

Abstract
Many complex mechanisms underlying the fascinating functionalities provided by tetrapyrrolic macrocycles in biochemistry have been already unraveled. Light harvesting, molecular transport, and catalytic conversion are some of the processes performed by tetrapyrrole-based centers embedded in protein pockets. The main function is determined by the single atom species that is caged in the macrocycle, while a finer tuning (band gap, chemical selectivity etc) is granted by the geometric and electronic structure of the tetrapyrrole, including its residues, and by the proximal and distal structures of the protein surroundings that exploit the molecular trans-effect and direct weak interactions, respectively. Hence, a scientific and technological challenge consists in the artificial replication of both structure and functionality of natural reaction centers in 2D ordered arrays at surfaces. Nano-architected 2D metalorganic frameworks can be indeed self-assembled under controlled conditions at supporting surfaces and, in the specific, porphyrin- and phthalocyanine-based systems have been widely investigated in ultra-high vacuum conditions by means of surface science approaches. Deep insight into the geometry, electronic structure, magnetic properties, ligand adsorption mechanisms, and light absorption has been obtained, with the strong experimental constraint of vacuum. Especially in the case of the interaction of tetrapyrroles with ligands, this limit represents a relevant gap with respect to both comparison with natural counterparts from the liquid environment and potential applicative views at both solid–liquid and solid–gas interfaces. Thus, a step forward in the direction of near-ambient pressure is strongly necessary, while maintaining the atomic-level detail characterization accuracy. Nowadays this becomes feasible by exploiting state-of-the-art experimental techniques, in combination with computational simulations. This review focusses on the latest advances in this direction.

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction: tetrapyrroles in nature
Nature exploits metal-organic architectures to perform a large variety of fascinating biological processes, including light-harvesting, energy conversion, and chemical synthesis [1, 2]. Life is essentially based on electronic and molecular evolution occurring at specific active sites that are synthetized by functional self-assembly [3], in the same way as polypeptide chains fold into proteins or complementary DNA strands amalgamate [1]. Many of these reactors consist in caged single metal atoms embedded in a protein backbone, like in some enzymes, catalyzing several chemical reactions essential for life (about a quarter of the genes in the human genome encodes enzymes [2]). The active sites selectively stabilize a transition state (i.e. the highest-energy species in a reaction pathway) to speed-up the process by means of the heteroleptic coordination occurring at the single metal centers (i.e. the bonding of two or more ligands). This is the case of 'intramolecular' multidentate coordination by exploiting the protein backbone as a superligand, and the case of dynamic coordination in solution with free, exchanging ligands [1]. In the specific, a very common architecture consists in the coordination of a single metal atom in a planar tetrapyrrolic cage (figure 1) [3, 4], yielding both steric [5] and electronic [2, 6] tuning advantages (as in heme-like systems) since the unsaturated metal atom can bind ligands on both sides of the molecular plane [3]. In this way, tailoring potentiality of the chemical reactivity is achieved.

Figure 1. (a) Chemical structures of different tetrapyrrole families and (b) various natural tetrapyrroles (Adapted from [3] under the Creative Commons CC BY 4.0 license); (c) crystal structure of selected enzymatic catalytic sites (Adapted from [4]. Copyright © 2019 Elsevier.).
Download figure:
Standard image High-resolution imageIn a mimetic approach [7, 8], a tailoring trans-effect [9–12] can be obtained by growing 2D self-assembled metal-organic heterostructures at surfaces, yielding ordered 2D crystals of equally spaced single metal atom reaction centers (figure 2) [8]. In this case the substrate-molecule charge transfer can influence and actually determine the electronic and structural configuration of the tetrapyrrole [13, 14], driving ligand adsorption and yielding electron density and spin tuning capabilities (figure 3) [10, 15, 16].

Figure 2. STM images of the ordered self-assembly of 2D metalorganic networks hosting different single atom metal centers. Reprinted with permission from [8]. Copyright © 2015 American Chemical Society.
Download figure:
Standard image High-resolution image
Figure 3. (a) Spin quenching and decoupling induced by the adsorption of axial ligands (NO, CO) at the Fe atom of FePc molecules adsorbed on the Au(111) termination in ultra-high vacuum and at T = 20 K (Adapted with permission from [16]. Copyright © 2011 American Chemical Society https://pubs.acs.org/doi/10.1021/jp204461k.); (b) calculated spin-resolved partial density of d-states for a free FePc molecule, after adsorption at the Au(111) surface, and upon axial ligation with NH3, Py, CO, and NO (Reproduced with permission from [15]. Copyright © 2010 Institute of Physics.); (c) calculated, optimized geometries and ligand binding energies of Co- and Fe-TPP molecules upon axial ligation with NO and CO, evidencing the role of the Ag surface trans-effect, represented by a single trans Ag atom (Reprinted with permission from [10]. Copyright © 2011 American Chemical Society.).
Download figure:
Standard image High-resolution imageAn impressive number of scientific papers and reviews has been written already, in order to shed light onto the working mechanisms of synthetic, biomimetic, and artificial homologue systems [1, 3, 8, 17–30]. Therefore, we come to the point questioning about the need for this additional review that may necessarily suffer from incompleteness and redundancy at the same time. Self-assembled monolayers of porphyrins and phthalocyanines have been investigated extensively by means of experimental surface science approaches under ultra-high vacuum (UHV) conditions or at the solid–liquid interface. In the view of potentially applicative purposes of biomimetic, synthetic 2D metal-organic heterostacks [14, 31], a strategic issue is the pressure gap in the case of solid–gas interfaces [32]. This work will focus for the first time on a very recent, emerging approach to the experimental, fundamental atomic-level characterization of the ligands interaction with 2D metal-organic heterostructures at surfaces. Indeed, by means of state-of-the-art techniques it is now possible to extend the detailed atomic-level characterization approach typical of surface science to the domain of the near-ambient pressure (NAP) regime, thus bridging the gap [33, 34]. This review, after introducing tetrapyrroles and some examples of standard UHV and ex situ approaches, will focus exactly on this point, by reporting recent advances in the characterization at close to ambient pressure conditions (room temperature and gas pressures in the mbar range) of self-assembled porphyrins and phthalocyanines monolayers. The change in chemical potential due to the several orders of magnitude higher pressure with respect to UHV studies yields access to new phenomena that can be characterized in situ at gas partial pressures up to hundreds of mbar. An example helps understanding why to focus on this specific pressure regime. Myoglobin (the protein that stores oxygen in tissues) hosts a heme prosthetic group with a central Fe atom. While Fe forms four bonds with the nitrogen atoms of the tetrapyrrole, the imidazole ring of a histidine (proximal) residue from the protein occupies the fifth coordination site, and the sixth site is available for oxygen bonding. Upon O2 adsorption, both the electronic and geometric configuration of the site change, together with the magnetic properties of the protein. In the case of hemoglobin, a similar picture is found, even if more complex cooperative mechanisms influence oxygen adsorption. However, a common aspect is that for both cases a distal histidine and the molecular trans-effect through the direct bonding of a proximal residue stabilize oxygen (figure 4) [35], with ligand saturation uptakes that develop in the 10–100 mbar range [2]. Self-assembled metalorganic monolayers at surfaces aim at producing ordered 2D arrays of artificial active coordination sites that resemble the catalytically active sites of metallo-enzymes, yielding the possibility of ligand adsorption and fine tuning of reactions by exploiting the surface-trans effect as the 2D counterpart of the molecular trans-effect. Citing a recent review concerning the enzymatic catalytic activity, 'the question arises whether these reactions also take place using metal-organic networks as catalysts' [8], and here the scope of the present work as a first step towards a fundamental view into this direction at the gas–solid interface.

Figure 4. O2 binding pocket in myoglobin, evidencing the role of the proximal histidine in the trans-effect and the O2 interaction with the distal His64 residue (Reprinted with permission from [35]. Atoms color legend: red—oxygen, grey—carbon, blue—nitrogen, white—hydrogen, yellow—iron. Copyright © 2003 Institute of Physics.).
Download figure:
Standard image High-resolution image1.1. Structure and functionality
As anticipated in the introduction, tetrapyrroles are widespread in Nature and essential for life, since many critical biological processes are based on these functional centers, including photosynthesis (chlorophyll) and respiration (heme). In the cyclic form of a tetrapyrrole (figure 1(a)), four pyrrole subunits are linked by methine bridges (apart from corrins). The macrocycle can host a single metal atom (Fe, Ni, Mg, Co ...), while attached residues (figure 1(b)) can tune the electronic configuration of the unit as well as the chirality [3]. Indeed, both the insertion of a metal atom and the interaction with the surrounding environment can induce a deformation of the molecular plane, thus generating a chiral system. The distortion of a tetrapyrrole (figures 5(a)–(c)) can be obtained in a 2D artificial system by means of metalation, adsorption on and interaction with a surface, and by ligand adsorption [24, 25, 36]. This is extremely relevant since the deformation of the tetrapyrrolic plane can be associated with its functionality. The macrocycle conformation can control its physicochemical properties by influencing the π-system, yielding modified light absorption bands, redox properties, fluorescence yields, Stokes shifts, lifetimes of electronic excited states, and intersystem crossing and internal conversion mechanisms [3, 37]. In the biology framework, the concept of 'conformational control' has been developed in order to exploit the macrocycle conformation to fine-tune its physicochemical properties (figures 5(d)–(f)) [38]. This can be achieved via steric interactions with the surrounding protein chains, via axial coordination of the central metal, and via π–π interactions [3]. This approach represents at the same time a rationale for explaining the different behavior of a same chromophore in different protein environments [38], and a starting point for the development of bottom-up 2D active systems based on tetrapyrrolic tectons at surfaces. As an example, already the simple mechanical deformation plays a role in the case of hemoglobin (figure 6) [39]. Oxygen binding shifts the Fe atom from outside the plane of the porphyrin into the plane and the proximal histidine residue gets pushed with it. The same occurs upon ligation with other adducts. Consequently, the structural transition at the Fe ion in one of the four subunits of the protein is transmitted directly to the other subunits. This mechanism is at the basis of the nonlinear, cooperative absorption of oxygen in hemoglobin [2, 39]. More in general, similar mechanisms are exploited in many metalloenzymes, representing approximatively one third of the known enzymes, and promoting a variety of reactions well beyond just absorption and transport, including bond cleavage and formation, electron transfer, atom transfer, and radical chemistry [40]. The general protein scaffold includes the primary coordination sphere of ligating atoms directly attached to the metal ion (including sulfur, oxygen, and nitrogen depending on the cases). The coordination complex plays a major role since the coordinating ligands often specify which metal ion can bind to the enzyme and control which oxidation and/or spin state can be accessed. The secondary coordination sphere, which includes atoms that are near (but not directly attached to) the metal ion, can be also considered as part of the scaffold, since the properties of these atoms can strongly influence the reactivity. The second coordination sphere strongly influences the kinetics and thermodynamics of enzymatic catalysis [40, 41]. Deeper comprehension of the working mechanisms of these complex systems has been obtained in the recent years. In a recent achievement, a reconstituted artificial metalloenzyme containing an iridium porphyrin exhibits kinetic parameters similar to those of the natural counterpart [41]. In the specific case, variants of the P450 enzyme CYP119 containing iridium in place of iron catalyze insertions of carbenes into C–H bonds with up to 98% enantiomeric excess and 2550 h−1 turnover frequency. In a second example, a chlorin tetrapyrrole containing an Mg ion is exploited in chlorophyll for light harvesting purposes. In the PS I complex, 12 polypeptides bind 96 light-harvesting chlorophyll and 22 carotenoid pigment molecules, yielding extremely efficient charge transport properties [42]. After photoexcitation of the special pair of chlorophyll (P700), an electron is transferred to a monomeric chlorophyll (Chl) within ∼1 ps (figure 7) [42]. The excited electron relaxes via two intermediate phylloquinones to three [4Fe–4 S] iron–sulfur centers. The first iron cluster (FX) is located just outside the molecular electron transfer chain, and the transfer rate from the phylloquinones to FX is biphasic with time constants of 15–150 ns. Lastly, the electron is transferred to the two iron clusters FA/FB in less than 500 ns.

Figure 5. (a)–(c) DFT computed structure for a CoTPP molecule adsorbed on the Cu(111) surface before (a) and after single (b) and double (c) ligation of a CO adduct (Reprinted with permission from [36]. Copyright © 2011 Springer Nature.); (d)–(f) linear displays of the deformation shifts along the z axis of the tetrapyrrolic plane for hemes in the α- and β-subunits of deoxyhemoglobin (d)–(e) and for the heme in isoenzyme−1 of cytochrome c from baker's yeast (f) (Reprinted with permission from [38]. Copyright © 1998 Royal Society of Chemistry.).
Download figure:
Standard image High-resolution image
Figure 6. Molecular models for carboxyhemoglobin, deoxyhemoglobin, oxyhemoglobin, and glycated hemoglobin: ligation of an adduct to the Fe atom in the porphyrin macrocycle induces structural deformations propagating to the whole protein. Adapted with permission from [39]. Copyright © 2018 Taylor & Francis.
Download figure:
Standard image High-resolution image
Figure 7. Electron funneling path and recombination times for the reaction center transfer chain in the chlorophyll P700 special pair. After photoexcitation, the electron is transferred to three iron-sulphur cubane clusters. Reprinted with permission from [42]. Copyright © 2012 Springer Nature.
Download figure:
Standard image High-resolution imageThe above examples, chosen as representative of a large number of cases, show as a common denominator that it is a combination of elements that actually determines the functionality of a tetrapyrrole-based system, including the metal ion, the primary, and the secondary coordination spheres. These ingredients yield tailored electronic, magnetic, and chemical properties, i.e. the functional character that synthetic, self-assembled 2D metalorganic monolayers aim at mimicking [8].
1.2. Electronic, chemical, and magnetic properties
The aromatic nature of tetrapyrroles provides the basis for their unique light absorption properties in the UV/Vis region (figure 8) [43], exploited by Nature for light harvesting, and of extreme interest in the view of single- and multi-exciton (ME) generation and charge separation in organic photovoltaics [3, 37, 44]. Absorption occurs in two regions termed the Soret or B-band (380–420 nm) and the Q-bands (500–800 nm), respectively. These properties originate from the splitting of the two Highest Occupied Molecular Orbitals (HOMOs—a1u and a2u) and Lowest Unoccupied Molecular Orbitals (LUMOs—degenerate egx and egy), well described by the Gouterman's four-orbital model [3]. The absorption bands in porphyrin systems arise from transitions between these orbitals, yielding two excited states, and it is the identities of the metal center and of the substituents on the ring that affect the relative energies (figures 8(a)–(b)). Orbital mixing splits the degenerate LUMOs, creating a higher energy state (Soret or B band) and a lower energy state (Q-bands). The absorption spectrum of a typical porphyrin consists therefore of two distinct regions. The former involves the transition from the ground state to the second excited state (S0 → S2), while the latter region consists of a weak transition to the first excited state (S0 → S1). These spectroscopic features of porphyrins stem from the conjugation of 18 available π-electrons and provide the advantage of easy and precise monitoring of guest-binding processes by UV-visible spectroscopic methods [43]. A key spectral difference between metalated and non-metalated porphyrins is the number of Q-bands (figure 8(c)). The lower symmetry of the reduced free-base (D2h) reflects in the non-equivalence of the two axes defined by the opposite pyrrolic nitrogen atoms, yielding non-degenerate Qx and Qy excitations. A generalized metalated porphyrin exhibits instead the highest symmetry for a porphyrin (D4h) [3].

Figure 8. (a) Representation of the four Gouterman orbitals in porphyrins; (b) energy levels of the four orbitals upon symmetry reduction from D4h to C2v; (c) UV–vis spectrum of a porphyrin (the inset enlarges the Q region in the 480–720 nm range). Adapted with permission from [43] under the Creative Commons CC BY 4.0 license.
Download figure:
Standard image High-resolution imageBesides the optical properties of tetrapyrroles associated with light harvesting functions, another biologic function is molecular transport and catalytic chemical conversion. As we have seen, porphyrins are indeed widespread as active centers in many enzymes and other functional biomolecules. Among many, hemoproteins represent the most studied class due their wide range of functions, including oxygen transport and storage (hemoglobin, myoglobin, neuroglobin, cytoglobin), catalysis (cytochrome P450s, cytochrome c oxidase, peroxidases), electron transfer (cytochromes a, b, and c) and defense (catalase) as examples [24]. Many complex mechanisms are responsible for the adsorption, conversion, and desorption of molecular ligands/adducts at the metal center in the tetrapyrrolic pocket. In the specific, the reactivity is mainly determined by four properties: (i) the embedded metal species, (ii) the peripheral structure of the tetrapyrrolic cage, (iii) the nature of the axial ligand (exploiting the trans-effect for tuning purposes), and (iv) the nature of the distal superstructure of the hosting protein [4]. These elements are of paramount importance, since they can be exploited for tailoring purposes in biomimetic systems (figure 9) [10, 45]. At surfaces, the tuning role of axial ligands is played by the surface trans-effect [10], where hybridization of the LUMO with the substrate states takes place [46, 47] while the surrounding metalorganic coordination environment (lateral interactions among tectons) governs the self-assembly [9, 26, 30, 48, 49], the charge delocalization [37], and the chemical/thermal stability [50]. In order to draw a characterizing picture of natural systems in the view of steering the surface synthetic counterpart, we can make further representing examples [4], referring to heme-based enzymes all containing a single Fe atom. Peroxidase enzymes, which catalyze the reaction RORR' + 2e− + 2 H+ → ROH + R'OH, have a histidine axial ligand (figure 1(c)). Catalase enzymes (converting H2O2 to O2) have instead a tyrosine ligand with a phenolate head group. In cytochrome P450 the thiolate head group of cysteine plays instead the role. The different bonding environments yield in this way a fine-tuning of the activity and selectivity of the 'same' Fe atom by stabilizing different intermediates. For hemoglobin and myoglobin the distal histidine stabilizes the dioxygen molecule via bonding to the imidazole head, while cysteine, showing an anionic thiolate head, activates molecular oxygen in P450, making O–O bond cleavage more favorable [4]. The residues around the active site also determine the electron and proton transfer channels, as well as the entrance and exit routes for reactants and reaction products. A last, very interesting example is again cytochrome P450. In the specific, benzene hydroxylation is a fundamental process catalyzed in Nature by P450. However, the oxygen transfer mechanism is of considerable complexity and it has been found that the whole tetrapyrrolic macrocycle takes part in the process [6]. Interestingly, a proton shuttling picture was proposed (figure 10) [6], in which a protonated nitrogen atom of the porphyrin ring plays the role of the intermediate state. Within this picture, the porphyrin protonation mechanism suggests that there is an enzymatic pathway to convert benzene directly to phenol and ketone, in addition to the post-enzymatic production by conversion of benzene oxide [6, 51]. The porphyrin ring acts as a proton shuttle, thus bypassing benzene oxide as a reaction intermediate, and benzene conversion is favored by a double low-spin mechanism (S = 1/2) with a calculated activation barrier (in vacuo) of 17.5 kcal mol−1 [6].

Figure 9. Role of the trans-ligation effect in biology as well as in the case of tetrapyrroles at surfaces: (a)–(b) deoxy- and oxy- myoglobin and hemoglobin (Adapted with permission from [45]. Copyright © 2017 National Academy of Sciences.); (c)–(d) DFT optimized structure of CoTPP/Ag72 before (left) and after (right) NO ligation (Adapted with permission from [10]. Copyright © 2011 American Chemical Society.). For both cases it can be readily observed that ligation of an adduct (O2 and NO, respectively) yields profound structural changes in the macrocycle, as well as in the residues. The trans-effect acts, thus influencing ligation, through the proximal histidine in the case of myoglobin, and through the Ag surface in the case of the CoTPP molecule. In the latter case, the adsorbed CoTPP molecule undergoes a saddle-shape distortion of the porphyrin core, accompanied by the rotation of the peripheral phenyl rings toward the molecular plane. Upon NO coordination to the Co center, the Co–Ag distance increases significantly.
Download figure:
Standard image High-resolution image
Figure 10. Doublet low-spin reaction pathways for the oxidation of benzene to benzene-oxide, phenol, and ketone at the active site of cytochrome P450. The red arrow evidences the proton-shuttling intermediate. Adapted with permission from [6]. Copyright © 2003 American Chemical Society.
Download figure:
Standard image High-resolution imageBesides electronic and chemical properties, tetrapyrroles show also interesting magnetic character when the caged transition metal atom possesses unpaired electrons and, thus, non-zero magnetic moment [24]. Coordination [15], as well as adsorption on a surface [52, 53], can influence the magnetic moment of the complex, with surprising magnetic behavior like for example spin-crossover [54–56] and singlet-fission [37, 57], associated with the possibility of exploiting metalated tetrapyrroles in spintronic devices (figure 11) [54, 58, 59]. Spin-crossover (figure 11(a)) is a thermal transition between electronic states with different numbers of unpaired electrons, accompanied by changes of a material's properties like magnetic moment, color, dielectric constant, and electrical resistance [56]. It is most commonly observed in six-coordinate iron(II), iron(III), and cobalt(II) molecular complexes, but several other types of compounds can exhibit the phenomenon, including organometallics. In practice, six-coordinate iron(II) complexes of nitrogen-donor ligands are most often used in spin-crossover research, since these exhibit the greatest structural differences between their high- and low-spin states. Thus, materials with that metal/ligand combination usually afford the most cooperative spin transitions with the most unusual structural chemistry [56]. As an example, iron(III) porphyrins and related homologues show a complex spin-state behavior. High/low spin (S = 5/2–S = 1/2), high/intermediate spin (S = 5/2–S = 3/2) and intermediate/low spin (S = 3/2–S = 1/2) equilibria have been demonstrated in synthetic ferric tetrapyrroles, while spin-state transitions are important to the functioning of several biologic heme proteins [56, 60, 61]. The low spin state can be converted into the metastable high spin state not only by temperature, but in some cases also by light (figure 11(b)) [54]. This phenomenon is known as the Light-Induced-Excited-Spin-State-Trapping (LIESST) effect. The reverse process is also possible (reverse LIESST), and is a common feature of most Fe(II) spin crossover systems. Singlet fission is instead a process that allows creation of two or more electron–hole pairs from the absorption of one photon that initially excites a fundamental singlet state S0 into a higher energy one, S1 (figure 11(c)). The process involves then a ME intermediate state, also called a correlated triplet pair 1(TT), with an overall singlet-spin multiplicity, that eventually relaxes into two independent triplet states (T1). One general principle is that the energy of 2 × T1 must be equal to or less than that of S1. Under this condition, the correlated triplet pair can relax to two triplets, which leads to a high singlet-fission efficiency [62]. Singlet-fission shows attracting applicative potentialities, since the efficiency of a conventional solar cell may be enhanced if one incorporates a molecular material capable of singlet fission, yielding the production of two triplet excitons from the absorption of a single photon [63]. Spin transitions can also affect the ligand adsorption properties of single metal atom reactive centers, thus playing an important role in catalysis by means of cooperative binding mechanisms. In the latter case, an initial binding event facilitates the uptake of additional substrate molecules, a mechanism that is common in biological systems such as hemoglobin. Selective adsorption of carbon monoxide (CO) in a series of metal-organic frameworks (MOFs) featuring coordinatively unsaturated iron(II) sites was found to be mediated by a mechanism by which neighboring iron(II) sites undergo a spin-state transition above a threshold CO pressure (figure 12) [64]. These materials exhibit large CO separation capacities with only small changes in temperature.

Figure 11. (a) Change of electron distribution between HS and LS states of an octahedral iron(II) coordination compound exhibiting thermal spin crossover. The orbitals eg* and t2g arise from splitting of the 3d orbitals in an octahedral ligand field. Depletion of charge in the antibonding eg* orbitals during HS to LS transition shortens the metal-to-ligand bond distances and reduces the molecular volume. The condition to fulfill in order to observe thermal spin crossover is ΔEHL ∼ kBT. Increasing the temperature favors the HS state, decreasing it favors the LS state (Adapted with permission from [54]. Copyright © 2000 Royal Society of Chemistry. Adapted with permission from [59] under the Creative Commons CC BY license.). (b) Jablonski diagram showing the photophysical processes of the LIESST and reverse-LIESST effects (Reproduced with permission from [54]. Copyright © 2000 Royal Society of Chemistry.). (c) Singlet fission in pentacene: initial photon absorption occurs at the left-hand side to access the optically bright single-exciton states, S1–S3; subsequent geometric relaxation can localize such states to an excited-state dimer unit (right side), which can lead to a nonadiabatic transition to the ME state, D (middle); the dark state D directly connects to a pair of triplet states, T1 (Adapted with permission from [58]. Copyright © 2011 American Chemical Society.).
Download figure:
Standard image High-resolution image
Figure 12. Cooperative spin-transition in a MOF: at a CO partial pressure below the step region of the isotherm, a phase consisting of all high-spin iron(II) (orange spheres) is observed. As the pressure is increased through the step pressure, some high-spin iron(II) is converted to low-spin iron(II) (purple spheres). When further increasing the CO pressure, beyond the step pressure, full conversion to a low-spin phase is observed. Grey, green, orange, purple, blue and red spheres represent C, Cl, high-spin iron(II), low-spin iron(II), N and O atoms, respectively; H atoms are omitted for clarity. Reproduced with permission from [64]. Copyright © 2017 Springer Nature.
Download figure:
Standard image High-resolution image2. Tetrapyrroles and MOFs at surfaces under UHV conditions: a fundamental approach
As we will see briefly in the following, the surface science of metal-organic supramolecular assemblies has come to a point at which deep fundamental insight has been already obtained. Self-assembly, growth, and metalation mechanisms have been described for a large variety of surface systems under UHV conditions, providing a toolbox to prepare superstructures with tailored and controlled properties [8]. Single metal atoms can be caged in regular arrays within these superstructures, thus yielding 2D functionalized surface periodic patterns that resemble the active sites of metallo-enzymes. Several chemical and photochemical reactions take place at the embedded metal ions in enzymes and the very question is whether these reactions can take place efficiently also using metal-organic networks as catalysts [8]. Moreover, a biomimetic approach to the artificial synthesis of model structures that reproduce the active sites and organic ligands of enzymes may yield a step forward towards a deeper understanding of efficiency and selectivity issues of enzymatic reactions [8, 26, 65]. Of course, both similarities and differences occur between 2D networks and metalloenzymes. In the specific, the role of the supporting, templating surface is extremely relevant since it also works as a charge reservoir within the picture of a surface-trans effect. This provides effective electronic, chemical, and (electro)-catalytic tuning capabilities [8, 10, 14], together with selected electron-dynamics properties in the case of donor/acceptor interfaces for light harvesting and charge separation purposes [37, 66]. As an example, a strategic reaction from both environmental and technological points of view is the reduction and conversion of carbon dioxide [67]. A few surface science examples are reported in the literature [8, 14], in which a biomimetic approach is exploited to engineer the first, most difficult step consisting in the activation of the molecule. CO2 is indeed a closed shell, very stable molecule and its activation is obtained by injecting negative charge in it, thus yielding a bent, 'V' shaped precursor that is chemically reactive [68–72]. In Nature this step is performed by the enzyme Ribulose−1,5-bisphosphate carboxylase oxygenase (RuBisCO) where the active conversion center incorporates a Mg2+ ion. The modular, ordered self-assembly of Mg atoms and TPA molecules on a Cu(100) surface (figure 13) constitutes a first 2D synthetic approach in this direction [8]. The point remains however if what is observed under model conditions (low temperature, UHV) or ex situ, can be directly transferred to applicative, realistic catalytic working environments. The answer is generally that this is not the case, thus pinpointing the need for in situ and operando approaches, while still maintaining a fundamental, atomic-level insight (sections 3.2 and 3.3). Reducing the pressure and material gaps, and interestingly even the terminology gap (table 1) separating surface science from biochemistry [73], may contribute in the development of a merged forma mentis and approach, a necessary starting point for the effective investigation of 2D biomimetic systems.

Figure 13. (a)–(b) Model of the RuBisCO enzyme and of its reactive center, hosting a single Mg++ ion; (c) STM image of the Mg-TPA metalorganic network on Cu(100). Adapted with permission from [8]. Copyright © 2015 American Chemical Society.
Download figure:
Standard image High-resolution imageTable 1. Common terminology pertaining to the fields of chemistry/biochemistry and surface science: differences and similarities.
| Chemistry/Biochemistry and surface science | |
|---|---|
| Absorption | Chemistry/biochemistry: Physical or chemical phenomenon or process in which atoms, molecules or ions enter a bulk phase; at variance with adsorption, absorption is a bulk-specific process; the process of absorption refers also to light when the photon's energy is captured by matter |
| Surface science: Analogously, sticking/diffusion/entering of single atoms or small molecules from the gas phase, through the surface, into the interstitial subsurface or bulk sites of a solid | |
| Adsorption | Chemistry/biochemistry: Adhesion of atoms, ions or molecules from a gas, liquid or dissolved solid to a surface; binding of a protein or a cell to a surface |
| Surface science: Analogously, sticking of a small molecule at a surface, a cluster, or at a single metal atom at a solid–gas interface; adsorption is a surface-specific process | |
| Ligand | Chemistry/biochemistry: Ion, molecule, or functional group that binds to a central metal atom to form a coordination complex |
| Surface science: Analogously, small molecule adsorbed at the single metal atom center (as in a tetrapyrrole) | |
| Residue | Chemistry/biochemistry: An atom or a group of atoms that forms part of a molecule (like a methyl group); a specific monomer within a polymeric chain of a protein (like histidine) |
| Self-assembly [26] | Chemistry/biochemistry: bonding of complementary molecular units forming a stable complex; this mechanism plays a fundamental role in the function and proliferation of living organisms |
| Surface science: Synonym of molecular self-assembly, a primary strategy for noncovalent synthesis of complex 2D monolayers or supramolecular assemblies/networks, typically integrating multiple components with molecular recognition capabilities | |
| Self-organization [26] | Chemistry/biochemistry: It indicates a cooperative or collective behavior; it addresses the emergence of order from cosmic to macromolecular dimensions; interestingly, self-organization represents the transition from inanimate matter to living organisms |
| Surface science: Spatiotemporal order phenomena in open systems, governed by competing energy supply, diffusion, and chemical interactions constraints | |
| Self-organized growth [26] | Chemistry/biochemistry: Arrangements in processes accompanied by matter accumulation |
| Surface science: Mesoscale evolution, in molecular-beam epitaxy, of regular patterns in semiconductor or metal-epitaxial growth, governed by the delicate interplay between thermodynamics and kinetics | |
| Substrate | Chemistry/biochemistry: Typically, the chemical species of interest, often a precursor, which reacts with a reagent to generate a product; in enzymatic catalysis, the substrate is the material upon which the enzyme acts |
| Surface science: Surface (generally a single-crystal termination) at which growth, deposition, or reactions take place | |
| Template | Chemistry/biochemistry: A template reaction is a ligand-based reaction that occurs between two or more adjacent coordination sites on a metal center; in coordination chemistry, the template effect emphasizes the pre-organization provided by the coordination sphere |
| Surface science: Surface with a regular pattern offering guidance for the regular adsorption or self-assembly of a geometrically ordered adlayer |
2.1. Experimental and theoretical insight at the atomic level
2.1.1. Molecular assembly
Supramolecular, surface-confined coordination chemistry dates back to the early 1980s [18], and exploits self-assembly protocols at surface atomic lattices employing metal centers to steer the organization of molecular ligands, together with the template-induced organization of pre-engineered tectons [19]. Molecular self-assembly at surfaces is governed by the fine balancing between intermolecular and molecule-surface interactions. The admolecules experience indeed the potential energy surface of the templating substrate and surface mobility is a key parameter for the assembly process. The strength and nature of the competing molecule-molecule lateral interactions are similarly important. Finally, the thermal energy governs the level of ordering within both energetic and kinetic constraints. Experimental observations reveal a unique variety of low-dimensional coordination architectures that can be synthesized in vacuo at well-defined single crystal terminations. It is suggested that striking achievements may be expected when complementary expertise in synthetic chemistry, surface preparation and nanoscale characterization, and theoretical modeling will come together [18]. The most convenient synthesis routes, pertaining to bottom-up or top-down approaches, should also be addressed in the view of a large scale production of molecularly engineered 2D heterostacks [17]. However, an extension of the fundamental, atomic level detail approach typical of surface science beyond the vacuum gap will be necessary in this sense. Achieving long-range order over multiple length scales is also important when the assemblies' properties are to be harnessed at a macroscopic level. Organizational hierarchies are abundant in biological systems and biomaterials like proteins and viruses, but this is still far from being translated to 2D systems [26].
Both covalent and non-covalent bonding can be exploited to bind molecular tectons in 2D ordered superstructures (figure 14) [30, 74–76]. Under UHV conditions, covalent synthesis or coupling [77] can be achieved by means of the thermal activation of building blocks and their subsequent chemical reaction at predefined connection points (figures 14(a)–(c)). This can be obtained within the framework of two approaches. In the first one, molecular building blocks are pre-deposited on a surface, where they get successively activated by dissociation of the substituent atoms upon annealing. In the second case, instead, activation occurs in the thermal evaporator and the already activated molecules are deposited on the substrate [30]. The non-covalent synthesis under UHV conditions exploits instead planar π-tectons with peripheral functional groups that adsorb, on appropriate substrates, in flat-lying geometry, thus favoring lateral group recognition (figure 14(d)). The cooperative geometrical complementarity (zwitterionic coupling) promoting the formation of multiple weak linkages (H-bonding, Van der Waals interactions) is an important factor in 2D supramolecular engineering [26, 78]. Moreover, hydrogen-bonded networks assembled at low temperatures on a surface can be transformed into metal-organic arrangements by exploiting the catalytic activity of the substrates upon annealing. Hydrogen bonding provides selectivity and directionality, is abundant in biological systems, and stable in solution, molecular crystals, and in 2D layers. In the head-to-tail hydrogen bonding scheme a head pyridyl group and a tail carboxyl group are ideal [27, 49]. The coordination of transition metal atoms to tailored ligands at surfaces provides an alternative versatile approach for the synthesis of highly organized molecular arrangements (figure 14(e)). The relatively high metal-ligand bond energies yield robust structures and the same stabilizing metal atoms bestow specific electronic, magnetic, or catalytic functions [26]. A representative example is the self-assembly of guanine on Au(111), where the formation of multiple H bridges enhances the single bond strength from 0.22 to 0.42 eV/bond (figure 14(f)). Interestingly, the stabilized guanine 2D quartets resemble the guanine-based tetramers in cyclic biological supramolecular complexes [26, 76]. Bifunctional 2D networks have also been synthetized, including non-equivalent monometallic centers. Coordination and metalation with Cu of 2H-TPyP molecules on Au(111) between room temperature and 450 K allow the formation of a network that includes mixed-valence single Cu atoms, where Cu(II)TPyP complexes are coordinated and stabilized by Cu(0) centers [79].

Figure 14. (a)–(c) Mechanism for the activated covalent bonding of Br4TPP/Au(111), together with STM images collected after deposition of the tectons and thermal activation of the bonding (Adapted with permission from [30]. Copyright © 2007 Springer Nature.); (d) terephthalic acid self-assembly on Au(111) (Adapted with permission from [74]. Copyright © 2004 American Chemical Society.); (e) STM imaging of a Co-biphenolate honeycomb network on Ag(111) (Adapted with permission from [75]. Copyright © 2007 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim.); (f) cooperative hydrogen bonding in the self-assembly of 2D guanine networks on Au(111) yielding a regular network of flat-lying molecules comprising quartet arrangements (Adapted with permission from [76]. Copyright © 2005 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim.).
Download figure:
Standard image High-resolution image2.1.2. Metalation
The metalation of tetrapyrroles at surfaces under UHV conditions represents an effective route to the synthesis of metal-organic active complexes, thus adding biomimetic functionalities [28]. In the biologic environment, free-based species undergo metalation in the homogenous liquid phase by reaction with dissolved metal salts. Enzymes catalyze the process by a multi-step reaction pathway involving a significant geometric distortion of the macrocycle. At surfaces, at least two common aspects are shared with the biologic counterpart. A sitting atop complex plays a relevant role as an intermediate metalation step, in which the metal center already forms a coordination bond to the macrocycle, but the central H atoms are not yet released. Secondly, the molecule-surface interaction may induce geometric distortions of the tetrapyrrole macrocycle in analogy to the enzymatic process [38]. This is obtained thanks to the conformational adaptation of the tetrapyrrole to the local adsorption environment through the flexibility of the porphyrin plane and the rotational degrees of freedom of the meso-groups [80]. Significant deformations of the molecular shape occur upon both adsorption at a surface and metalation [81–83], generating buckled and 'umbrella'-shaped profiles [84]. Under UHV conditions, metalation has been addressed by means of x-ray photoelectron spectroscopy (XPS), near edge x-ray absorption fine structure (NEXAFS) spectroscopy, scanning tunneling microscopy and spectroscopy (STM and STS), temperature programmed desorption (TPD), sum-frequency generation (SFG) spectroscopy, and modeled by means of ab initio density functional theory (DFT) calculations.
Metalation routes. Several metalation routes can be exploited at surfaces in UHV (figure 15) [28], including physical vapor deposition (PVD), self-metalation, tip manipulation, and chemical vapor deposition (CVD) [24, 25, 28, 29, 80, 84–91]. For PVD, both free-base macrocycles and single metal atoms are deposited on a supporting surface, and temperature-dependent metalation occurs, in some cases even at ambient temperature [92]. Since strong molecule-substrate interactions are to be avoided in this case, relatively inert surfaces are preferred like in the case of Ag(111) and Au(111) terminations [28]. In order to promote the actual metalation, the molecular layers need often to be annealed, depending on the chosen free-base compound, up to 550 K. This leads to stability issues related to the tetrapyrroles. For 2H-TPP/Ag(111), annealing to 550 K induces indeed parallel cyclodehydrogenation, yielding both rotation and fusion of the peripheral phenyl rings [93]. Therefore, on-surface metalation is limited by the chemical/thermal stability of the selected molecular species. Self-metalation consists instead in the deposition of the tetrapyrroles in vacuo on a metal termination, followed by a thermal treatment that induces incorporation of a surface metal atom of the underlying surface into the tetrapyrrolic cage. The sitting atop complex, the formation of which may also coincide with the rate-limiting step, is again the most common candidate intermediate. The extraction of the metal atom from the top-most surface layer may determine the activation barrier that can be lowered in some cases by oxygen pre-adsorption. However, best candidates for the self-metalation process are diffusing single metal adatoms detaching from steps and largely available already at room temperature [94]. Thus, the 2D phase equilibrium between adatoms and steps (2D vapor pressure) of the metal surface may represent the limiting factor [85]. The latter vapor pressure can be modified by tuning the surface temperature or by oxygen coadsorption, so explaining the observed phenomenology. Whatever the picture, however, strong kinetic limitations contribute and may even prevail the thermodynamic energy balance [85]. Local tip manipulation by means of an STM can also be exploited to induce metalation. The 2H-Pc → AgPc + H2 reaction was induced on a Ag(111) surface by initial hydrogen extraction upon application of voltage pulses. Two-fold symmetric molecules are converted to symmetric four-fold appearance, showing a central depression associated with N bonding to the substrate. An Ag-covered W tip was then used to transfer a single Ag atom into the dehydrogenated macrocycle, thus finally obtaining the AgPc [95]. Precursor complexes (like e.g. metal carbonyls) can be instead used in the CVD metalation process, a variant of the co-deposition method [28].

Figure 15. Summary of the major on-surface metalation methods described in the text. Adapted with permission from [28] under the Creative Commons CC BY NC 3.0 license.
Download figure:
Standard image High-resolution imageExperimental metalation markers. Several surface science techniques can provide experimental metalation markers, i.e. measurable fingerprints of the presence of a central single metal atom in the tetrapyrrolic cage of the macrocycle on a surface (figure 16) [85, 90, 91, 96–99]. XPS of the N 1s core level generates local and collects spatially averaged information about the N bonding environment (figure 16(a)). In particular, free-base tetrapyrroles exhibit two nonequivalent nitrogen species in the core of the macrocycle, namely the pyrrolic (–NH–) and the iminic (–N=) nitrogen atoms. A significant core level shift allows clear distinction between the two species, contributing with adiabatic N 1s peaks at 399.8–400.3 and 397.6–398.5 eV, respectively, depending on the molecular species and on its interaction with the metal substrate. Upon metalation, the N 1s associated with the M-N4 center grows around 398.6–398.9 eV, depending on the metal, at the expense of the former doublet [28, 29, 89, 90, 96, 100–102]. Local geometric information of tetrapyrroles adsorbed at surfaces can be obtained by angle- and/or energy-resolved photoemission experiments in photoelectron diffraction based approaches, allowing distinction between metalated and non-metalated molecules [84, 103]. Experimental data, complemented by simulations, revealed precious information about the deformations of 2H-P and CuP on Cu(111) induced by molecule-substrate interactions [84]. Another spectroscopic approach that can provide both electronic and geometric structure information at the same time is NEXAFS (figure 16(b)). By combining polarization-dependent measurements with ab initio simulations, resonant electronic transitions from the C and N 1s core levels to outer LUMOs can be resolved and identified. Thus, the presence of a central metal species and the geometric alignment of both the macrocycle and the peripheral residues can be inferred on average for a 2D mono- or multi-layer. In the case of 2H-TPP and CuTPP on Cu(111), the investigation of π and σ N 1s transitions up to the LUMO+26, together with the C 1s resonances, allowed the thorough characterization of the –NH–, =N–, N–Cu local environments and of the orientation of the peripheral phenyl rings [90]. Accurate TPD and temperature programmed reaction measurements (figure 16(c)), associated with isotope-exchange labelling, can provide indirect metalation evidence by following the temperature-dependent evolution of the H2, DH, and D2 desorption rates [96, 104]. Indeed, after formation of the sitting atop complex, the metal atom actually enters the macrocycle and a hydrogen molecule is released. At temperatures above 550–660 K, dehydrogenation of the external moieties of the tetrapyrrole takes place, contributing in hydrogen desorption features up to 1000 K [96, 104]. When the surface metalation reaction is oxygen-assisted, water is produced instead [99–101]. STM can be exploited as a local probe to distinguish between metalated and non-metalated single tetrapyrroles. However, the interpretation of the image appearance is not always straightforward and the intuitive assignment of a corresponding geometric topography may be sometimes misleading (figures 16(d)–(e)). Indeed, while in most cases metalated tetrapyrroles at a surface show a central bright protrusion upon STM imaging, in selected cases the opposite occurs. The latter is the case, for example, of NiTPPs adsorbed at the Cu(100) termination (figure 16(e)). The strong molecule-surface interaction, associated with a significant charge transfer, yields the rotation of the phenyl residues, the consequent bright appearance of the peripheral, distorted terminations, and a dim molecular macrocycle, despite the presence of the central Ni atom [97]. Information about the local density of states close to the Fermi level can be obtained by means of STS, allowing distinction between metalated and non-metalated tetrapyrroles, as well as identification of possible ligands [10]. Raman spectroscopy can also be exploited for the investigation of tetrapyrroles at surfaces (figure 16(f)), yielding spectroscopic information about the internal vibrational modes of the adsorbed molecules. In the specific, lateral resolution can be obtained by the aid of a tip (tip-enhanced Raman spectroscopy—TERS), as in the case of 2H-TBPP and ZnTPP on Ag(111) [98]. Combined vibrational and vibronic insight can be obtained by means of nonlinear optical approaches, like IR–Vis SFG (IR–Vis SFG) spectroscopy (figure 16(g)). The intrinsic interface sensitivity of the technique makes this a fundamental approach that can be employed for the investigation of metalorganic monolayers at surfaces in situ and operando well beyond UHV, up to ambient pressure and even at the solid–liquid interface in electrochemical environments. Few examples have recently appeared in the literature concerning porphyrin and phthalocyanine monolayers at single crystal terminations investigated by means of this technique both in UHV and at NAP conditions [37, 99, 105–108], and the self-metalation of 2H-TPPs on the Pd(100) surface has been specifically addressed [99, 108].

Figure 16. Experimental techniques delivering metalation markers of tetrapyrroles in surface science: (a) N 1s core level evolution upon metalation with Fe atoms of a TPP monolayer on Ag(111) (Reproduced with permission from [85]. Copyright © 2008 American Chemical Society.); (b) self-metalation of 2H-TPP/Cu(111) followed by means of NEXAFS (Reproduced with permission from [90]. Copyright © 2012 AIP Publishing.); (c) self-metalation and progressive dehydrogenation of 2H-TPP/Cu(111) followed by means of TPD spectroscopy (Adapted with permission from [96]. Copyright © 2014 WILEY‐VCH Verlag GmbH & Co. KGaA, Weinheim.); (d) STM imaging of 2H-Pc and FePc molecules on Ag(111) where the central metal atoms appear as bright protrusions (Adapted with permission from [91]. Copyright © 2008 American Chemical Society.); (e) STM imaging of NiTPP/Cu(100) where the whole macrocycle hosting the single Ni atom appears as a dim structure (Reproduced from [97] under the Creative Commons CC BY license.); (f) TERS spectroscopy of tetrapyrroles on Ag(111) (Adapted with permission from [98]. Copyright © 2017 Springer Nature.); (g) IR–Vis SFG spectroscopy of PdTPP/Pd(100) [99].
Download figure:
Standard image High-resolution imageActivation barriers. The quantitative evaluation of the barriers for the metalation of tetrapyrroles at surfaces is far from being straightforward. From the computational point of view, ab initio approaches result in demanding efforts due to the complexity of the system, since both molecule-molecule and molecule-substrate interactions should be accounted for, requiring very large computational unit cells. Therefore, reaction barriers have been computed mainly for gas phase reactions (figure 17(a)) [89], and the direct comparison with the experiment (figures 17(b)–(c)) is quite puzzling [85, 99]. On the other side, we have seen that many metalation markers can be exploited in surface science experiments to follow the single metal atom inclusion in the tetrapyrrole. Temperature- and time-resolved measurements can therefore provide thermodynamic information about the metalation process. However, kinetic hindrance plays a relevant role due to: (i) the 2D constrains of the system [85], (ii) the single metal adatom diffusion at the surface [94], (iii) the ballistic cross section of the tetrapyrrole core upon PVD, thus questioning the obtained barriers numbers. In general, depending on the substrate, on the molecule, and on the metal atom, metalation temperatures between RT and 570 K have been observed under UHV conditions at single crystal surfaces [24], and even just below RT (285 K) as in the case of the oxidized copper (100) surface [109]. Activation energies of 1.48 ± 0.12 and 1.35 eV were measured for the self-metalation of 2H-TPP on Cu(111) [110], and for the metalation of 2H-TPP/Ag(111) with Zn [89], respectively. The computational barrier values, obtained via ab initio methods, strongly depend on the computational framework. Concerning the mechanism, it is confirmed that the reaction starts with the coordination of the metal atom by the intact porphyrin, resembling the sitting-atop complex proposed for the metalation in solution (figure 17(d)) [89, 111], accompanied by a distortion of the macrocycle as induced by dedicated biological metalation enzymes (figure 17(e)) [112–114]. The pyrrolic hydrogen atoms are then transferred to the metal atom, and successively released in the form of H2. The H-transfer step represents the rate-limiting configuration, within a single (as for Cu and Zn) or a double (as for Fe, Co, Ni) step metal insertion mechanism in the formation of the sitting atop complex. Alternative pathways include the release of water when pre-adsorbed oxygen is necessary to promote self-metalation (figure 17(c)), as in the case of 2H-TPP/Pd(100) [99, 108], 2H-TPP/MgO, and 2H-TPP/MgO/Ag(100), for which also the formation of surface hydroxyl groups is proposed [115, 116].

Figure 17. (a) Calculated metalation mechanism of a 2H-P with Zn in gas phase (Reprinted with permission from [89]. Copyright © 2007 American Chemical Society.); (b) PVD metalation with Fe of 2H-TPP/Ag(111) with hydrogen desorption in UHV (Adapted with permission from [85]. Copyright © 2008 American Chemical Society.); (c) self-metalation of 2H-TPP/O/Pd(100) with water desorption in UHV [99]; (d) sitting atop complex calculated for the Fe2+ ion for decreasing numbers of H2O ligands (from left to right) in solution (Adapted with permission from [111]. Copyright © 2004 Elsevier.); (e) tetrapyrrole distortion mechanism catalyzed by enzymes for the synthesis of metalated tetrapyrroles in biology: in the specific, bonding at the ferrochelatase of a N-MeMP porphyrin inhibitor is represented (Adapted with permission from [112, 114]. Copyright © 2000 and 2008 Elsevier.).
Download figure:
Standard image High-resolution image2.2. Reactivity: ligands, charge transfer, and magnetic tuning
We have already seen how the coordination of axial ligands to metalated tetrapyrroles is a key for the biological functionality of these complexes, as well as for their technical applicative potentiality as gas sensors, single metal atom catalysts [21, 22], and for light harvesting and spintronic purposes. Within the model framework of surface science, several fundamental studies dedicated to ligand adsorption at the single metal atom sites of tetrapyrroles at surfaces have been reported, yielding profound insight into the reversibility of charge, spin, and chemical switching mechanisms [24, 25]. However, the majority of these case studies refers to UHV conditions and cryogenic temperatures, which are often mandatory in order to 'freeze' small, loosely bound ligands at the coordination site.
Adsorption of an axial ligand competes with the metal interaction with the underlying supporting surface (figure 18) [10, 36, 97, 117], well interpreted by the surface trans effect [10], in perfect analogy with the biological molecular trans-effect. In the cytochrome P540 family the cleavage of the O–O bond and other reactions at the Fe site are selectively tuned by an opposite (trans) axial thiolate ligand. Similarly, the binding between NiTPPs and the Cu(100) surface results in the filling of LUMOs up to the +3 level (figure 18(a)) [7, 97], consequently stabilizing NO2 even at room temperature under UHV conditions [118]. Also CoTPP and FeTPP undergo a reduction of the metal oxidation state, when deposited at the Ag(111) termination, yielding the formation of new valence states [10, 119]. Upon adsorption of NO at the metal ions at 140 K, the Co–Ag and Fe–Ag interaction is suppressed, thus reversing the surface-induced effects and yielding an increase in the oxidation states and the disappearance of the new, surface-induced valence states (figure 18(b)). Annealing to 500 K induces NO desorption and restores the initial situation. Weak binding is instead observed for NO and NH3 with ZnTPP/Ag(111), while NO–Co and NO–Fe binding energies as high as 1.29 and 1.75 eV are obtained, respectively. Despite the strong interaction in the latter case, saturation of the single metal sites can be achieved by exposing the metalorganic monolayers to as much as 300 L of NO [10]. This suggests the participation of activated adsorption mechanisms, associated with the metal-substrate trans-interaction, or of kinetic and steric effects. The same applies indeed to the adsorption of O2, NO2, and NH3 on the CoTPP/Au(111) monolayer, where 600 L are necessary to obtain saturation of the sites at 80 K [120]. In this case, the presence of the ligand is investigated by means of STM, allowing its direct imaging. NO stability at room temperature in UHV conditions has been observed upon ligation at the central metal atom in a tetrapyrrole on a surface in a few cases. After exposure to 6000 L NO of a CoTPP/O/Ni(100) termination, the stable ligand could be sharply imaged in the 6–300 K temperature interval, thanks to the strong decoupling of the molecular adsorbates and their orbitals from the metallic substrate associated with the oxygen reconstruction [12]. The same NO exposure was necessary to saturate the CoTPP/Ni/Cu(100) system [117]. The binding of ligands was observed also for the case of covalently bound tetrapyrroles: a new class of biomimetic 2D tetrapyrrole systems, namely graphene-anchored AgP molecules, can indeed bind CO at low temperature in UHV [121].

Figure 18. Tetrapyrrole-surface charge transfer influencing axial ligation (surface trans-effect) for (a) NiTPP/Cu(100) as measured by μ-ARPES experiments (Adapted with permission from [97]. Copyright © 2017 Springer Nature) and (b) CoTPP/Ag(111) systems (Adapted with permission from [10]. Copyright © 2011 American Chemical Society); (c) CO mono- and di-ligation (dashed and solid circles, respectively) to CoTPP as imaged with STM (Adapted with permission from [36]. Copyright © 2011 Springer Nature); (d) calculated optimal geometry for the cis-dicarbonylation of a saddle-shaped CoTPP molecule (Adapted with permission from [36]. Copyright © 2011 Springer Nature.); (e) NO-controlled CoTPP/Ni/Cu(100) spin switch (Adapted with permission from [117]. Copyright © 2010 Springer Nature).
Download figure:
Standard image High-resolution imageWe have discussed how the geometric distortion of the macrocycle of a tetrapyrrole molecule is exploited in Nature within the framework of enzymatic catalysis, both for the tetrapyrrole metalation reactions and to achieve reactivity tuning and selectivity. Similarly, the macrocycle can undergo geometric distortion upon adsorption at a metal surface due to the strong interaction with it, resulting in saddle-shape conformations [36]. Bending of the molecular plane provides more space for the bonding of ligands (figures 18(c)–(d)), so that cis-di-carbonyl binding was observed upon exposure of CoTPP and FeTPP on Ag(111) and Cu(111) in the 6–20 K range [36, 122]. The reactive cleavage of a small ligand was also observed, as for O2 adsorption on MnTPP/Ag(111) at room temperature [123]. Exposure of the metalorganic monolayer up to 104 L of O2 converts 90% of the tetrapyrroles from Mn(II)TPP to Mn(III)OTPP. Interestingly, the conversion proceeds in couples of molecules, suggesting that the two cleaved O atoms bind to adjacent tetrapyrroles. The same reasoning can be extended also beyond 2D systems based on tetrapyrroles, achieving biomimetic charge-transfer, cooperative, and chemical tuning capabilities in 2D (metal)-organic/metal interfaces in general [124–126]. A bioinspired magnesium-organic network assembled on the Cu(100) surface resembles the RuBisCO enzyme, the most abundant protein on earth, and is able to mimic the Mg-carboxylate RuBisCO active site [127]. CO2 binds at room temperature (470 L) on the Mg2+ reactive centers, inducing a charge imbalance that catalyzes a phase transition into a different 2D crystal configuration. O2 (450 L) interacts instead with Mg2+ giving rise to an irreversible distortion in the metal-organic bond that leads to the collapse of the structure. A second example exploits di-iron centers in a MOF grown on Cu(100) that are able to catalyze the dissociation of molecular oxygen thanks to the cooperative catalytic action of two adjacent Fe2+ atoms, resembling both structural and functional analogies with the cofactors of non-heme enzymes [128]. It is found in this case that the active response of di-iron centers contrasts with the behavior of mono-iron centers, which selectively bind and stabilize oxygen molecules but do not undergo a structural transition, nor induce O2 dissociation.
Magnetic tuning is also possible by ligand adsorption (figure 18(e)) [129]. The interplay between the adsorption of NO (S = 1/2) or NH3 (S = 0) and supporting Ni and Co ferromagnetic surfaces affects indeed the spin of Co(II)TPP (d7, S = 1/2), Fe(II)TPP (d6, S = 1), Mn(II)TPP (d5, S = 5/2), and Mn(II)Pc (d5, S = 3/2) molecules [12, 117]. The structural trans-effect governs the molecular spin state and the sign and strength of the exchange interaction with the substrate. The phenomenon is addressed in terms of a spin trans effect, and is reversible upon desorption of the ligand. Thus, ligand coordination can be exploited as a chemical spin switch. The same applies also in the case of CO and NO coordination at 20 K to FePcs/Au(111) [130]. Four level switches can instead be obtained by exploiting the proton transfer within the empty tetrapyrrole, locally controlled by means of an STM tip in UHV at 6 K [131].
3. A fundamental approach beyond UHV
In the previous sections, we have seen that similar reasoning can be done for both biologic enzymatic tetrapyrroles and for their 2D synthetic counterparts, at least concerning a few of their properties and some aspects of their behavior. Charge and spin can be carefully tuned in surface-confined metal-organic networks, at least in UHV, and we tackle here the issue whether this fundamental bottom-up approach can be extended beyond the surface science UHV environmental constraints. Recently, the reactivity of tetrapyrrole monolayers at surfaces has been investigated in situ at the solid/liquid interface under electrochemical conditions, mainly by STM methods [20, 132–142]. Very recently, instead, surface science spectroscopic approaches combining SFG and NAP-XPS methods allowed extension of the investigations of the ligand-macrocycle interaction up to the mbar pressure regime at room temperature, thus close to applicative relevant conditions [14, 37, 99, 105, 108]. We recall that by definition an 'in situ' approach allows the collection of experimental data (spectra, images ...) under the same conditions relevant to the system operation, while 'operando' combines the in situ characterization of a working system/surface/catalyst during genuine reaction conditions with the simultaneous measurement of the system's performance (activity, selectivity, efficiency ...) [20].
3.1. In situ and ex situ characterization of tetrapyrroles at surfaces: tackling the solid/liquid interface
In perspective, the advances in the in situ/operando characterization techniques represent an important step towards the better understanding of the role of single-atom catalysts (SACs) [20–22] as model systems for sharpening our insight into the nature of catalytic sites [20]. Tetrapyrrole-based heterostacks and, more in general, SACs have become one of the most active new frontiers in heterogeneous catalysis [22], at least for a number of processes. Some applicative prototype reactions like CO2 electroreduction can be excellently run for example on model CuPc-based electrodes, yielding good selectivity and excellent stability [143]. The C–C coupling from carbon dioxide via electrochemical reduction routes represents a big challenge [142]. In situ XANES and NEXAFS experiments, combined with in situ XPS allowed the characterization of a nanostructured Fe(III) oxyhydroxide complex on nitrogen-doped carbon yielding high Faraday efficiency and selectivity to acetic acid synthesis. It was found that Fe(II) coordination to the N sites directly correlates with the high reactivity of the system. The presence of the N-species on the carbon substrate both stabilizes the active Fe(II) centers hindering further reduction of Fe, thus inhibiting the hydrogen evolution, and allows coordination of the reactive CO2-related species related with the liquid phase [142]. Interestingly, it is found that the latter (H2CO3, HCO3−, CO3−) consistently differ with respect to their counterpart in the case of the solid/gas reaction [68–72, 144–146]. Metal-nitrogen coordinated electrocatalytic systems offer the capability of electron donation/withdrawal to/from the carbonaceous ligand, thus with improved and tunable activity [147–150]. A single-atom and single-molecule insight can be obtained by means of in situ STM, like in the case of 2H-OEP, NiOEP, and CuOEP on Au(111) (figures 19(a)–(c)) [132]. The same approach allowed imaging of FePc/Au(111) under electrochemical conditions catalyzing the oxygen reduction reaction [138]. By shifting the electrode's potential, high- and low-contrast FePc species can be reversibly interconverted. The former species are associated with the O2-FePc complex (figures 19(d)–(k)) [138]. Depending on the potential, a rearrangement of the molecular network is also observed, witnessing a weaker molecule-substrate interaction at negative potentials (50 mV). On a manganese atomic network, STM allowed the detection of different oxidation states of individual tetrapyrroles. The reduction of the Mn center of the porphyrin from Mn(III) to Mn(II) with loss of the counterion is a requirement for the binding of an O2 molecule to give a dioxygen adduct. The O–O bond can then be cleaved following two alternative routes: (i) the reaction with Mn(II) to form a Mn(IV)-oxo complex, or (ii) the reaction with an electron and two protons to water and an Mn(V)-oxo ionic species [140]. On a Highly Oriented Pyrolytic Graphite (HOPG) support, STM imaging allowed distinction among a variety of different chemical states, and the O transfer mechanism was also investigated in situ. The mechanism involving the homolytic dissociation of a Mn–O–O intermediate would require the simultaneous conversion of two adjacent porphyrins, which was not observed. The surface-mediated diffusion of atomic oxygen and the solvent may instead contribute in a more consistent picture [140]. Finally, in situ STM combined with ex situ spectroscopies allowed the investigation of the metalation mechanism of tetrapyrroles. In the specific case, the metalation with Zn of protoporphyrin IX (PPIX) molecules bonded directly to the Au(111) surface or to a self-assembled monolayer on Au(111) was studied at the solid–liquid interface, revealing an important role of the underlying substrate in the metalation process. PPIX molecules could be fully metalated already at room temperature when in direct interaction with the Au surface [134].

Figure 19. (a) Scheme of the electrophotochemical STM approach and (b), (c) imaging at the solid–liquid interface of a NiOPE (b) and CuOPE (c) monolayer on the Au(111)/mica substrate; (d)–(g) sequential STM images and cross-section profiles of a FePc monolayer on Au(111) in 0.1 M HClO4 saturated by oxygen at different potentials; (h)–(k) top/side views of the computed density of states for the O2-FePc and FePc molecules. (a)–(c) Adapted with permission from [132]. Copyright © 2018 American Chemical Society. (d)–(k) Adapted with permission from [138]. Copyright © 2016 American Chemical Society.
Download figure:
Standard image High-resolution image3.2. In situ characterization of tetrapyrroles on surfaces at NAP
The progressive extension beyond UHV conditions of the experimental approaches adopted in the examples reported above reveals itself as a necessary step towards a fundamental understanding of nanostructures based on tetrapyrroles under operative conditions. We will review in the following very recent examples in this sense, a summary of which is listed for best clarity in table 2.
Table 2. List of discussed 2D adsorption systems and heterostacks based on tetrapyrroles that were recently investigated at near-ambient pressure conditions and room temperature.
| Ligand | P range (mbar) | Support | Tetrapyrrole | Effects | Reference |
|---|---|---|---|---|---|
| Carbon monoxide | 100–101 | Ir(111) | FePc | Ligation, monocarbonylation | [105] |
| Carbon monoxide | 100–101 | GR/Ir(111) | FePc | Monocarbonylation, singlet-fission, excitons | [37] |
| Carbon monoxide | 101 | Al2O3/Ni3Al(111) Cu/ Al2O3/Ni3Al(111) | FePc | Monocabonylation, geometric tunability | [50] |
| Carbon dioxide | 101 | GR/Ir(111) GRO/Ir(111) | FePc | Selective adsorption, bond tuning, trans-effect | [14] |
| Molecular oxygen | 10–5–10–3 | Au(111) | CoTPyP | Stabilization of dioxygen | [107] |
| Molecular oxygen | 10–7–100 | Pd(100) | H2TPP | Gas phase induced self-metalation | [108] |
3.2.1. Reactivity: ligands adsorption, charge transfer
Now we know that since metallotetrapyrroles possess in general two axial coordination sites available as centers for catalytic activity or sensor functionality, the coordinatively unsaturated character of the metal ions is the key to their specific reactivity and biological importance [10]. Therefore, in 2D, the competition between the two axial ligands (molecules from the gas phase form one side, and the underlying surface metal atoms on the other one) plays a central tuning role. The charge transfer between adsorbed tetrapyrroles and the surface actually determines the electronic configuration of the binding pocket, including both the single metal atom and the whole macrocycle [14]. Moreover, the time-dependent evolution of local charges (excitons) generated by the absorption of visible photons in a single tetrapyrrole is related to the molecule-substrate interaction and by the interconnections among the molecules in the 2D organic framework. This is extremely interesting in view of potential applicative ideas in which charge generation by impinging light (light harvesting, exciton formation) and charge separation (photovoltaics) are strategic properties. Lateral interactions can yield intersystem crossing and singlet-fission processes in a two-for-one picture, where two long-lived excitons are created upon absorption of a single photon [37].
3.2.1.1. Carbon monoxide as a probe ligand
A first experimental spectroscopic evidence obtained in situ for the adsorption at room temperature and NAP conditions of a small ligand at the solid–gas interface is represented by the carbonylation, at equilibrium with the carbon monoxide gas phase, of iron phthalocyanines [37, 50, 105]. For FePcs adsorbed on the (111) termination of an iridium single crystal [105], the adsorption process occurs at CO pressures above the mbar yield. CO binds to the molecules giving origin to a C–O stretching feature at 2004 cm−1 (figures 20(a), (f)). The low-energy region of the measured IR–Vis SFG spectra (1400–1800 cm−1) delivers instead information about the internal vibronic modes of the Pc. In the specific, small but interesting changes are observed also in the vibrational fingerprint of the metalorganic molecules upon adsorption of CO, i.e. in the resonances at 1529 and 1597 cm−1, associated with the in-plane stretching of the porphyrazine (P) and benzene (B) groups, respectively. A small blue-shift is observed for the B feature, while its phase varies by 40°, consistently with a re-alignment of the benzenic rings of the molecule. A small phase shift (20°) is observed also for the P resonance, witnessing a deformation of the macrocycle and/or a charge redistribution. A simple Langmuir description of the adsorption mechanism yields a CO–Fe binding energy of about 0.3 eV, depending on the assumptions about the pre-exponential factor (figure 20(h)). The same internal C–O stretching energy is observed upon adsorption of a single CO ligand on the same FePc molecule supported on an ultra-thin oxide film (figure 20(a)). The formation of the FePc metalorganic monolayer on the ultrathin alumina film grown on the Ni3Al(111) termination can be steered, since it is possible to artificially divert from substrate- to laterally-driven self-assembly mechanisms by properly tailoring the corrugation of the potential energy surface of the growth template [50]. By exploiting the capability of the Al2O3/Ni3Al(111) supercell dot sites to host metallic nanoparticle seeds, the symmetry of the FePc 2D crystal is tuned, thus showing that it is possible to switch from trans to lateral dominating interactions in the controlled growth of the organic/inorganic heterostack (figures 20(b), (d)). By selecting the size of the metallic clusters, the FePc-metal interaction strength is also controlled and the FePc bonding geometry is influenced (figures 20(c), (e)). A different picture appears instead upon single carbonylation of a FePc monolayer grown on an almost free single graphene sheet [37]. STM imaging (figure 20(g)) of the FePc/GR/Ir(111) system reveals the presence of central bright protrusions at the Fe sites and an almost square lattice, characterized by an angle of 87° and lattice vector lengths of 14.1 and 13.6 Å. The FePc monolayer lattice is rotated by 13° with respect to the underlying GR sheet, while the molecules are rotated by 18° with respect to the FePc lattice. The CO uptake curve in the mbar range yields again a CO–Fe binding energy of 0.3 eV, in line with the COFePc/Ir(111) case (figure 20(h)), and in remarkable agreement with ab initio theoretical calculations (red line in figure 20(h)). Surprisingly, the vibronic data obtained by means of IR–Vis SFG spectroscopy allow the clear identification of not one, but four non-equivalent internal C–O stretching modes at 1986, 1992, 2005, and 2011 cm−1, respectively (figure 20(a)). DFT calculations unequivocally confirm instead adsorption of a single CO molecule at each Fe site, thus excluding multiple-carbonylation [37], as observed in UHV and cryogenic temperature for CO adsorption on Co and Fe porphyrins [36]. The puzzling scenario is explained only within a picture that considers that the visible beam (532 nm) employed for the SFG measurements generates short-lived localized excitons (figure 21(a)) [37]. The latter singlet excited states decay within a few hundreds of fs to long-lived triplet states (figure 21(c)) via a singlet fission mechanism thanks to an intersystem crossing process associated with the tetrapyrrole lateral interactions mediated by the supporting graphene sheet (figure 21(b)). Thus, in addition to the fundamental low-spin ground state, upon adsorption of visible photons, three non-equivalent high-spin triplet states are populated, with a lifetime (28 ± 8 ps) of the order of the experimental laser probe pulse duration. The non-equivalent electronic configuration of the excited and ground state tetrapyrroles reflects in a different (and dynamic) Fe to CO charge transfer, affecting the C–O bond strength (within a Blyholder picture) that can be measured by the induced shift in the internal C–O stretching frequency. The CO ligand is therefore exploited as a vibrational fingerprint of the excitonic configuration of the tetrapyrrole center, so that excitons are written by visible light and read in situ by a vibrational probe at room temperature in a mbar pressure environment [37].

Figure 20. Monocarbonylation of FePcs at NAP: (a) SFG spectra of the C–O internal stretching region collected at room temperature in situ for FePc monolayers self-assembled on (from top to bottom) the ultrathin alumina film grown on Ni3Al(111) [50], the Cu-seeded alumina film [50], the bare Ir(111) termination [105], and a single graphene sheet on Ir(111) [37]; (b) STM image of the FePc/Al2O3/Ni3Al(111) superstructure [50]; (c) DFT structure of the FePc at the dot sites of the alumina ultrathin film, showing that no binding occurs [50]; (d) STM image of the FePc/Cu10/Al2O3/Ni3Al(111) superstructure [50]; (e) best DFT structures for the FePc molecule adsorbed at the dot sites of the alumina film pre-seeded with 6- and 9-atom Cu clusters [50]; (f) cartoon of the FePc molecule adsorbed at the Ir(111) surface [105]; (g) STM images and structural models of the FePc monolayer self-assembled on a single graphene sheet grown on the Ir(111) termination [37]; (h) SFG CO uptake curves measured in situ at room temperature for the systems shown in (f) and (g), shown together with the best fit (dashed lines) and the ab initio predicted theoretical curve (red line) [37, 105]. Adapted with permission from [37, 50, 105]. Copyright © 2018 Springer Nature under the Creative Commons CC BY license, 2018 American Chemical Society, and 2016 American Chemical Society, respectively.
Download figure:
Standard image High-resolution image
Figure 21. CO-FePc/GR/Ir(111) at room temperature at NAP conditions [37]: (a) FePc TR-2PPE integrated intensity for the short-lived state plotted as a function of the pump-probe delay (spectra were recorded with hν1 = 2.4 eV—pump—and hν2 = 4.8 eV—probe); (b) Jablonski diagram of the exciton formation mechanisms in the CO-FePc/GR system: the visible radiation yields excitation of the fundamental low-spin (LS) state into an excited singlet configuration, followed by fast relaxation into long-lived triplet high-spin (HS) states through inter-system crossing (ISC) and singlet fission (SF) mechanisms; (c) FePc TR-2PPE integrated intensity for the long-lived state plotted as a function of the pump-probe delay collected as in (a). Adapted from [37] under the Creative Commons CC BY license.
Download figure:
Standard image High-resolution image3.2.1.2. Activation of carbon dioxide
In a biomimetic approach, the activation of carbon dioxide at a 2D crystal of phthalocyanines supported by graphene (the same system reported in the previous section—FePc/GR/Ir(111)) can be controlled by chemically tuning the position of the Dirac cones of the support through oxygen adsorption [14]. The CO2-Fe chemical bond is stabilized at room temperature and characterized in situ by means of IR–Vis SFG spectroscopy, corroborated by computational simulations. The CO2-Fe bond is tailored by tuning the charge transfer across the interface between the graphene and the metalorganic layer. This is accomplished through the oxidation of graphene at close to ambient conditions. Only slight p-doping is observed indeed for the bare GR/Ir(111) system, where Dirac points are only 0.067 eV above the Fermi level. A strongly binding site can be obtained from a weakly binding one in this 2D heterostack. Indeed, an electronic bandgap, associated with a shift of the Dirac cones below or above the Fermi level, can be artificially induced by oxidizing GR already under model UHV conditions, or by intercalating oxygen beneath it, thus by introducing n- or p-doping, respectively. At NAP, n-doping of the FePc-graphene heterostack can be obtained by oxidation [14]. CO2 adsorption becomes favored thanks to the charge transfer at the organic/inorganic interface to the FeN4 centers. We will see that, consequently, on the doped system, carbon dioxide binds to Fe at 0.05 mbar at room temperature in a 'V' shaped, highly reactive configuration.
In the undoped system only the physisorbed, linear CO2 configuration is stable and no adsorbed state is detected. As from figure 22(a), the GR optical mode at 1614 cm−1 dominates the IR–Vis SFG vibronic spectrum of the clean, as grown FePc/GR/Ir(111) heterostack in UHV. Two internal FePc modes are observed at lower energy: the benzene scissoring at 1335 cm−1 and the B2 mode, involving the tetrapyrrole and the isoindole moieties, at 1398 cm−1. No significant modifications of the vibronic spectrum of the FePc are detected in situ when exposing the undoped system at room temperature to 5 mbar CO2 (figure 22(b')), indicating that CO2 adsorption does not take place.

Figure 22. (top) DFT results for the negatively charged FePc (+2e) and its interaction with adsorbed CO2 molecule: from left to right, the 'V'-shaped CO2 molecule adsorbed on the FePc, additional electronic density in the charged FePc molecule in absence of CO2, charge transfer from the FePc to the adsorbed CO2 molecule; (bottom) IR–Vis SFG spectra including the range of the FePc intramolecular modes, of the optical G mode of graphene, and of the internal CO2 stretch. The spectra were collected in situ sequentially from (a) to (d): (a) FePc 2D crystal on GR/Ir(111) as prepared, under UHV conditions; (b') in 5 mbar CO2; (b) after oxidation in 100 mbar O2 at room temperature; (c) FePc 2D crystal on GRO/Ir(111) in 0.05 mbar CO2; (d) FePc 2D crystal on GRO/Ir(111) after evacuation, back to UHV. The low wavenumber part of the spectra (left) has been magnified for clarity. The best fit curve of the data is also shown (continuous lines), together with the deconvolution (colored, filled curves) of the FePc internal modes. Adapted with permission from [14]. Copyright © 2019 American Chemical Society.
Download figure:
Standard image High-resolution imageWhen the as-grown 2D system is exposed to 100 mbar O2 the non-resonant background and the resonance associated with the GR phonon are strongly amplified, the latter resonance undergoing a redshift from 1614 to 1609 cm−1 (figure 22(b)). IR–Vis SFG data combined with XPS core level spectra support the formation of graphene oxide (GRO). The shape change of the IR–Vis SFG spectral contribution of the FePcs (figures 22(a)–(b)) witnesses a phase shift of −60 ± 5°, attributed to the oxidation inducing the doping. A pronounced energy bandgap (>0.35 eV) is introduced indeed at the Fermi level, yielding a deformation of the dispersion from linear to parabolic, and a downshift of the valence band below the Fermi edge. The FePc/GRO heterostack treated in this way is then exposed carbon dioxide at 0.05 mbar. Two peaks grow this time at 1535 and 1729 cm−1 (figure 22(c)), associated with an internal mode of the FePc (porphyrazine stretching), and with the antisymmetric internal stretching mode of an activated, adsorbed carbon dioxide species, respectively. CO2 binds in a bent, 'V'-shaped configuration at the Fe metal center, because of the transfer of charge from the metal to the molecule (figure 22, top panel). The SFG spectral features associated with the internal FePc modes (figure 22(c), left) indicate adsorption of the ligand at the macrocycle. The deformation of GR and of the tetrapyrrole macrocycle, the reorientation of the benzene rings, and the charge transfer from the macrocycle to the ligand induce the observed increase of the SFG amplitudes, as well as the phase shifts. An additional phase shift by −260 ± 5° with respect to the pristine FePc/GR heterostack and by −200 ± 5° with respect to the modified FePc/GRO is detected in the vibronic fingerprint of the macrocycle. The CO2 uptake is reversible, so that CO2 desorbs from the Fe sites when recovering UHV conditions (figures 22(c)–(d)) and all spectral features are accordingly restored. The picture is corroborated and confirmed by ab initio DFT calculations. The additional electronic charge available on the FePc is initially mainly located at the Fe center and at the macrocycle, while it is distributed to a less amount to the remaining atoms. Upon bonding of CO2, a dipole forms across the macrocycle plane and some electronic charge is transferred from the latter to the ligand according to the cited trans-effect. As mentioned, carbon dioxide is stabilized at the Fe atom in the 'V' shaped configuration (CÔC = 144°), in line with what generally observed for CO2 chemisorption on metal surfaces. The CO2-Fe bond (2.26 Å) takes place via the C atom (figure 22, top panel) with a calculated energy of 0.28 eV, in agreement with the experimental data.
3.2.1.3. Ligation of di-oxygen
The production of oxygen from water using homo- and hetero-bimetallic catalysts can be boosted by the suitable insertion of metal centers in an organic environment. Recently, both the stability and the activity of M1TPyP-M2 monolayers (with M1,2 = Co, Fe) grown on Au(111) were probed at the solid–liquid interface [137, 151]. In addition to the metal atom M1 in the center of the macrocycle, a second single metal atom M2 is incorporated by the four pyridyl groups between surrounding M1TPyPs, thus yielding a bimetallic ordered superstructure. The bimetallic frameworks can be prepared in all different metal combinations and show the same STM topography [151]. Ex situ topographic and spectroscopic characterization reveals structural changes of the molecular coordination network after oxygen reduction, and its decomposition and transformation into catalytically active Co/Fe (oxyhydr)oxide during oxygen evolution [137]. The spectroscopic characterization of the CoTPyP/Au(111) monolayer was performed in situ, instead, at the solid–gas interface at room temperature. The results indicate that reversible O2 adsorption occurs already in the low 10–3 mbar range [107]. In the specific, in the 1200–1300 cm−1 region several resonances pertaining to the tetrapyrroles are resolved at 1215, 1233, and 1264 cm−1 (figure 23(a)), and at 1560, 1570, 1589, and 1595 cm−1, associated with both macrocycle and pyridyl vibrational modes. Upon exposure to molecular oxygen, a resonant feature progressively grows at 1276 cm−1, reversibly disappearing upon pumping out oxygen and recovering UHV conditions. Thus, this latter vibronic contribution is associated with the O2 interaction with the metalorganic monolayer. More in detail, the measured value is in between that of the internal O–O stretching of an adsorbed superoxide species (1049 cm−1) and that of a physisorbed O2 molecule (1565 cm−1) [152]. The mode at 1276 cm−1 is therefore compatible with the internal O–O stretch of an adsorbed O2 species, stabilized by the charge transfer from the Co atom to the dioxygen adduct. While the resonance amplitude grows with the oxygen background pressure (figure 23(c)), the amplitude of the non-resonant SFG background diminishes (figure 23(b)), witnessing substantial changes in the electronic configuration of the system close to the Fermi level (λVis = 532 nm, hν = 2.33 eV) during the O2 uptake. The tetrapyrroles are not significantly further distorted, since the resonances' phases do not evolve (figure 23(a)) [107].

Figure 23. Room temperature di-oxygen ligation to a CoTPyP metalorganic framework on Au(111): (a) IR–Vis SFG spectra collected in situ at low (bottom) and high (top) O2 background pressure together with the best fit and the deconvolution of the resonances; (b) amplitude of the non-resonant background as a function of the O2 background pressure (the dashed line is drawn to guide the eye); (c) saturation curve obtained from the SFG resonant amplitude of the mode at 1276 cm−1 indicated by the dashed line in (a) (the continuous line in (c) represents the best fit with a Langmuir uptake model) [107].
Download figure:
Standard image High-resolution image3.2.2. Self-metalation
Self-metalation is one of the most promising synthesis routes to include a single metal atom in a tetrapyrrolic dye macrocycle supported by metal surfaces. The molecule-surface interaction may provide the charge transfer and the geometric distortion of the molecular plane that are necessary for metal inclusion, thus mimicking the enzyme-mediated metalation of porphyrins in biology. However, while the latter process proceeds at room temperature, at a metal surface the presence of an activation barrier can represent a technical obstacle that cannot be overcome by compensating with a higher temperature without affecting the layer integrity. As we have already discussed, the formation of the intermediate state, the sitting-atop complex that weakens the two N–H bonds in the tetrapyrrole preceding the metal insertion, can be facilitated in some cases by oxygen pre-adsorption at the supporting metal surface in UHV, like in the case of the H2-TPP/O/Pd(100) system. In such cases, the activation barrier can be overcome by mild annealing, yielding the formation of desorbing products (hydrogen, water) and of the metalated tetrapyrrole [99]. It has been recently shown [108], instead, that the self-metalation of H2-TPP molecules at the bare Pd(100) surface can be induced already at room temperature by the presence of a high-pressure oxygen gas phase. At close-to-ambient conditions an Eley–Rideal mechanism, resembling the asymmetric interactions available at an enzymatic pocket, opens the way to novel routes to MOFs at surfaces (figure 24(c)). An ordered H2-TPP/Pd(100) monolayer can be prepared by deposition of the porphyrins on the substrate at room temperature, followed by annealing in UHV to 410 K to induce best ordering [99]. The IR–Vis SFG spectrum of the as prepared H2-TPP monolayer, collected at room temperature in UHV conditions (figure 24(a), bottom) resolves eight distinct vibronic features in the low energy spectral region (filled profiles). In the specific, the resonances are measured at 1327, 1369, 1436, 1442, 1492, 1499, 1558, and 1600 cm−1 accounting for several stretching, bending, and rocking modes [99, 108]. Annealing of the organic layer in UHV up to 470 K does not induce significant spectroscopic modifications, thus indicating good stability and poor reactivity of the molecules. Indeed, the layer does not self-metalate upon annealing in UHV. The intensity ratio of the resonances at 1327 and 1369 cm−1 can be exploited as a metalation fingerprint when the H2-TPP monolayer is deposited on the oxygen pre-covered Pd(100) termination and subsequently annealed to 410 K, thus inducing metalation and water production at variance with the H2-TPP/Pd(100) case. By exploiting this metalation marker, self-metalation was found to occur at room temperature in 1 mbar O2 (figure 24(a), top). The feature at 1369 cm−1 undergoes a blue shift of 6 cm−1 and almost disappears, yielding a lower relative amplitude with respect to the resonance at 1327 cm−1. Three modes actively contribute to the former resonance: the Cβ–Cα–NH asymmetric stretching, the H rocking of the macrocycle pyrrolic moieties, and the Cα–N–Cα' stretching. On the contrary, the lowest energy feature mainly originates from the phenyl H rocking modes. The spectroscopic information suggests therefore a strong local deformation of the C–N bonds and the loss of the central H atoms, compatibly with the metalation to PdTPP molecules. XPS confirms the picture (figure 24(b)). The N 1s spectrum of the as-prepared system corresponding to the vibronic spectrum in the bottom part of figure 24(a) is reported in the bottom of figure 24(b). Two symmetric components of equal intensity can be clearly resolved at binding energies of 399.8 and 397.8 eV, associated with the pyrrolic (N–H) and iminic N, respectively, of H2-TPPA adsorbed at the Pd surface. Room temperature exposure of the system to NAPs of oxygen yields the N 1s spectrum shown in the top part of figure 24(b). A new peak doublet and two additional, intense singlet features grow at the expense of the initial peaks, as evidenced by the deconvolution (filled profiles). In addition to the pristine H2-TPPA species (light yellow), a new H2-TPPB species (dark yellow) can be spectroscopically distinguished, associated with pyrrolic and iminic N 1s components at 399.2 and 397.0 eV, respectively. The intense peaks are instead associated with the metalated form of the porphyrin, and grow at 397.5 eV (PdTPPA—orange) and at 398.2 eV (PdTPPB—light orange). A consistent kinetic picture shows that impinging O2 molecules undergo dissociation at the center of the tetrapyrroles, contributing at the same time to the formation of the metalation transition state (first O atom, sitting-atop complex) and to the oxidation (second O atom) of the Pd surface. Exposure to oxygen yields also formation of the B species (both H2TPPB and PdTPPB), interpreted as porphyrins whose phenyl groups have undergone partial dehydrogenation due to reaction with adsorbed oxygen originating from the O2 dissociation in the formation of the sitting-atop complex.
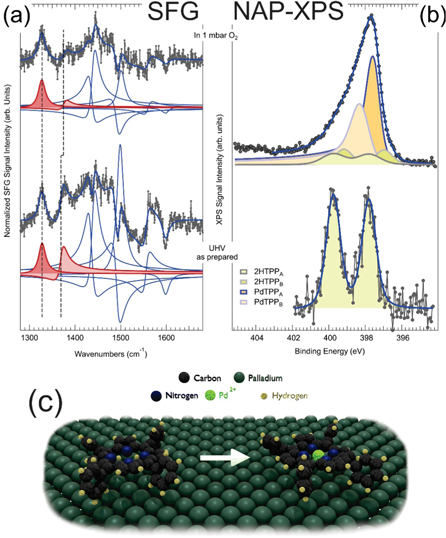
{kind=link}
{kind=link}
{kind=link}
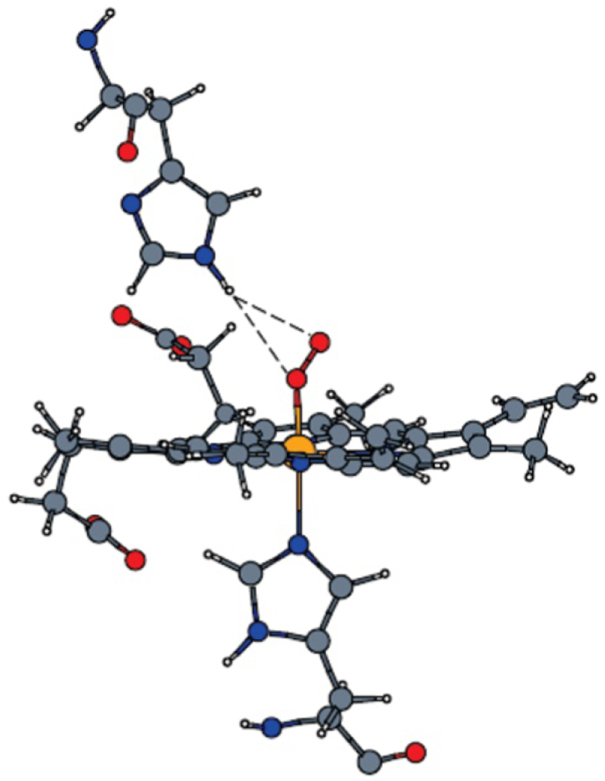{kind=link}
{kind=link}
{kind=link}
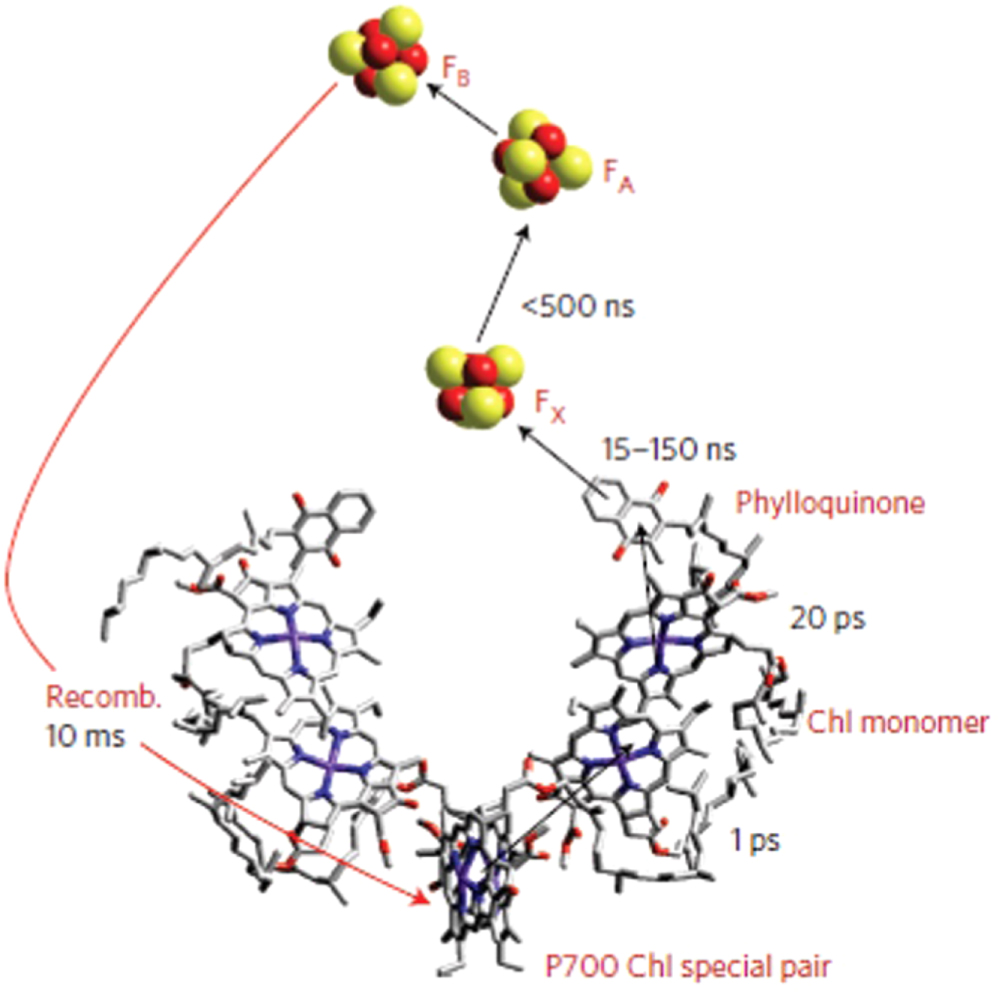{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
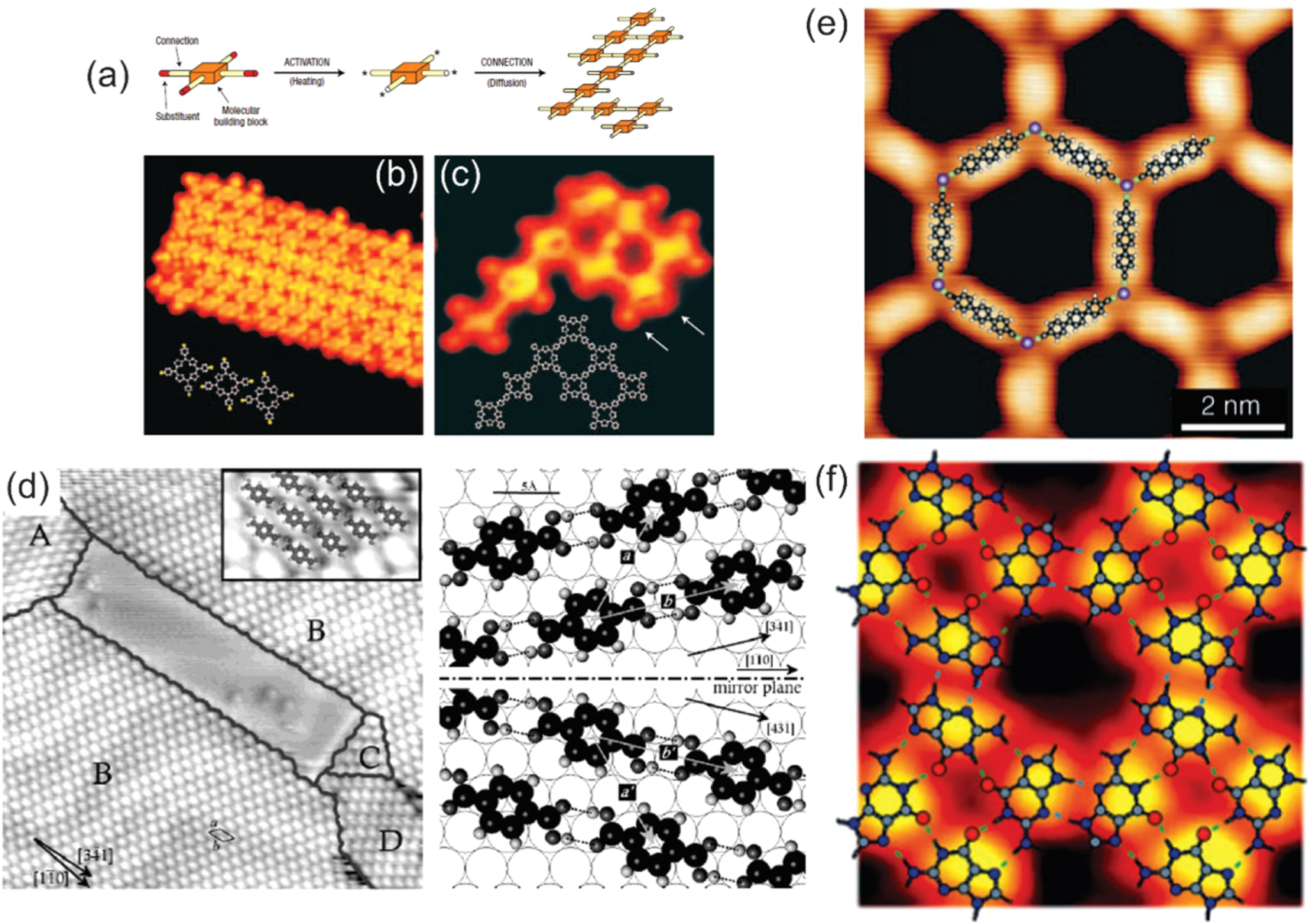{kind=link}
{kind=link}
{kind=link}
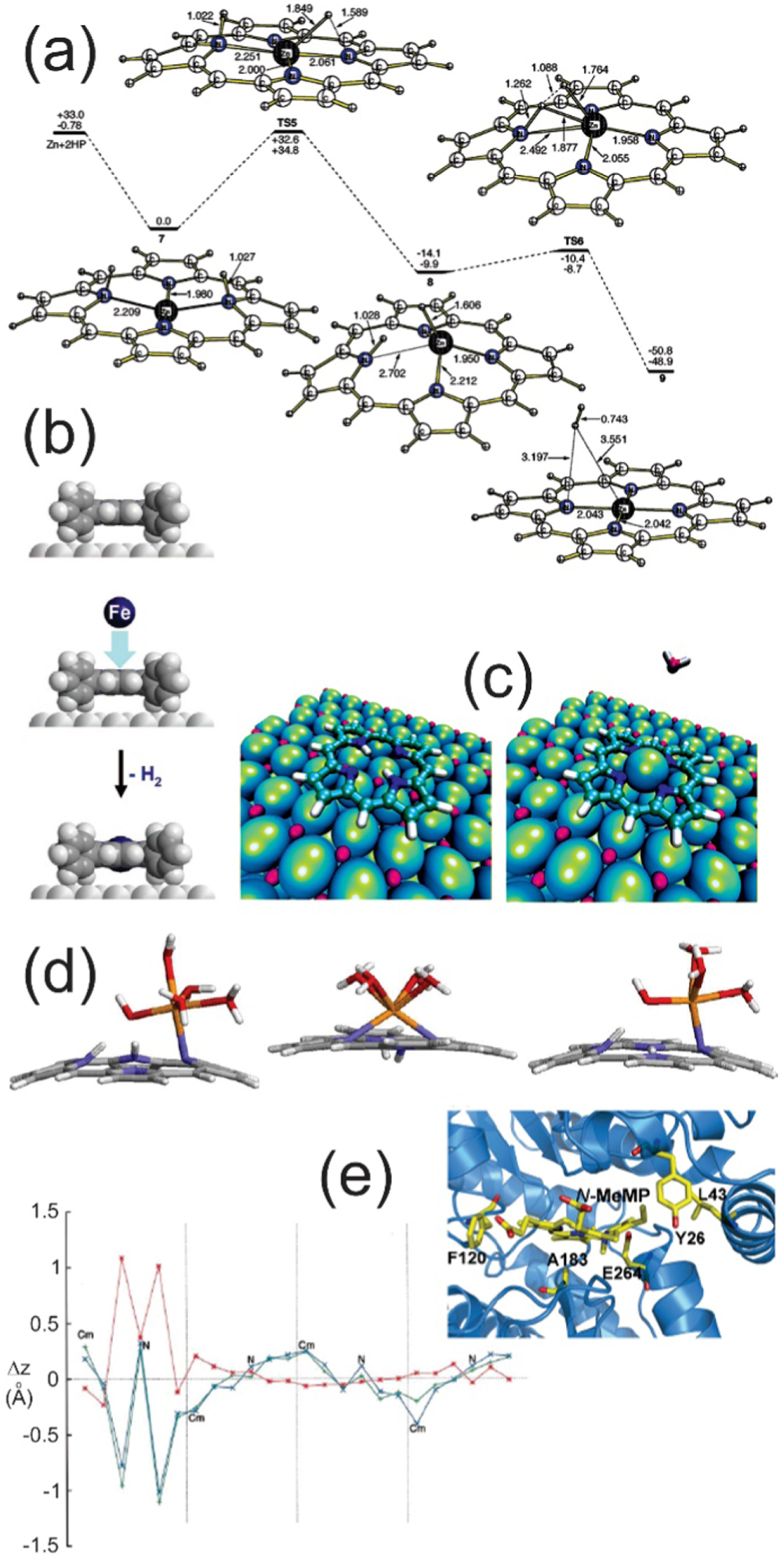{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 24. Self-metalation of a tetrapyrrole at a surface can be induced at room temperature by the gas phase at NAP; in the specific, 2H-TPP molecules are converted to PdTPP on the Pd(100) termination upon exposure to 1 mbar O2: (a) IR–VIS SFG spectra of the as prepared 2H-TPP/Pd(100) system collected at RT in UHV (bottom) and in situ in 1 mbar O2 showing the vibrational fingerprint for metalation to PdTPP; the spectroscopic data are shown together with the best fit and the deconvolution of the resonances; dashed lines indicate metalation markers; (b) N 1s XPS spectra of the as prepared 2H-TPP/Pd(100) system collected at RT in UHV (bottom) and in situ in 1 mbar O2 showing the spectroscopic core level fingerprint for metalation to PdTPP; best fit and deconvolution are also shown; (c) cartoon of the metalation process [108].
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusions and outlook: biomimetic perspectives
We have shown the deep insight that has been achieved progressively in the last decades into the atomic-level understanding of the behavior of tetrapyrrole-based 2D self-assemblies at surfaces. Many fundamental aspects have been characterized, including geometric ordering and self-assembly mechanisms, chemical reactivity (metalation processes and ligand adsorption), electronic structure, lateral interactions, interactions of the tectons with the templating substrate, and magnetic properties. The knowledge attained under model surface science UHV conditions is progressively being extended to solid–liquid and solid–gas interfaces. In the latter case, both the evolution of several experimental techniques and the introduction of new ones have allowed researchers to bridge the pressure gap, extending the fundamental investigation approach up to NAPs. Especially, with respect to the activity of 2D MOFs hosting single metal atoms, great attention has been devoted to the role of charge transfer between MOF and support. The tuning and tailoring capability of the charge redistribution at the interface has allowed the selective tuning of the adsorption properties with respect to the ligands, like in the case of FePcs supported on graphene. This achievement, well-described by what is known as surface trans-effect, may be considered as the biomimetic, synthetic homologue of the molecular trans-effect occurring at the reaction pockets of proteins in Nature (like for example the role of the proximal histidine in myoglobin and hemoglobin). A further parallelism with Nature should consider also the third dimension, i.e. the biomimetic caging of the reactive centers. The idea is that confining reactive components in well-defined synthetic binding pockets would lead to additional and novel enzyme mimicking tunable properties [65]. This is because Nature tailors the activity and selectivity of active sites by exploiting first of all the structure of the porphyrin center, but also the axial (proximal) ligand and the distal superstructure [4]. There is still much to do to engineer the latter distal coordination at surfaces. The homologue of the cis-coordination to distal residues is indeed still lacking a synthetic counterpart in the 2D systems, although some first efforts have already been reported [153], offering challenging prospects in this sense. Another interesting route may instead exploit double-decker porphyrin and phthalocyanine complexes stabilized by a central single lanthanide atom (LnP2, LnPc2) [154–158]. Interesting preliminary results have been obtained under UHV and applicative conditions with respect to gas sensing, electronic, chemical, and optical properties. In general, it is evidenced that the challenges associated with applicative perspectives of MOFs need to expand the field beyond the realm of synthetic chemistry and surface science, involving expertise in solid-state physics, lithography, process design, materials integration and device engineering [159].
Acknowledgments
I am grateful to many former and present colleagues, students, and collaborators who made my contribution to the surface science of tetrapyrroles at NAP possible: F Armillotta, F Bournel, C Cepek, G Comelli, M Corva, R Costantini, E D'Incecco, M Dell'Angela, C Dri, Z Feng, A Ferrari, V Feyer, J J Gallet, M Jugovac, A Knop-Gericke, F Mohamed, S Moro, G Pastore, C Rameshan, M Rinaldi, M Roiaz, A Sala, N Seriani, M Stredansky, E Tomsic, A Verdini, and G Zamborlini. I am indebted and grateful to G Rupprechter for having introduced me into the world of SFG. I thank M Corva, M Panighel, and A Verdini for reading carefully and critically the manuscript. Financial support from Italian MIUR within the framework of the project PRIN 2017KFY7XF is acknowledged.