shows two antiferromagnetic transitions at  K and
K and  K, and displays a giant dielectric constant with a characteristic of the dielectric relaxation at TN2. It does not show the evidence of the electric polarization for the crystal used for this study. The transition at TN1 corresponds with a paramagnetic to antiferromagnetic transition with a magnetic propagation vector doubling the unit cell along three crystallographic axes. Upon cooling, at TN2, the commensurate spin ordering transforms to a spiral magnetic structure with a propagation vector of (
K, and displays a giant dielectric constant with a characteristic of the dielectric relaxation at TN2. It does not show the evidence of the electric polarization for the crystal used for this study. The transition at TN1 corresponds with a paramagnetic to antiferromagnetic transition with a magnetic propagation vector doubling the unit cell along three crystallographic axes. Upon cooling, at TN2, the commensurate spin ordering transforms to a spiral magnetic structure with a propagation vector of (

 ), where
), where  ,
,  , and
, and  are odd, and the incommensurability δ is temperature dependent. Around the transition boundary at TN2, both commensurate and incommensurate spin ordering coexist.
are odd, and the incommensurability δ is temperature dependent. Around the transition boundary at TN2, both commensurate and incommensurate spin ordering coexist.
Computational and experimental methods
Show article list
Open access
Quantitative atomic force microscopy
A variety of atomic force microscopy (AFM) modes is employed in the field of surface science. The most prominent AFM modes include the amplitude modulation (AM) and the frequency modulation (FM) mode. Over the years, different ways for analyzing data acquired with different AFM modes have been developed, where each analysis is usually based on mode-specific assumptions and approximations. Checking the validity of the seemingly different approximations employed in the various analysis methods can be a tedious task. Moreover, a straightforward comparison of data analyzed with different methods can, therefore, be challenging. Here, we combine the existing evaluation methods which have been separately developed for the different AFM modes and present a unifying set of three equations. These three AFM equations allow for a straightforward analysis of AFM data within the harmonic approximation, regardless of the AFM mode. The three AFM equations provide the three and only pieces of information about the tip-sample force available within the harmonic approximation. We demonstrate the generality of our approach by quantitatively analyzing three-dimensional AFM data obtained in both the AM and FM mode.
Computational methods for 2D materials: discovery, property characterization, and application design
View article, Computational methods for 2D materials: discovery, property characterization, and application design
PDF, Computational methods for 2D materials: discovery, property characterization, and application design
The discovery of two-dimensional (2D) materials comes at a time when computational methods are mature and can predict novel 2D materials, characterize their properties, and guide the design of 2D materials for applications. This article reviews the recent progress in computational approaches for 2D materials research. We discuss the computational techniques and provide an overview of the ongoing research in the field. We begin with an overview of known 2D materials, common computational methods, and available cyber infrastructures. We then move onto the discovery of novel 2D materials, discussing the stability criteria for 2D materials, computational methods for structure prediction, and interactions of monolayers with electrochemical and gaseous environments. Next, we describe the computational characterization of the 2D materials' electronic, optical, magnetic, and superconducting properties and the response of the properties under applied mechanical strain and electrical fields. From there, we move on to discuss the structure and properties of defects in 2D materials, and describe methods for 2D materials device simulations. We conclude by providing an outlook on the needs and challenges for future developments in the field of computational research for 2D materials.
The atomic simulation environment—a Python library for working with atoms
View article, The atomic simulation environment—a Python library for working with atoms
PDF, The atomic simulation environment—a Python library for working with atoms
The atomic simulation environment (ASE) is a software package written in the Python programming language with the aim of setting up, steering, and analyzing atomistic simulations. In ASE, tasks are fully scripted in Python. The powerful syntax of Python combined with the NumPy array library make it possible to perform very complex simulation tasks. For example, a sequence of calculations may be performed with the use of a simple 'for-loop' construction. Calculations of energy, forces, stresses and other quantities are performed through interfaces to many external electronic structure codes or force fields using a uniform interface. On top of this calculator interface, ASE provides modules for performing many standard simulation tasks such as structure optimization, molecular dynamics, handling of constraints and performing nudged elastic band calculations.
Advanced capabilities for materials modelling with Quantum ESPRESSO
View article, Advanced capabilities for materials modelling with Quantum ESPRESSO
PDF, Advanced capabilities for materials modelling with Quantum ESPRESSO
Quantum ESPRESSO is an integrated suite of open-source computer codes for quantum simulations of materials using state-of-the-art electronic-structure techniques, based on density-functional theory, density-functional perturbation theory, and many-body perturbation theory, within the plane-wave pseudopotential and projector-augmented-wave approaches. Quantum ESPRESSO owes its popularity to the wide variety of properties and processes it allows to simulate, to its performance on an increasingly broad array of hardware architectures, and to a community of researchers that rely on its capabilities as a core open-source development platform to implement their ideas. In this paper we describe recent extensions and improvements, covering new methodologies and property calculators, improved parallelization, code modularization, and extended interoperability both within the distribution and with external software.
Open access
A proposed simulation method for directed self-assembly of nanographene
View article, A proposed simulation method for directed self-assembly of nanographene
PDF, A proposed simulation method for directed self-assembly of nanographene
A methodology for predictive kinetic self-assembly modeling of bottom-up chemical synthesis of nanographene is proposed. The method maintains physical transparency in using a novel array format to efficiently store molecule information and by using array operations to determine reaction possibilities. Within a minimal model approach, the parameter space for the bond activation energies (i.e. molecule functionalization) at fixed reaction temperature and initial molecule concentrations is explored. Directed self-assembly of nanographene from functionalized tetrabenzanthracene and benzene is studied with regions in the activation energy phase-space showing length-to-width ratio tunability. The degree of defects and reaction reproducibility in the simulations is also determined, with the rate of functionalized benzene addition providing additional control of the dimension and quality of the nanographene. Comparison of the reaction energetics to available density functional theory data suggests the synthesis may be experimentally tenable using aryl-halide cross-coupling and noble metal surface-assisted catalysis. With full access to the intermediate reaction network and with dynamic coupling to density functional theory-informed tight-binding simulation, the method is proposed as a computationally efficient means towards detailed simulation-driven design of new nanographene systems.
Construction of crystal structure prototype database: methods and applications
View article, Construction of crystal structure prototype database: methods and applications
PDF, Construction of crystal structure prototype database: methods and applications
Crystal structure prototype data have become a useful source of information for materials discovery in the fields of crystallography, chemistry, physics, and materials science. This work reports the development of a robust and efficient method for assessing the similarity of structures on the basis of their interatomic distances. Using this method, we proposed a simple and unambiguous definition of crystal structure prototype based on hierarchical clustering theory, and constructed the crystal structure prototype database (CSPD) by filtering the known crystallographic structures in a database. With similar method, a program structure prototype analysis package (SPAP) was developed to remove similar structures in CALYPSO prediction results and extract predicted low energy structures for a separate theoretical structure database. A series of statistics describing the distribution of crystal structure prototypes in the CSPD was compiled to provide an important insight for structure prediction and high-throughput calculations. Illustrative examples of the application of the proposed database are given, including the generation of initial structures for structure prediction and determination of the prototype structure in databases. These examples demonstrate the CSPD to be a generally applicable and useful tool for materials discovery.
Long-time atomistic dynamics through a new self-adaptive accelerated molecular dynamics method
View article, Long-time atomistic dynamics through a new self-adaptive accelerated molecular dynamics method
PDF, Long-time atomistic dynamics through a new self-adaptive accelerated molecular dynamics method
A self-adaptive accelerated molecular dynamics method is developed to model infrequent atomic-scale events, especially those events that occur on a rugged free-energy surface. Key in the new development is the use of the total displacement of the system at a given temperature to construct a boost-potential, which is slowly increased to accelerate the dynamics. The temperature is slowly increased to accelerate the dynamics. By allowing the system to evolve from one steady-state configuration to another by overcoming the transition state, this self-evolving approach makes it possible to explore the coupled motion of species that migrate on vastly different time scales. The migrations of single vacancy (V) and small He-V clusters, and the growth of nano-sized He-V clusters in Fe for times in the order of seconds are studied by this new method. An interstitial-assisted mechanism is first explored for the migration of a helium-rich He-V cluster, while a new two-component Ostwald ripening mechanism is suggested for He-V cluster growth.