Abstract
Anion exchange membranes (AEMs) are of interest as hydroxide conducting polymer electrolytes in electrochemical devices like fuel cells and electrolyzers. AEMs require hydroxide stable covalently tetherable cations to ensure required conductivity. Benzyltrimethylammonium (BTMA) has been the covalently tetherable cation that has been most often employed in anion exchange membranes because it is reasonably basic, compact (limited number of atoms per charge), and easily/cheaply synthesized. Several reports exist that have investigated hydroxide stability of BTMA under specific conditions, but consistency within these reports and comparisons between them have not yet been made. While the hydroxide stability of BTMA has been believed to be a limitation for AEMs, this stability has not been thoroughly reported. We have found that several methods reported have inherent flaws in their findings due to the difficulty of performing degradation experiments at high temperature and high pH. In order to address these shortcomings, we have developed a reliable, standardized method of determining cation degradation under conditions similar/relevant to those expected in electrochemical devices. The experimental method has been employed to determine BTMA stabilities at varying cation concentrations and elevated temperatures, and has resulted in improved experimental accuracy and reproducibility. Most notably, these results have shown that BTMA is quite stable at 80°C (half-life of ∼4 years), a significant increase in stability over what had been reported previously.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Alkaline membrane fuel cells (AMFCs) employing anion exchange membranes (AEMs) are of increasing interest in fuel cell research as they potentially enable the use of non-Pt fuel cell catalysts, a primary cost limitation of proton exchange membrane fuel cells.1 Beyond fuel cells, AEMs are also finding interest in the areas of electrolysis, flow batteries, and photoelectrochemical water splitting.1,2 A major concern for AEMs has been stability at high temperature and high pH which are the target operating conditions for many of the devices.2,3 Current anion exchange membranes suffer from both polymer backbone degradation4 as well as degradation of the hydroxide conducting cation.5,6 While backbone degradation may be avoided by alternating backbone chemistry amongst a large number of choices, a cation is necessary in these systems for conductivity and must remain stable in the presence of hydroxide.
The benzyltrimethyl ammonium (BTMA) cation, in particular, is the most commonly employed covalently tetherable cation7 and is commonly used in commercial AEMs.8 AEMs have largely been developed for ion exchange applications where cation stability toward hydroxide attack is not a significant concern. In electrochemical devices employing anion exchange membranes, the presence of hydroxide is typically required for the energy conversion reactions taking place, and high temperatures are desirable to decrease catalytic over potentials, increase conductivity, and aid in the removal of carbon dioxide from the system (carbonates form in the presence of hydroxide). Degradation of BTMA has been the subject of several past studies.5,9–13 However, methodologies of degradation were not standardized or fully validated. Several reports that include BTMA degradation are presented in Table I to highlight the differences in reaction conditions and use of various experimental techniques.
Table I. Variability of methodologies employed to investigate BTMA hydroxide stabilities.
| Experimental Conditions | |||||
|---|---|---|---|---|---|
| Ref. | Temp °C | BTMA | Base | Analytical Methods | BTMA Degradation Results |
| Bauer9 | 160 | 0.1M | 2M KOH in glycol | UV/vis adsorption at 520 nm | 29.1 min ½ life |
| Einsla5 | 80 | 1M | 1, 2, 3M NaOH (in D2O) | NMR | 90% remaining after 700 hours |
| 120 | 10–30 wt% | No excess base | 10wt% (0.55M) ½ life = 35 h | ||
| 20wt% (1.1M) ½ life <5h | |||||
| 30wt% (1.65M) ½ life <2h | |||||
| Marino27 | 80–160 | 0.1M | 1-10 M NaOH | UV/vis | 4.18 h ½ life at 160°C (6M) |
| Deavin10 | 60 | 1M | 1M OH− | FT-Raman | No significant degradation |
| Noonan11 | 80 | 0.1M | 1M NaOD in CD3OD | NMR, GC-MS | 66% degraded after 20 days |
| Nunez12 | 60 | 30 mM | 0.6 M KOD in 3:1 | NMR w/ in situ heating | ½ life = 2887 h |
| CD3OD:D2O | 99.5 ± 0.6 remaining after 779 min (∼13 h) | ||||
| Price13 | 80 | 0.1M | 1M NaOD in D2O | NMR | ½ life longer than limit of detection for experiment (>400 h) |
Several of the listed stability studies were run at 80°C or lower, which would represent target operating temperatures for many electrochemical devices. 80°C, in particular, has been suggested as a target operating temperature as it reflects a temperature often used in proton exchange membrane (PEM) systems and may allow for more efficient self-purging of CO2 from ambient air.14,15 However the reported stabilities in Table I at 80°C are inconsistent and confusing as they range from insignificant degradations after several days5,10,13 to 66% degradation after 20 days.12 Other studies have reported that BTMA is less stable at elevated temperatures (>120°C).5,9 Direct stability comparisons cannot be made however as the experimental methods (including reaction vessels, heating methods, internal standards, concentrations, and solvent systems) vary from one study to another, and in many cases details may be lacking. Still, general trends like decreasing stability with increasing temperature or the introduction of methanol can be inferred and make sense, but the quantitative rates of degradation are fairly inconsistent. NMR has been the most commonly employed technique to monitor degradation; however, other techniques have also been reported (Raman, UV/Vis, GC-MS). Our group has been working in this area for several years, including some of the data presented in Table I. During the course of our studies we were somewhat perplexed by the difficulties we encountered reproducing degradation rates and getting agreement with literature values.
In order to obtain repeatable, relevant measurements of cation degradation, we investigated a number of experimental variables and explored their impact on observed degradation rates. Experimental variables probed included: reaction vessel, heating method, the use of internal (NMR) standards, (cation and base) concentration, and different aqueous environments (most notably D2O vs H2O). From these efforts, we have developed a method that reliably measures hydroxide stability in aqueous 2M KOH utilizing Teflon lined Parr reactors heated in a conventional oven. NMR analysis is employed using an external standard sealed in a capillary tube that is not exposed to reaction conditions of the degraded BTMA. Gas chromatography - mass spectrometry (GC-MS) analysis has been used to verify product distribution. This experimental setup has resulted in more accurate/reliable rates being observed than rates obtained in our previous attempted approaches for the degradation of BTMA in hydroxide.5 The degradation rates observed in this study have been found to be slower than those previously reported in Table I for quantified values. Specifically, at 80°C, BTMA has been found to have a half-life of > 4 years, a stability that is likely to be sufficient for a number of potential applications.
This observed high stability of BTMA is important to the AEM community because BTMA is a relatively compact, easily synthesized, and relatively low cost cation. Any other cations being pursued for use in AEMs should offer substantial advantages over BTMA for consideration for use in AEMs, and the stability of BTMA under current suggested operating conditions has been a concern that has inspired research into other potential AEM cations including imidazolium,8,10,13,16–19 phosphonium,11,20,21 guanidinium,22,23 and others.24–26
Zhang et al. synthesized an anion exchange membrane utilizing an imidazolium cationic group that exhibited promising results in terms of ion exchange capacity (IEC) and conductivity.17 However upon exposure to caustic conditions (3M NaOH at 60°C) membrane conductivity decreased by 23.3%. This loss of conductivity was also accompanied with slight weight loss (6.8%) suggesting that although the polymeric backbone remained intact, the imidazolium head group degraded. Thomas et al. found that in poly(dialylbenzimidazolium) salts the cation has a large effect on both IEC and conductivity. However they also report hydroxide instability as attempts to prepare the hydroxide form of the material resulted in decomposition of the polymer.16 Chen and Hickner later compared the degradation of AEMs functionalized with BTMA hydroxide conducting head groups to AEM functionalized with imidazolium.8 The BTMA head group was found to be slightly more stable than imidazolium; however in each case both head groups as well as polymeric backbone degraded in 1M NaOH at 60°C over 48 h. Varcoe and coworkers also confirm that BTMA head groups are also more stable than imidazolium in caustic operating conditions.10 Later Varcoe and coworkers compared the hydroxide stability of substituted imidazolium (benzyldimethylimidazolium) to that of benzylmethylimidazolium and BTMA.18 Although the dimethylimidazolium was more stable than methylimidazolium, both imidazoliums were less stable than BTMA. Price et al. investigated the relationships between structure and alkaline stability of free imidazolium cations, specifically probing the effect of steric hindering groups.13 They report that 1,2,3-trimethylimidazolium is quite stable in 1M NaOH at 88°C (half-life ≥1000 h). However the phenyl-substituted analogue (1,3-dimethyl-2-benzylimidazolium) degrades under these same caustic conditions. In a theoretical study, Long and Pivovar used DFT calculations to estimate hydroxide degradation barriers of imidazolium cations.19 They also report that the benzylimidazolium derivatives are less stable than their imidazolium counterparts. The substantial interest in imidazolium in these reports and their comparisons to BTMA, demonstrate the concern over BTMA stability that has existed in the research community.
Beyond imidazolium, Noonan et al. directly compared the alkaline stability of a quaternary phosphonium cation to that of BTMA by subjecting both cations to 1M NaOD in CD3OD at 80°C.11 After 20 days 66% of the BTMA was degraded, in contrast no degradation of the phosphonium cation was observed. In their studies, Yan and coworkers have synthesized hydroxide exchange membranes utilizing quaternary phosphonium cations as hydroxide conductors.20,21 When comparing their membranes with quaternary phosphonium head groups to quaternary ammonium head groups (similar to BTMA), they found that the phosphonium based material exhibits both higher conductivity and better alkaline stability. They also report that the structure of the cation was important, contributing enhancement of both stability and basicity to electron-donating groups attached to the quaternized phosphonium. Structural trends were also observed in ionomers containing a guanidium head group.23 A sulfone guanidinium functionalized ionomer was nearly completely degraded after soaking in 0.5M NaOH at 80°C for 24 h. However a conjugated phenylguanidinium functionalized ionomer showed no signs of degradation under the same conditions for 72 h. Very recently, Marino and Kreuer have performed a wide systematic study of different quaternized ammonium groups primarily using UV-vis spectroscopy to measure hydroxide degradation rates of tens of different cations, primarily reporting degradation at 6M NaOH and 160°C.27 For BTMA, they report a number of different experimental conditions between 1–10M NaOH and 80 and 160°C.
A number of the references cited have put an emphasis on employing cations with improved alkaline stability relative to BTMA in AEMs. Some of these studies presented were performed on free cations in solution and others on polymers with covalently tethered cationic groups, an area where there are still disconnects between the alkaline stabilities of the polymeric membrane materials and the free cationic head groups. While the stability of the free cation in solution investigated here may be different than that of the tethered cation in an AEM, these studies provide a basis for comparison and a preliminary screening tool in order to determine cations that have the highest promise. Still, these findings are at least qualitatively relevant/consistent,2 and for all cases an alkaline stability benchmark is currently lacking. An accurate value for the stability of the free BTMA cation in solution allows for the appropriate consideration of BTMA for use under specific conditions to be properly weighed against the need for alternative cations with potentially enhanced stability.
The study presented here holds value beyond just the more accurate quantification of BTMA degradation, as it also establishes a standardized method that can be applied across different cation systems and labs allowing for more valid comparisons to be made. Such a standardized method does not currently exist in the community and has led to difficulties in making comparisons between different cations. Additionally, having accurate, consistent data for degradation routes available between different cations would be of high value for the computational efforts which require these for model validation. The work presented here has value both for the quantification of BTMA stability and the establishment of a standardized method for reporting cation stability.
Experimental
BTMA hydroxide (40 wt% in H2O) (Sigma Aldrich), KOH (Fisher Scientific), and 3-(trimethylsilyl)propionic acid-d4 sodium salt (TMS) (Sigma Aldrich) were all used as received. A 1M stock solution of BTMA hydroxide was prepared by dilution of the 40 wt% solution in aqueous 2M KOH. Serial dilution of the stock was utilized to make up 0.5M, 0.1M, and 0.01M solutions in 2M KOH. These solutions were then immediately used in the degradation studies. These studies were carried out in sealed Teflon lined stainless steel Parr reactors (vessel #4749 23 mL capacity), which were charged with the BTMA solution and placed in a preheated oven.
The reactors used for these studies had significant (thermal) mass, weighing just over 1 kg, and even more importantly, the relatively thick, sealed, Teflon liner (low heat transfer coefficient) is expected to result in thermal equilibrium being reached slowly. For this reason, transient heating rates of the reactors were investigated. Heating rates of the reactors were measured in-situ utilizing a ThermoWorks high temperature data logger (HiTemp 140-1) probe that was placed into a Teflon lined Parr reactor containing 1 mL of water. The reactor containing the data logger was sealed and placed into the preheated oven and subjected to reaction temperatures for a minimum of 24 h. The 23 mL reactor vessels were too small to fit the temperature probe. Therefore, in order to accommodate the temperature probe a larger Parr reactor was used (vessel #4744 45 mL capacity). Based on the geometry of the devices and masses, we believe that the temperature data obtained from the larger vessel is reasonably consistent.
Suitable external NMR standards were made by preparing a 1 wt% TMS in D2O solution. This solution was then charged into 6' capillary tubes that were subsequently flame sealed; creating an inert isolated standard that could be added to an NMR tube containing sample and reused. NMR analysis was conducted on a Bruker Advance 400. GC analysis was conducted on a Thermo Scientific Trace GC Ultra equipped with a Thermo TG-5SILMS column coupled to an ISQ mass spectrometer. Analytes were matched and identified via the NIST/EPA/NIH/Mass Spectral Library. Aqueous reaction mixtures were extracted with 1 mL diethyl ether to obtain samples suitable for GCMS analysis.
A known amount (1 mL) of reactant solution, consisting of BTMA at a desired concentration in 2M KOH was charged into the Parr reactor and sealed. Multiple charged Parr reactors (at least 10) were used in each experiment. The reactors would then be placed in a calibrated oven at a specified temperature (80°C, 120°C, 140°C, and 160°C) without stirring. A sample of the reactant solution would be set aside to represent an initial (zero) time point in each degradation study. Initial time points were measured from the freshly prepared BTMA solution for each concentration. Periodically (depending on temp. and concentration) single reactor vessels would be removed from the oven and carefully quenched in a room temperature water bath (∼30 min). Each reactor represents a single data point on the data obtained and reactors were not returned to the oven for further degradation once removed. The analyte from each reactor was removed and charged into an NMR tube containing a sealed external standard for immediate analysis.
Results and Discussion
In the development of a standardized degradation method we desired accuracy and reproducibility. We were also interested in speed (accelerated conditions), under the assumption that the test conditions would remain relevant. One specific target approach was using elevated temperatures to accelerate rates, however this also required that we accurately understand thermal response. Ideally, thermal transients would be much shorter than the half-lives for the degradation reactions probed. Figure 1 shows the thermal transient of a data logger in a DI water equivalent of our degradation solutions in a Parr reactor to track the reactant temperature as a function of time. The oven was placed at 140°C and the temperature probe asymptotically approached the oven temperature, reaching near equilibrium after approximately 120 minutes. For lower temperatures, this time was slightly faster (higher temperatures could not be probed due to thermal stability of the temperature probe), but 2 hours serves as a fair gauge of the time it takes for the sample temperature to reach the target degradation temperature of 140°C. Because of the thermal transient of the system, we targeted degradation times that would have half-lives greater than 24 hours to minimize impact of thermal transients. Half-lives of one to two days were seen as ideal compromises between sample throughput and experimental accuracy.
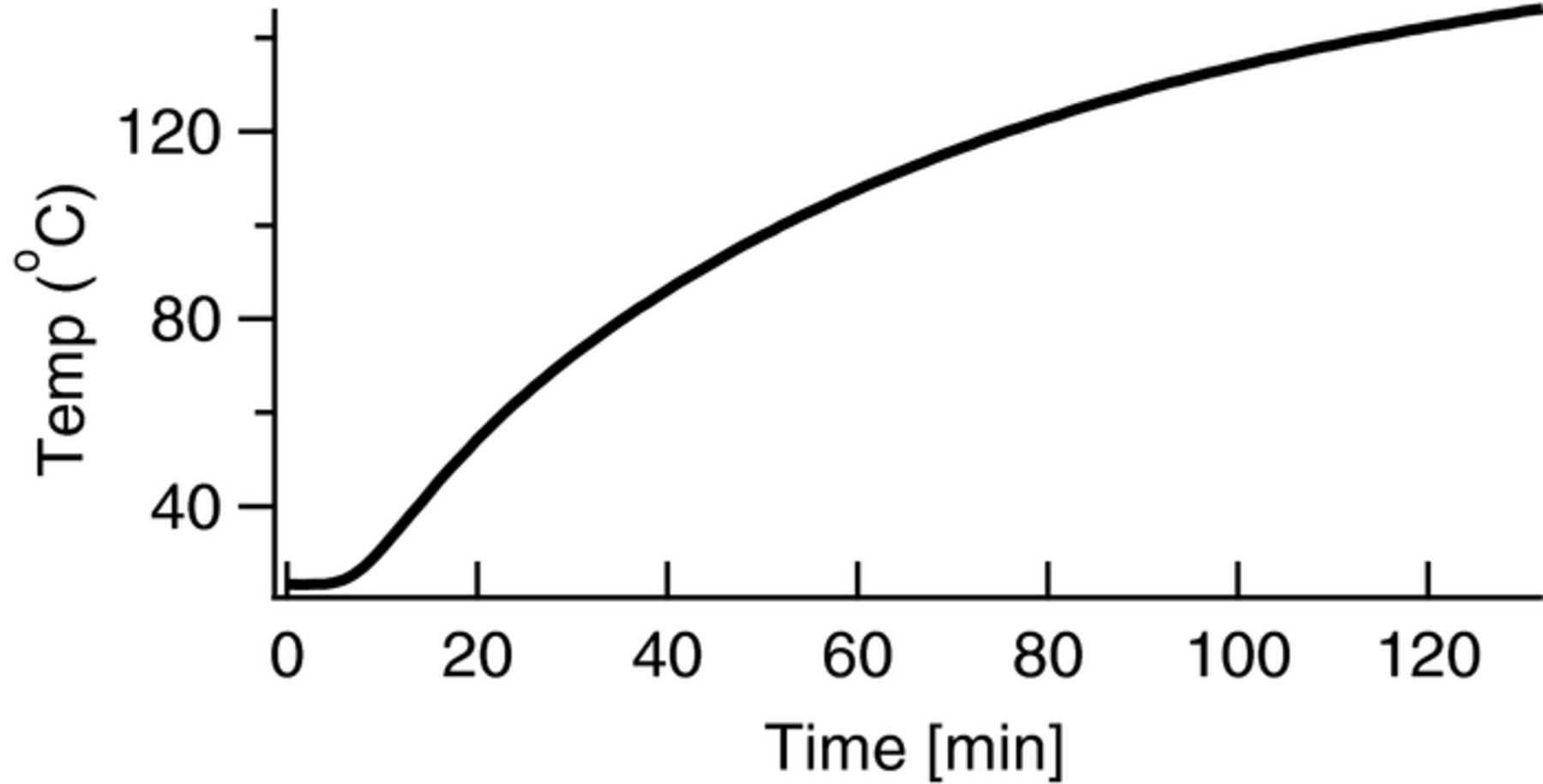
Figure 1. Heating rate obtained from a temperature probe contained in a Parr reactor placed in an oven at 140°C.
The measurement of cation degradation rates at high temperature in caustic environments (high pH) has a number of challenges beyond thermal transient response. The methods employed in the studies listed in Table I highlight the wide disparity of results that might be obtained for such studies. Several potential areas of concern include the use of glass reaction vessels, internal (NMR) standards, and heating methods (including aspects that go beyond the previous paragraph). It was determined that glass reaction vessels should be avoided under caustic reaction conditions as they can also be attacked by hydroxide ions, forming a visible precipitate that can complicate sample analysis (as also reported by Deavin et al.).10 When we have performed degradation studies in glass reaction vessels (including NMR tubes), an unknown white precipitate is observed, see Figure 3A. Additionally, a shift of the H2O proton peak in the 1H NMR spectrum is observed, presumably due to changing pH as hydroxide is consumed via reaction with glass (Figure 2). In our efforts we investigate quartz NMR tubes, as compared to our baseline borosilicate glass tubes, we found the quartz tubes exhibited decreased precipitate formation, but were still to be susceptible to hydroxide attack. To prevent hydroxide attack on glass vessels and associated complications arising from the use of glass, we employed Teflon lined Parr reactors (23 mL volume) in the degradation studies reported here. An additional concern in the degradation reactions, particularly those that occur at high temperatures or over long periods of time, is that of the quality of the seal and potential leaking of water through evaporation. Any water that leaks from the system results in an artificial increase in concentration of cation in the remaining solution and can be misinterpreted as a slower degradation rate than is actually occurring (as well as adding complications with increased KOH concentration). The use of the Teflon lined Parr reactors is seen as a strong benefit in this case as well as high compression forces are applied and the Teflon liners are believed to have exceptional sealing characteristics, reflected by the high temperature (250°C) and high pressure (1800 psi) reactions that these reaction vessels are designed for.28
Figure 2. 1H NMR showing H2O proton peak shift due to changing pH during cation degradation in a glass vial.

Figure 3. Illustration of common experimental pitfalls. A: Hydroxide etches glass reaction vessel creating a white precipitate while changing pH. B: Internal standard in tube 2 degrading when exposed to caustic conditions (no standard present in tube 1). C: Common heating set up in which reflux can occur at unheated head space. D: Observed phase separation in reaction mixture of 1M BTMA in 2M KOH after heating.
Another potential complication that arises during NMR studies occurs due to the addition of an internal (NMR) standard to the reaction mixture. We have found multiple common standards, used to quantify NMR signals, which lack sufficient stability under the thermal and caustic conditions of the degradation studies, see Figure 3B where 3-(trimethylsilyl)propionic acid-d4 sodium salt (TMS) was employed. Rather than employ an internal standard which is contained in the degradation solutions, we have instead employed an internal standard contained in a sealed capillary tube which is added to samples just prior to NMR analysis and is not subject to the degradation environment. Isolating the standard in this manner also prevents direct interaction with the analyte where other reactions have the possibility of occurring.
Uneven heating of reaction vessels can also pose a problem. Placing sample vessels in a heating bath (sand, oil, heating block) can leave significant head space exposed to ambient temperatures; upon which solvent reflux occurs and desired temperatures are not reached, see Figure 3C. The entire sample vessel should be evenly heated and at thermal equilibrium. Thus a calibrated convection oven was used as the heating source, ensuring that the entire sample vessel was evenly heated once equilibrium was obtained.
BTMA concentrations and the solvent systems in which the degradations are conducted were found to have dramatic impact in some cases on observed cation stability, and are therefore highlighted as areas of potential major concern. BTMA concentrations between 0.01M and 1M were probed and concentrations of 0.5M and 1.0M were observed to produce a phase separated solution with an organic phase on top of the aqueous phase, see Figure 3D. The systems that were found to phase separate, also exhibited vastly increased degradation rates, discussed later in the context of data presented in Figure 5. Hydroxide degradation rates increase with decreasing dielectric constant,29 like those found in organic solvents.11,12 The nucleophilicity of the hydroxide anion increases in the lower dielectric constant media and the observed increase in degradation rate may simply be due to partitioning of some fraction of BTMA and hydroxide into the organic phase accompanied by accelerated degradation rates. Additionally the possibility exists that the organic phase components also participate in degradation reactions thereby increasing degradation rates.

Figure 5. Different rates of BTMA degradation based on integration of: aromatic (×), benzyl protons (●), and trimethyl (+) when alkaline degradation is run in D2O (0.1M BTMA in 2M KOD in D2O at 160°C).
We performed GC-MS on the organic and aqueous phase for a few cases where phase separation was observed to occur. A representative sample result from these limited studies is given in Table II with the 4 primary species identified in the organic phase. Trimethylamine and benzyl alcohol, the primary degradation products expected from the major degradation route previously identified7,30,31 (see scheme 1), were both observed in the organic phase. Benzyl alcohol, the stoichiometric degradation product associated with trimethylamine, was partitioned between both the organic and aqueous phase. Trimethyl amine was not present in the aqueous phase; reflecting a higher solubility of benzyl alcohol in water, and at least partially explaining the non-stoichiometric ratio of benzyl alcohol to trimethyl amine in the organic phase. Although small amounts of condensation products arising from benzyldimethyl amine were observed in the organic phase, no appreciable amounts of methanol nor benzyldimethyl amine were found in either the aqueous or organic phase; suggesting the demethylation degradation reaction shown in scheme 1 was perhaps non-existent. A current limitation in interpreting these findings arises because we are uncertain which species may exist in the gas phase above the solution due to difficulties obtaining gaseous samples for analysis.32 Further reaction of the initial products may also account for their absence in the GC-MS analysis. The balance of the species found in the organic phase consisted of (methoxymethyl)benzene and dibenzyl ether at very similar concentrations. The origin of these species is not absolutely determined at this time, but it seems certain they are the result of the further reaction of degradation products, and may have the ability to react with BTMA.
Table II. Chemical structures of major and minor products observed in the organic phase of phase separated reaction solution from 1M BTMA in 2M KOH reacted at 160°C.
| Major Products | Minor products |
|---|---|
| Trimethylamine | (Methoxymethyl)benzene |
| Benzyl alcohol | Dibenzyl ether |
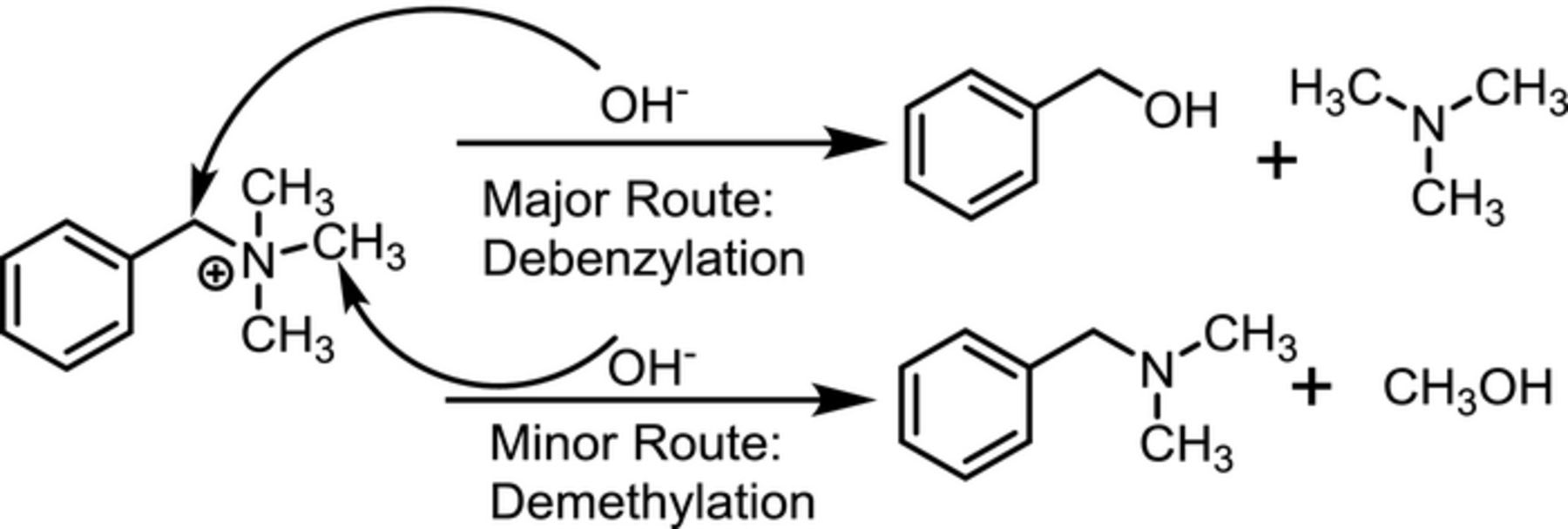
Scheme 1. Degradation reactions expected for OH- attack on BTMA.
The concentration effects reported due to the formation of a second phase had a major impact on observed degradation rates, but we also found that using deuterated water could result in data interpretation that resulted in an even larger impact on observed degradation rates. Often times degradation experiments which utilize NMR techniques as the primary method of analysis will conduct the study in deuterated solvents in order to prevent solvent peaks from interfering with proton signals of target reactions. Figure 4 presents NMR spectra as a function of time for a 0.1M BTMA solution in 2M KOH at 160°C for samples measured in deuterated water (4a) and non-deuterated water (4b). The initial NMR spectra for both cases clearly show aromatic, benzyl, and trimethyl protons from BTMA (denoted on the Figures 4A and 4B). The non-deuterated sample shows a much more pronounced water peak (near 5.0 ppm), but this peak did not interfere with the analysis of BTMA peaks (nor did the several product degradation peaks that grow as the BTMA peaks diminish). Both conditions show a loss of BTMA peaks, but the deuterated sample shows inconsistent peak loss between the different protons in BTMA (Figure 5).

Figure 4. 1H NMR showing degradation of aromatic (×), benzyl (●), and trimethyl (+) protons in BTMA when alkaline degradation is run in: A: D2O (0.1M BTMA in 2M KOD in D2O at 160°C) and B: H2O (0.1M BTMA in 2M KOH in H2O at 160°C).
Figure 5 shows the % BTMA remaining vs time based on integration of the 3 distinct BTMA protons (aromatic, benzyl, and trimethyl) for the degradation of BTMA in D2O (i.e. integration of peaks shown in Figure 4A). Protons in BTMA have been shown to exhibit H/D exchange.5,10,11,13,33 The rate of H/D exchange differs from one type of proton to another. The observed rate of H/D exchange in BTMA is: benzyl > methyl > aromatic (as reflected by the data in Figure 5). When the % BTMA remaining is determined by integration of the benzyl protons, a half-life of less than 2 h is observed; integration of the methyl protons show a half-life of ∼4h, and integration of the aromatic protons give rise to a half-life on the order of 6h. As these protons are subject to H/D exchange, degradation studies should be conducted in non-deuterated systems to prevent isotopic exchange from being misinterpreted as degradation. In this case, we cannot assume that even the data from the aromatic peaks is immune from the impact of isotopic exchange. The data presented in the rest of the paper is performed in non-deuterated water with the only drawback being the large water peaks in the NMR spectra and the slight decrease in signal to noise in the sample. Still, for the systems studied, signal to noise was a minor issue, even for samples of only 0.01M concentration.
After fully refining this experimental method and eliminating or minimizing to the best of our abilities all potential challenges of performing high temperature, high pH degradation studies, we applied this method to the study of BTMA degradation at concentrations of 0.01M, 0.1M, 0.5M, and 1M in 2M KOH at 80°C, 120°C, 140°C and 160°C. The results of these studies are summarized in Figure 6. Multiple samples were probed in many cases and standard deviations are reported where relevant. For these non-deuterated water studies, all three integrated BTMA peaks have been averaged to calculate BTMA degradation rates. As a representative example, the data from Figure 4B is presented in Table III showing very good agreement between the 3 integration peaks (benzyl, aromatic, and trimethyl).
Table III. % degradation of BTMA from integration of aromatic protons (×), benzyl protons (●), trimethyl protons (+) in the 1H NMR at 0, 1, 2, 4, 10, and 15 h. 0.1M BTMA in 2M KOH at 160°C (average % used in Fig. 6A).
| Time (h) | Aromatic (X) | Benzyl (●) | Trimethyl (+) | Average |
|---|---|---|---|---|
| 0 | 100 | 100 | 100 | 100 |
| 1 | 81.3 | 78.0 | 80.7 | 80.0 |
| 2 | 74.7 | 75.6 | 74.3 | 74.9 |
| 4 | 58.2 | 58.5 | 56.7 | 57.8 |
| 10 | 27.8 | 28.3 | 24.4 | 26.8 |
| 15 | 12.3 | 13 | 10.3 | 11.9 |
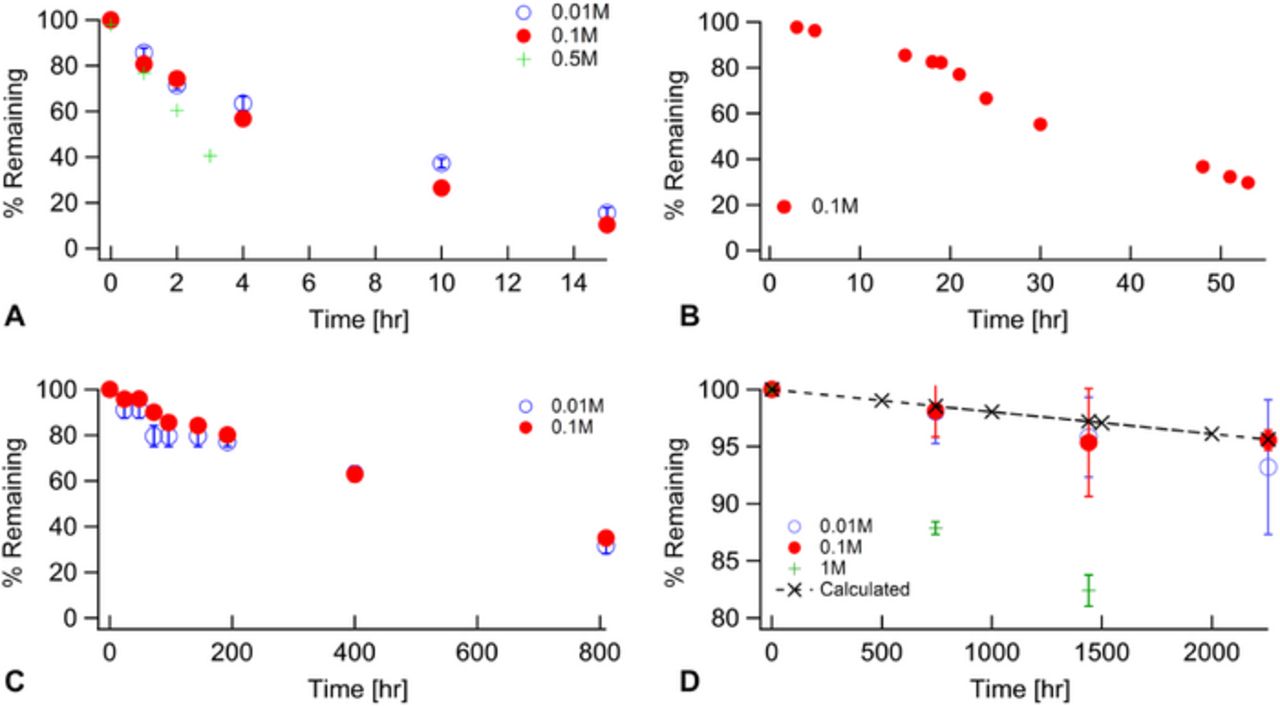
Figure 6. Degradation rates of various concentrations of BTMA in 2M KOH at 160°C (A), 140°C (B), 120°C (C), and 80°C (D).
80°C has been noted as a target operating temperature for devices and was investigated for BTMA stability. Unfortunately for the purposes of performing timely degradation studies, but fortunately for the possible utility of BTMA under high temperature conditions, even after more than 2000 hour far less than 10% of the BTMA degraded when tested at 0.1 and 0.01M concentrations, see Figure 6D. The degradation rates for both 0.1M and 0.01M were in relatively good agreement with each other, but as previously noted 1M degradation was significantly higher and a second organic phase was noted in the samples. As degradation rates were low, elevated temperatures were also investigated, see Figures 6A–6C. In all cases in Figure 6, 0.1M and 0.01M degradation rates show good agreement. Again, the higher concentration data for 0.5M BTMA at 160°C show a deviation to higher degradation rates at longer times, in this case the 2 and 3 hour samples showed a clear second organic phase, while the appearance of such a phase after 1 hour was less clear if it existed at all. Based on the higher concentration studies, we focused exclusively on 0.1 and 0.01M for the other temperatures investigated with 0.1M being the concentration we used at all 4 temperatures due to tradeoffs with experimental accuracy being slightly better at higher concentration while still avoiding phase separation issues.
As expected, a clear trend of increased degradation rates with increasing temperature is apparent, shown for 0.1M BTMA as a function of temperature in Figure 7. In comparing this data with our thermal transients (∼2 hours to achieve equilibrium), 160°C degradation occurs at a time scale (half-life of 4.8 hours, See Table III for half-lives of data in Figure 5 and associated degradation energy barriers) only about double that of where we would expect thermal equilibrium to have been achieved. The 140°C half-life, 25 hours, represents a time scale much longer than the thermal transient and also a time scale conducive to high throughput. The studies at 120°C and especially 80°C, take much longer. In fact, the 80°C data, in spite of running the experiments for over 2000 hours still have significant experimental uncertainty associated with it due to the low degradation that has occurred over that time frame.
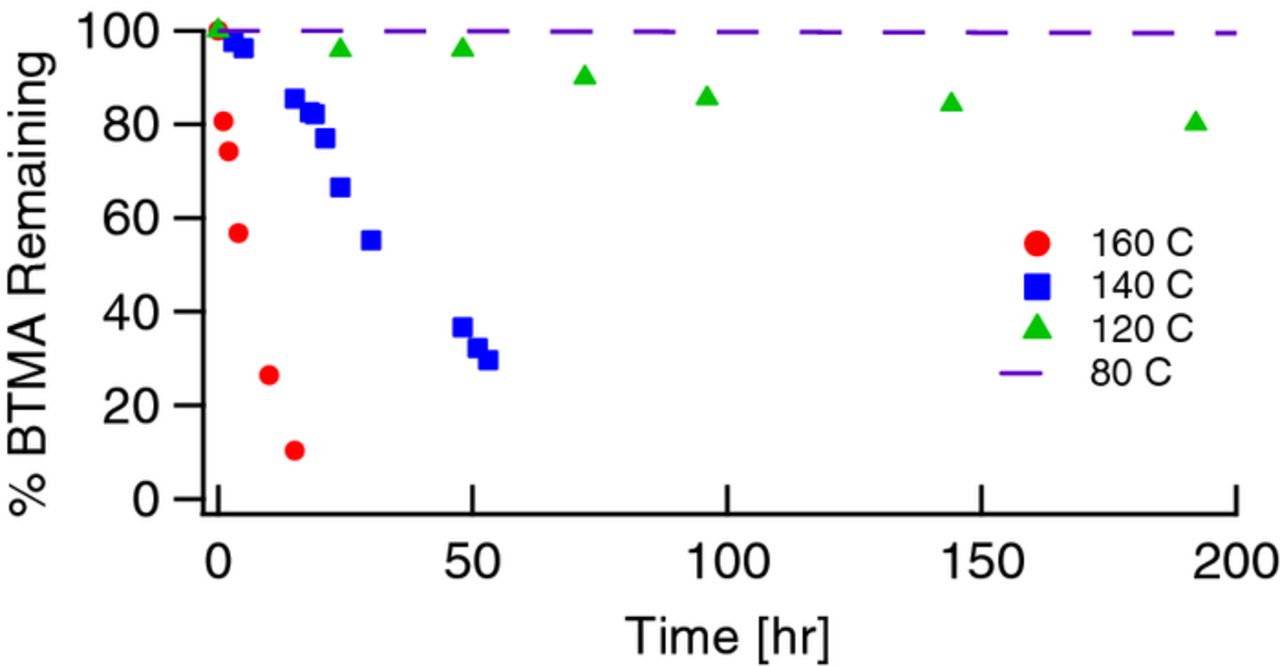
Figure 7. Degradation of 0.1M BTMA in 2M KOH at 80°C (–), 120°C (▲), 140°C (■), and 160°C (●).
In order to assess the viability of using higher temperature data to extrapolate down to lower temperatures, we used the data obtained in Table IV to compare energy barriers for degradation. At 0.1M, the degradation energy barriers at 120°C (136.4 kJ/mol), 140°C (132.6 kJ/mol), and 160°C (133.9 kJ/mol) are essentially unchanged. This shows that temperature at most has a very minor impact on observed energy barriers and that degradation rates obtained at higher temperatures can be used to extrapolate down to more relevant operating conditions. In addition, no additional products (due to thermal degradation) were observed at the higher temperatures. Taking the average energy barrier from these three temperatures and extrapolating down to 80°C results in a predicted half-life of greater than 4 years, with only 10% degradation occurring after 5300 hours. To further check the validity of this extrapolation, calculated values were overlaid onto the experimental data obtained for 80°C degradation in Figure 6D. It is clear that the calculated values agree well with the experimental data at 80°C and give further confidence that accelerated high temperature degradation rates can be extrapolated down to more relevant temperatures.
Table IV. Observed ½ lives and associated ΔG≠ of BTMA degradation at various concentrations (0.01M, 0.1M, and 0.5M) and temperatures (120°C, 140°C, and 160°C).
| T (°C) | Conc (M) | ½ life (h) | ΔG≠ (kJ/mol)§ | DFT ΔG≠ (kJ/mol)# |
|---|---|---|---|---|
| 160 | 0.5 | 2.41 | 131.4 | |
| 160 | 0.1 | 4.77 | 133.9 | 140.6 |
| 160 | 0.01 | 6.01 | 134.7 | |
| 140 | 0.1 | 24.7 | 132.6 | 138.1 |
| 120 | 0.1 | 558 | 136.4 | 135.6 |
| 120 | 0.01 | 529 | 136.0 |
§Calculated using ½ life values and equation 9 and 10 from Long 2012.7 #DFT calculation results using M06 method, 6-311++G(2d,p) basis set, and SMD solvation model.
When comparing the experimental data obtained in this study to that found in Table I, for the cases where degradation was actually quantified, the reported value of BTMA durability was much lower than that reported here; although in some cases degradation rates were not quantified and only reported as: below limit of detection; with the exception of the work of Marino and Kreuer27 which show good agreement with the BTMA data reported here, although identical conditions were not reported (use of KOH vs NaOH and lack of 2M base concentration). Some of the methods in Table I involved the inclusion of methanol or glycol, less polar solvents expected to accelerate degradation. Methanol has often been added due to solubility concerns of cations in solution. The use of lower cation concentrations in our experimental method has the potential advantage of alleviating solubility concerns. Additionally, the use of 2M KOH provides a significant excess of hydroxide so that hydroxide consumption during cation degradation can be ignored at these low concentrations. As a whole, the well quantified and reproducible BTMA degradation rates reported here suggest that BTMA is a potentially viable cation for a number of applications. Only for long lifetime applications (thousands of hours) at elevated temperatures (approaching 80°C) under well hydrated conditions (an additional caveat not specifically investigated here, but of potentially large impact in operating devices) will the stability of the free BTMA cation be a major concern. These results suggest that for a number of low temperature applications that the chemical stability of BTMA in the presence of hydroxide is likely to be sufficient for very long lifetime operations.
Conclusions
We have chosen BTMA as the most logical choice for establishing an appropriate baseline for free cation stability. In order to properly quantify degradation rates of cations in the presence of hydroxide we had to develop a novel method and isolate and minimize or eliminate several different sources of experimental error and uncertainty. The resulting method employing a relatively high base concentration (2M KOH), a relatively low cation concentration (< 0.1M), the removal of any deuterated solvents, and Teflon lined Parr reactors placed in convection ovens has allowed us to get reproducible degradation rates under thermally accelerated conditions. As we have applied our method to BTMA we have more accurately quantified its durability in alkaline environments and demonstrated that it has a higher durability than previously believed. This has strong implications for the range of operating conditions for which BTMA may be an acceptable cation for use in electrochemical devices.