

Abstract
Soft nanoparticles (NPs) have recently emerged as a promising material for intracellular drug delivery. In this regard, NPs derived from polydimethylsiloxane (PDMS), an FDA approved polymer can be a suitable alternative to conventional soft NPs due to their intrinsic organelle targeting ability. However, the available synthesis methods of PDMS NPs are complicated or require inorganic fillers, forming composite NPs and compromising their native softness. Herein, for the first time, we present a simple, robust and scalable strategy for preparation of virgin sub-50 nm PDMS NPs at room temperature. The NPs are soft in nature, hydrophobic and about 30 nm in size. They are stable in physiological medium for two months and biocompatible. The NPs have been successful in delivering anticancer drug doxorubicin to mitochondria and nucleus of cervical and breast cancer cells with more than four-fold decrease in IC50 value of doxorubicin as compared to its free form. Furthermore, evaluation of cytotoxicity in reactive oxygen species detection, DNA fragmentation, apoptosis-associated gene expression and tumor spheroid growth inhibition demonstrate the PDMS NPs to be an excellent candidate for delivery of anticancer drugs in mitochondria and nucleus of cancer cells.
Export citation and abstract BibTeX RIS
1. Introduction
For the past few decades, soft nanoparticles (NPs) have attracted great attention in the arena of drug delivery [1–7]. Due to their deformable nature, soft NPs can easily overcome the biological barriers for diffusion and penetrate deep into the tissues more efficiently than the analogous non-deformable hard NPs [4, 8–10]. Such distinct capability of soft NPs has recognized them as an invaluable asset for delivering therapeutic agents across the extracellular barriers [3, 11]. Soft NPs have also been explored for circumventing the intracellular barriers for drug delivery. For example, soft liposomal nanocarriers have been developed for delivering macromolecules into mitochondria via membrane fusion [12]. Wang et al postulated that NPs possessing size transformation ability will have a higher probability for nuclear entry [13]. Therefore, soft NPs are considered as a potential candidate for efficient intracellular delivery of biomolecules.
Several polymers have been investigated in the past for developing soft NPs with essential biocompatibility for drug delivery [3, 5–7, 14]. Soft hydrogel NPs exhibited better circulation time, lessened immunogenicity and enhanced tissue penetration [4, 15, 16]. Polyethylene glycol-based soft nanoparticles were encapsulated with dual imaging probe molecules to develop a safe diagnostic agent [17]. Sub-120 nm soft nanocarriers were prepared from co-polyester of polybutylene succinate and poly(butylene succinate-co-butylene dilinoleate) for delivery of paclitaxel [7]. In particular, liposome based soft nanoformulations have been commercialized as anticancer drug delivery platforms [18]. Nevertheless, the available biocompatible soft NPs are limited by their lack of long term stability and poor scalability of the manufacturing processes [19]. Furthermore, the complicated conjugation strategies involved in the design of the targeted drug delivery systems make the process of production more difficult [19].
Among the FDA-approved soft polymers, polydimethylsiloxane (PDMS) possesses immense possibility for being successful as a material for developing drug delivery vehicle. PDMS is a hydrophobic polymer and a soft material with mechanical properties very similar to human tissues [20]. Due to its low elastic modulus (0.5–4 MPa), it can easily deform and modulate its size for smooth penetration through pores having more or even similar sizes [21]. In addition, its hydrophobicity may be useful for localization within hydrophobic organelles [22, 23]. In fact, PDMS NPs have been successful in intrinsic targeting of selective intracellular organelles in the past [24, 25]. However, PDMS nanoparticles are prone to collapse into a film [26]. To restrict the film-forming propensity of PDMS, the conventional synthesis of PDMS NPs involves complicated surface modification steps or incorporation of silica (or its precursor) as filler to form hybrid NPs [24, 25, 27]. Such composite NPs might attenuate the inherent flexibility of PDMS and surface modification would involve reduction in the innate hydrophobicity of PDMS. Also, the larger size of these NPs (diameter > 50 nm) might restrict their free diffusion into intracellular organelles [13, 28–30]. Overall, the existing literature data suggest the need to develop a simple and scalable methodology to fabricate stable NPs (preferably smaller than 50 nm) from PDMS without compromising with its native softness/hydrophobicity and explore their potential as a carrier for intracellular drug delivery.
Herein, for the first time, we propose a simple, robust and scalable one-pot synthesis strategy based on nanoprecipitation method to prepare virgin sub-50 nm PDMS NPs at room temperature. The synthesized NPs are about 30 nm in size, soft, hydrophobic and stable for at least two months in phosphate buffered saline (PBS). The NPs are biocompatible and possess an inherent ability to selectively localize in mitochondria and nucleus of cancer cells (without any targeting ligand) might be due to their inherent hydrophobicity, small size and easy elastic deformability. Such dual-targeting capability is a unique feature of sub-50 nm PDMS NPs that distinguishes them from conventional NPs. The efficacy of the synthesized soft PDMS NPs has also been explored for delivering anticancer drug doxorubicin (DOX) in various cancer cells. Owing to the intrinsic mitochondrial and nuclear dual-targeting ability of the NPs, the DOX delivered via the synthesized PDMS NPs induced an elevated level of apoptosis in cervical and breast cancer cells, resulting in drastic improvement in the therapeutic efficiency of DOX. Moreover, a rapid penetration and collapse of 3D multicellular tumor spheroids by DOX loaded NPs advocates for the tremendous potential of the NPs for effective delivery of anticancer drugs.
2. Experimental section
2.1. Materials
PDMS (SYLGARD® 184 silicone elastomer kit containing a base and a cross-linking agent) was procured from Dow Corning India Pvt. Ltd. Sodium dodecyl sulfate (SDS) and dimethyl sulfoxide (DMSO) were obtained from Merck India Pvt. Ltd. Tetrahydrofuran (THF) and gelatin were purchased from Loba Chemie. Cetyltrimethylammonium bromide (CTAB) was obtained from SD Fine Chemicals. Fluorescein isothiocyanate (FITC), rhodamine B isothiocyanate (RITC), 2´,7´-dichlorofluorescin diacetate (DCFH-DA), trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA), Dulbecco's modified eagle's medium (DMEM), 3-(4,5-dimethylthiazol-2-yl)−2,5-diphenyltetrazolium bromide (MTT), fluorescein diacetate (FDA), propidium iodide (PI) and doxorubicin hydrochloride (DOX) were procured from Sigma-Aldrich. Fetal bovine serum (FBS) was purchased from Invitrogen, USA. HeLa and NIH 3T3 cells were obtained from National Centre for Cell Science (NCCS), Pune, India. MCF-7 cells were received as a kind gift from Prof. Bushra Ateeq, IIT Kanpur, India. Human recombinant insulin (cell culture tested) and penicillin streptomycin antibiotic were bought from Himedia. Hoechst 33342, MitoTrackerTM Red CMXROS and MitoTrackerTM Green FM were purchased from Thermo Fisher Scientific. All the above-mentioned chemicals were utilized as received (without further purification steps). Deionized water was employed for all the necessary experiments.
2.2. Characterization
Field emission scanning electron microscopy (FESEM) and transmission electron microscopy (TEM) images were collected from SUPRA 40 VP Gemini, Zeiss, Germany and FEI Tecnai G2 U-Twin, respectively. PDMS NPs in the form of aqueous dispersion (10 μl; 10 μg ml−1) was drop-casted on a cleaned aluminium foil or copper TEM grid (carbon-coated; 300 mesh) with subsequent drying at room temperature for FESEM and TEM analysis, respectively. Prior to FESEM analysis, the samples were gold sputtered for 1 min. The deformability of the NPs was assessed via atomic force microscopy (AFM) instrument (purchased from Asylum Research by Oxford Instruments). The hydrodynamic size and zeta potential of NPs were determined by Zetasizer Nano series, Nano ZS90, Malvern. The surface functional groups were characterized by Fourier transform infrared (FTIR) spectroscopy (instrument: Tensor 27, Bruker, Germany) in the frequency range of 600–4000 cm−1. The contact angle values were determined by sessile drop technique employing a Goniometer purchased from Krüss GmbH in Germany. The thermal stability of the fabricated materials was analyzed by thermogravimetric analysis (TGA) instrument (SDT Q600) at a heating rate of 10 °C min−1 from 25 °C to 800 °C in a nitrogen environment. The absorbance measurements were performed by Thermo Scientific Multiskan Spectrum UV visible spectrometer. Zeiss LSM 780 confocal microscope and Olympus IX81 inverted microscope were used for imaging of cells in 2D and 3D cultures, respectively. Gel electrophoresis images were obtained from E-gel Imager, Life Technologies. Reverse transcriptase-polymerase chain reaction (RT-PCR) was performed using ProFlex PCR system (Applied Biosystems).
2.3. Synthesis of PDMS NPs
25 mg of PDMS base and 2.5 mg of PDMS cross-linking agent were dissolved separately in 2.5 ml of THF and mixed through stirring at a speed of 300–400 rpm for 5 min at room temperature (25 °C) in a round bottom flask. Around 15 ml of deionized water, containing dissolved SDS (5–8 wt% of PDMS polymer) was added to it while stirring. After 20 min, the flask was kept open and stirring was continued for 24 h at room temperature to ensure complete removal of THF from the dispersion. The nanoparticle dispersion was kept idle in aqueous dispersion form for another 24 h at room temperature before usage. For synthesis of PDMS NPs at gram-scale, the above protocol was followed using 1.125 gm of PDMS base, 0.1125 gm of PDMS cross-linking agent and 225 ml of deionized water.
2.4. Fabrication of PDMS film
Both the components of SYLGARD® 184 were mixed at a weight ratio of 10:1, degassed to remove the entrapped air-bubbles and then poured on a glass petri dish. The resulting film was cured at 100 °C for 2 h and then removed as a self-standing film.
2.5. DOX loading and release
3 mg of PDMS NPs were mixed with 1 ml of DOX solution in PBS (0.25 mg ml−1). After incubation for 5 min under dark condition, the DOX loaded NPs were collected via centrifugation and washed with PBS to remove free DOX. For evaluating the DOX loading capacity, the concentration of DOX in the supernatant solution was estimated by measuring absorbance at 484 nm using UV visible spectroscopy and then comparing with a standard calibration curve. In vitro DOX release experiments were carried out in PBS (pH 7.4 or 5.8). The DOX loaded PDMS NPs (3 mg) were dispersed in 1 ml of PBS and incubated at 37 °C under stirring condition in a dark environment. After specific time intervals, 100 μl of aliquot was withdrawn and the supernatant was collected via centrifugation. The precipitated NPs (collected at the bottom of the centrifuge tube) were re-dispersed in fresh PBS and added back to the main mixture. The amount of DOX release was determined from the calibration curve by measuring the absorbance via UV visible spectroscopy.
2.6. Cell culture
Human cervical cancer cells (i.e. HeLa), breast cancer cells (i.e. MCF-7) and mouse fibroblast cells (i.e. NIH 3T3) were cultured in DMEM comprising 10% FBS (10%), 100 U ml−1 penicillin and 100 μg ml−1 streptomycin in a humidified atmosphere with 5% CO2 at 37 °C. For culturing MCF-7 cells, human recombinant insulin (0.01 mg ml−1) was also added into DMEM to make the complete growth medium. For passaging of cells at regular intervals, trypsinization with 0.5% trypsin-EDTA was followed when the cells became 70%–90% confluent.
2.7. Intracellular localization
In each well of a 24 well culture plate, a gelatin coated coverslip was placed and 5 × 104 trypsinized cells (counted using a hemocytometer cell counter) were seeded onto it. The cells were then allowed to adhere and grow to their fully spread morphology for 7 h under cell culture conditions. Cells were then incubated with FITC labelled NPs (50 μg ml−1), DOX loaded NPs (50 μg ml−1) and the equivalent amount of free DOX for required time points (4 h and 12 h). Each slide was then washed with PBS for three times. For staining of mitochondria, the cells were incubated with 200 nM of MitoTrackerTM Green FM under cell culture conditions for 20 min, washed with PBS and then immediately observed under confocal microscope. For staining both mitochondria and nucleus, the cells were stained with 100 nM of MitoTrackerTM Red CMXROS for 20 min followed by washing with PBS thrice. The cells were then fixed under treatment of 4% formaldehyde at 4 °C for 20 min. After washing with PBS for three times, the nuclei were stained with 1 μg ml−1 of Hoechst 33342 for 15 min and washed with PBS again. The cell-seeded cover slips were then sealed with nail varnish on glass slides for observation in confocal laser scanning microscopy (CLSM). Adobe Photoshop CS5 software was used to process the CLSM images.
2.8. In vitro cytotoxicity
Cytotoxicity of PDMS NPs (with or without DOX) and free DOX was evaluated by MTT assay. Cells were seeded at a density of 2500 cells/well in a 96 well tissue culture plate. Further, the cells were treated with blank NPs, DOX loaded NPs and the equivalent amount of free DOX in the NPs concentration range of 0–150 μg ml−1. After the desired treatment time, the culture medium was removed and 200 μl of MTT solution (0.5 mg ml−1) was added. After 4 h, the medium was replaced with 200 μl of DMSO per well and after 20 min of incubation, the absorbance of the medium was monitored using a microplate reader at the wavelength of 570 nm. The cytotoxicity values were expressed as percentage of cell viability compared to untreated cells as control.
2.9. Imaging of reactive oxygen species (ROS) generation
MCF-7 cells were seeded at a density of 5 × 104 cells per well on a gelatin coated coverslip in 24 well tissue culture plate. Further, the cells were incubated with blank NPs (100 μg ml−1), DOX loaded NPs (100 μg ml−1) and the equivalent amount of free DOX for another 24 h. The cells were also treated with hydrogen peroxide (200 μM), a standard ROS inducer as a positive control for 45 min. The medium was then removed and DCFH-DA (10 μM) was added 30 min before visualizing fluorescence from cells (due to ROS generation) by CLSM. The images were processed using Adobe Photoshop CS5 software. For each set of samples, MFI of ROS generation was evaluated by measuring fluorescence intensity at ten different random locations within the cellular images using ImageJ (version 1.4.3.67) software.
2.10. DNA fragmentation assay
MCF-7 cells (2 × 105 cells) were seeded in a 6 well plate and treated with blank NPs (100 μg ml−1), DOX loaded NPs (100 μg ml−1) and an equivalent amount of DOX in free form for 24 h. The complete medium on the adhered cells was then centrifuged at 3000 × g for 10 min to collect the non-adherent cells. Adherent cells were lysed with hypotonic lysis buffer containing 10 mM Tris-HCl (pH 8.0), 10 mM EDTA and 0.5% Triton X-100. After complete lysis, the solution was pooled with the centrifuged non-adherent cells for homogenous lysis of cells. The solution was treated with RNase (0.1 mg ml−1) at 37 °C for 2 h followed by proteinase K at 50 °C for 2 h. Further, a mixture of phenol, chloroform and isoamyl alcohol (25:24:1) was added to extract DNA. The aqueous supernatant was removed and DNA was precipitated by adding an equal amount of isopropyl alcohol and stored overnight at −20 °C. Further, the solution was centrifuged at 12 000 × g for 10 min at 4 °C. The DNA pellet was air-dried and dissolved in TE buffer. The DNA was separated on 2% agarose gel electrophoresis and visualized by staining with ethidium bromide.
2.11. Total RNA isolation and RT-PCR analysis
MCF7 cells were cultured in 6 well plates at 37 °C and 5% CO2 conditions. The cells were exposed to blank NPs (100 μg ml−1), DOX loaded NPs (100 μg ml−1) and the equivalent amount of free DOX for 24 h. Further, total RNA was extracted by ReliaPrep™ RNA cell miniprep Kit (Promega) according to manufacturer's instructions. The concentration of the isolated RNA was determined by Nanodrop 2000c spectrophotometer purchased from Thermo Scientific. The first strand was synthesized from 1 μg of total RNA using High-capacity cDNA Reverse Transcription Kit manufactured by Applied Biosystems™ according to manufacturer's protocol. RT-PCR was performed with parameters including 10 min at 95 °C which was followed by 40 cycles involving denaturation step at 95 °C for 15 s, thereafter annealing at 60 °C for 30 s and finally elongation at 72 °C for 30 s. Specific primer sequences for the genes were used for RT-PCR analysis (table S1 (available online at stacks.iop.org/NANO/33/495102/mmedia)) [31–33]. The amplified gene products were analysed for gene expression levels on 2% agarose gel stained by ethidium bromide. The stained samples were detected under UV light.
2.12. Antitumor efficacy in 3D spheroid model
Breast cancer tumor spheroids were generated by modified hanging drop method. About 10 μl of trypsinized MCF-7 cell suspension containing 1500 cells was placed as drops on the lid of a 90 mm petri dish. The lid was then inverted over the petri dish containing PBS. The petri dish was then incubated for 7 d. During this period, the cells got accumulated at the bottom of the hanging drop due to gravity resulting formation of spherical aggregates of MCF-7 cells. The viability of the spheroids was assessed via staining with FDA and PI for marking live and dead cells, respectively. To visualize the penetration ability of RITC labelled NPs (100 μg ml−1), DOX loaded NPs (100 μg ml−1) and the equivalent amount of free DOX, the spheroids were incubated with these samples for 4 h. The medium was removed and the spheroids were washed with PBS before observing in CLSM. The inhibition of spheroid growth was monitored via optical microscopy by incubating blank NPs (100 μg ml−1), DOX loaded NPs (100 μg ml−1) and the equivalent amount of free DOX in spheroid containing growth medium for 10 d.
2.13. Statistical analysis
Statistical analysis data are presented as mean ± SEM. Univariate differences were assessed with Student's t-test method. All analyses were performed using GraphPad Prism software of version 8.2.0 (for Windows).
3. Results and discussion
3.1. Synthesis and characterization of PDMS NPs
PDMS is liquid at room temperature and has a natural tendency to get oriented towards air surface even after cross-linking due to its low surface tension resulting in the formation of a film [26]. Synthesis of NPs of such a soft material is extremely difficult because of two main reasons: these are prone to collapsing during solvent removal step through evaporation, centrifugation or drying, and additional chemical cross-linking step is required for their stabilization [34]. In the present work, PDMS NPs are prepared by a modified nanoprecipitation method at room temperature with careful selection of process parameters ensuring completion of cross-linking before removal of the entire dispersion medium.
A commercially available PDMS (SYLGARD® 184 Silicone Elastomer kit, Dow Corning Corporation) containing a vinyl-terminated PDMS (base polymer) including a platinum catalyst and a curing agent/cross-linker (containing a hydrosiloxane copolymer) was utilized for the synthesis of PDMS NPs. According to the manufacturer protocol, the components must be mixed with a proportion of 10:1 (base: curing agent) and cured at room temperature for 48 h for the fabrication of cross-linked PDMS. During the cross-linking reaction, the vinyl terminated PDMS is linked to the cross-linker through the action of the platinum catalyst to produce a cross-linked network of PDMS (figure S1) [35]. SDS was specifically chosen as a surfactant to stabilize PDMS NPs in water due to its low price, ability to stabilize PDMS oil droplets, biocompatibility and widespread use in cosmetics and biomedical industry [36].
As shown in Scheme

Scheme 1. Schematic representation of nanoprecipitation based synthesis of PDMS NPs.
Download figure:
Standard image High-resolution imageFigure 1(a) shows the FESEM images of the synthesized PDMS NPs deposited on aluminium foil. A uniform distribution of NPs was observed indicating the monodispersed nature of the NPs even in dried form. Some crack-like patterns are also visible which appear due to the sputter coated gold film developed during FESEM sample preparation. The presence of surfactant is necessary for maintaining the spherical morphology of the synthesized PDMS NPs until they get cured completely. In fact, the NPs collapsed and formed micron-sized islands in dried form when synthesized without surfactant (figure S2). The choice of surfactant is also a critical parameter to prepare highly stable monodispersed PDMS NPs. When CTAB was used for the synthesis as a cationic surfactant instead of anionic SDS, phase-separated polymer was observed at the bottom of the synthesis flask. The cationic surfactant might have reduced the inherent negative surface charge of surfactant-free PDMS NPs (data not shown), leading to aggregation followed by phase separation of PDMS.

Figure 1. (a) FESEM and (b) TEM image of PDMS NPs. AFM image of PDMS NPs (c) and corresponding height distribution of linear scans 1 and 2 (d). (e) Schematic representation of the soft and deformable nature of PDMS NPs during AFM analysis. The approximate size values are represented for illustration purpose only.
Download figure:
Standard image High-resolution imageThe robustness of the synthesis protocol was evaluated by varying several crucial process parameters. As shown in figure S3, the formation of NPs was observed even after variation of the PDMS base to curing agent ratio, amount of non-solvent addition and mixing time. Also, the size of the synthesized PDMS nanoparticles does not change when the concentration of PDMS (before nanoprecipitation) is in the range of 1–15 mg ml−1 (figure S4). Increasing the initial PDMS concentration led to an increase in the production rate and the highest value of PDMS concentration (i.e. 15 mg ml−1 in figure S4(c)) has been further scaled-up to produce around 1 gm of PDMS NPs from a single batch (figure S5). Such robust and scalable process for synthesis of PDMS NPs has not been reported in the past.
TEM image provides much clearer view of PDMS NPs. As shown in figure 1(b) and the corresponding histogram plot in figure S6, the size of the synthesized PDMS NPs deposited on carbon coated copper grid was around 30 nm. Such small size of the NPs may facilitate their entry into the intracellular organelles [13, 28–30]. The NPs got deformed when deposited on silicon wafer as analyzed by AFM (figures 1(c), (d) and figure S7). Soft materials e.g. liposomes are very sensitive to the scanning force of AFM cantilever tip and the adhesive contact forces of the substrate because of their low mechanical rigidity [37]. Hence, the decrease in height with the corresponding increase in width of the NPs during AFM analysis confirmed the soft and flexible nature of the synthesized PDMS NPs (figure 1(e)). Although hard silica NPs of 30 nm size (same as PDMS NPs) have not been able to enter into intracellular organelles [38], the unique soft/flexible nature of PDMS NPs might be helpful in their fusion with mitochondrial membrane (similar to MITO-Porter) [12] and nuclear entry through nuclear pores [13].
The as-prepared aqueous dispersion of PDMS NPs was analyzed through dynamic light scattering (DLS) technique and a narrow size distribution of PDMS NPs with an overall hydrodynamic diameter of 204 nm and polydispersity index (PDI) of 0.002 was observed (figure S8(a)). It may be noted that DLS measurement will not accurately refer the actual size of the NPs when the tested NPs deviate from spherical shape [39]. Thus, the hydrodynamic diameter was measured to be much higher than the actual size of the NPs, but extremely low PDI value reflected monodispersed nature of the synthesized NPs. Also, the NPs could retain their narrow size distribution in water even after a month (hydrodynamic diameter 203 nm, PDI 0.043) (figure S8(b)). The long-term stability of PDMS NPs was evaluated by dispersing the NPs in PBS of pH 7.4. Although PDI values increased slightly after two months of storage, no significant change was observed in hydrodynamic diameter and zeta potential values of the synthesized PDMS NPs, demonstrating excellent colloidal stability under physiological conditions (figure S9).
In order to compare the chemical and physical properties of the synthesized PDMS NPs with native PDMS, a cross-linked PDMS film was fabricated. As observed in FTIR spectra (figure 2(a)), all the surface chemical groups of PDMS film were present on the surface of PDMS NPs suggesting chemical similarity of the NPs with the film. The contact angle value for the PDMS film was 114° which is quite consistent with the data available in the literature (inset in figure 2(a)) [40]. The synthesized PDMS NPs exhibited hydrophobicity (contact angle of 108°) which is almost similar to that of the PDMS film (inset in figure 2(a)). Such hydrophobicity of the NPs will be suitable for targeting hydrophobic organelles of cancer cells [22, 23]. The thermal properties of the synthesized NPs were also compared with that of native PDMS film. TGA of PDMS NPs showed similar weight loss behavior like PDMS film (figure 2(b)). Also, the derivative thermogravimetric (DTG) profile of the NPs shifted slightly towards higher temperatures (figure 2(b)) indicating efficient cross-linking and improved thermal stability of the synthesized NPs even as compared to PDMS film. All these characterization results confirm that most of the native properties of PDMS were maintained after nanoprecipitation and solvent removal during the synthesis of NPs.

Figure 2. (a) FTIR spectra of PDMS film and PDMS NPs. Inset shows the water droplet contact angle measurements on a glass slide coated with PDMS film and PDMS NPs. (b) TGA and DTG spectra of PDMS film and PDMS NPs. (c) In vitro cumulative DOX release from DOX loaded NPs when dispersed in PBS of pH 7.4 (blue line) and pH 5.8 (red line). The temperature was set at 37 °C throughout the DOX release experiment. The data is represented as mean ± SEM (n = 3).
Download figure:
Standard image High-resolution image3.2. In vitro drug loading and release studies
To investigate the capability of PDMS NPs in drug delivery, DOX was chosen as a model anticancer drug [41–44] due to its potential to induce cellular apoptosis via intercalation with nuclear as well as mitochondrial DNA [45–47]. However, in free form, DOX enters only into the nuclei of cancer cells, losing its therapeutic potential through mitochondrial pathway [48]. In particular, delivering DOX selectively to the mitochondria and nucleus of the cancer cells can enhance its therapeutic efficiency [49]. DOX loaded PDMS NPs were prepared via incubation of the NPs with DOX solution followed by washing with PBS (pH 7.4) to remove weakly adsorbed drug from the surface of the NPs. The amount of drug loading was determined by UV/vis spectroscopy using a standard calibration curve for DOX (figure S10). The drug loading amount in PDMS NPs was measured as 0.58 ± 0.05% (w/w) (n = 3).
The in vitro release kinetics of DOX from the NPs was determined at different pH values at 37 °C under stirring condition in order to simulate the in vivo biological environment (figure 2(c)). In particular, PBS of pH 7.4 was utilized to simulate the blood circulation whereas PBS of pH 5.8 was applied to simulate the acidic tumor environment [48]. At physiological pH of 7.4, an initial burst release was observed which might be due to the diffusion of drug located near the surface of the NPs. However, the cumulative drug release percentage from the DOX loaded NPs was below 25% after 24 h, indicating a low proportion of premature release in the blood circulation system. After the initial burst release at pH 5.8, the DOX loaded NPs displayed sustained release profile revealing efficient delivery of DOX under acidic condition of tumor cells. This could be attributed to the protonation of amino group of DOX in the acidic environment which weakens the adsorption between DOX and the NPs, facilitating increased release in a sustained manner [48].
3.3. Intracellular localization of PDMS NPs
To understand the intracellular localization of PDMS NPs, they were labelled with a fluorescent dye (FITC) to be visualized through CLSM and then incubated with HeLa and MCF-7 cells for 12 h. It can be seen from figure 3(a) that the fluorescence from FITC labelled NPs almost completely overlapped with that of the mitochondria and nuclei staining dyes (MitoTrackerTM Red and Hoechst, respectively) displaying yellow and cyan colors in the merged image, respectively. The intracellular distribution of NPs in MCF-7 cells exhibited similar behavior as in HeLa cells as shown in figure 3(b). The corresponding line scanning profiles (figure S11) denoting superimposition of blue line (derived from Hoechst) and red line (derived from MitoTrackerTM Red) with green line (derived from FITC tagged NPs) further suggest that the PDMS NPs possess inherent tendency to selectively localize inside mitochondria and nucleus of cancer cells. Such intrinsic organelle-targeted ability of the NPs may be attributed to their inherent hydrophobicity, small size and easy elastic deformability, as determined before.
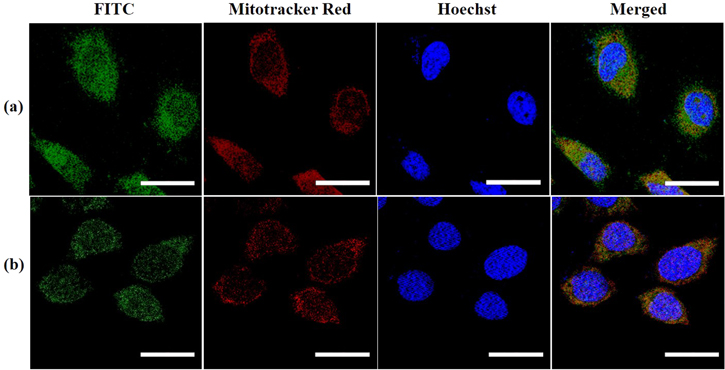
Figure 3. CLSM images of (a) HeLa and (b) MCF-7 cells after incubation with blank PDMS NPs for 12 h. NPs were tagged with FITC (green), mitochondria and nuclei were stained with MitoTrackerTM Red (red) and Hoechst (blue), respectively. Scale bars: 20 μm.
Download figure:
Standard image High-resolution imageThe dual-targeted PDMS NPs were explored for delivery of DOX into its action sites mitochondria and nucleus. After 4 h of incubation with HeLa cells, free DOX could rapidly enter into the cells and started accumulation inside the nuclei of HeLa cells (figure 4(a)). In contrast, DOX loaded NPs displayed a relatively slower and prolonged accumulation in the mitochondria and perinuclear region (figure 4(b)). An intense red line of demarcation denotes the accumulation of DOX loaded NPs along the nuclear boundary. After incubation of free DOX for 12 h, strong red fluorescence was observed throughout the nuclei of HeLa cells (without overlapping with the green fluorescent signal from mitochondria) indicating direct infiltration of free DOX into the nuclei by passive diffusion (figure 4(c)). The DOX loaded NPs also expressed strong red fluorescence in the nuclear region of HeLa cells after incubation for 12 h (figure 4(d)). In addition, the red fluorescence from DOX loaded NPs prominently overlapped with the green fluorescent signal of mitochondria suggesting complete accumulation in mitochondria and nucleus of HeLa cells. Similar kind of intracellular localization of free DOX and DOX loaded NPs was observed in MCF-7 cells further confirming the dual-targeting capability of the DOX loaded NPs (figures 4(e)–(f)). Since mitochondria and nucleus are the action targets of DOX [50], the dual-targeted DOX loaded PDMS NPs are expected to achieve synergistic therapeutic outcome. The nuclear localization of free DOX and DOX loaded NPs were further verified via staining of nucleus of HeLa cells with Hoechst dye (figure S12). Complete overlap of highly intense red fluorescence from DOX loaded NPs with the blue fluorescence from the nucleus after 12 h validates the capability of PDMS NPs in facilitating the delivery of DOX into the nucleus.
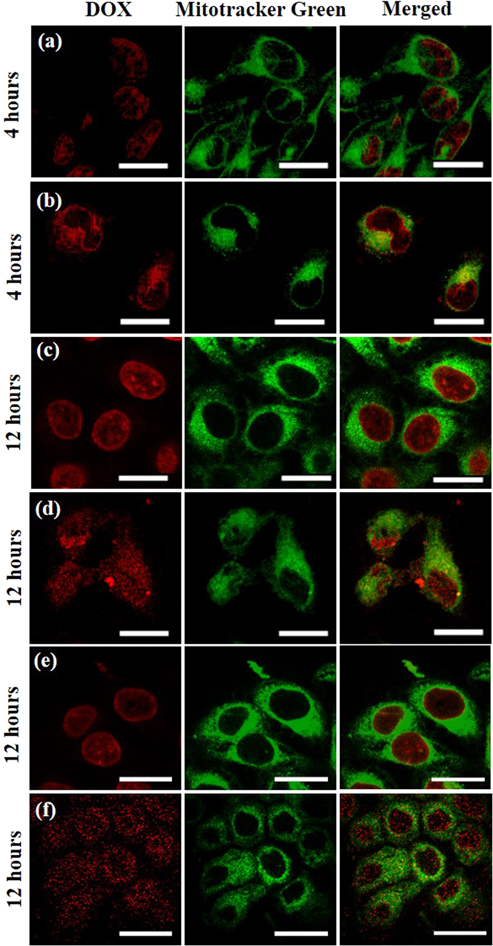
Figure 4. CLSM images of (a)–(d) HeLa and (e), (f) MCF-7 cells after incubation with free DOX (a), (c), (e) and DOX loaded NPs (b), (d), (f) for 4 h and 12 h. Mitochondria were stained with MitoTrackerTM Green while DOX expressed red fluorescence. Since MitoTrackerTM Green is not retained after fixation, simultaneous staining with nuclear markers (requiring fixation) was not performed. Scale bars: 20 μm.
Download figure:
Standard image High-resolution image3.4. Evaluation of cytotoxicity
A prerequisite of a successful nanocarrier for drug delivery is non-cytotoxicity so as to avoid any side effects in the cellular environment. Therefore, the cytotoxicity of PDMS NPs was first evaluated by in vitro MTT assay in cancerous (Hela and MCF-7) and healthy (NIH 3T3) cells as shown in figure S13. The viability of both types of cells remained above 85% when treated with blank PDMS NPs up to a concentration of 150 μg ml−1 for 24 h and even for 48 h. Thus, it can be concluded that PDMS NPs are not cytotoxic to the cells within the aforementioned concentration range.
The cytotoxicity induced by treatment with DOX was measured via MTT assay thereafter (figure 5). At equivalent DOX concentration, DOX loaded NPs significantly inhibited proliferation of cancer cells in a time and concentration dependent manner as compared to free DOX. When delivered in free form, the half maximal inhibitory concentration (IC50) values for DOX in HeLa and MCF-7 cells were found to be 0.474 μg ml−1 and 0.407 μg ml−1, respectively. However, the IC50 values for DOX, delivered via PDMS NPs were only 0.212 μg ml−1 and 0.091 μg ml−1 in HeLa and MCF-7 cells, respectively. The present data show more than four-fold decrease in the IC50 values of DOX, suggesting DOX loaded PDMS NPs possess enhanced inhibition ability of cancer cell proliferation than free DOX. The targeted delivery of DOX into its action site specific organelles (i.e. mitochondria and nucleus) inside cancer cells can enhance the therapeutic effects by inhibiting cell growth and ultimately leading to cell death. Owing to the mitochondria and nucleus dual-targeting ability, PDMS NPs were able to deliver DOX more efficiently than free DOX, resulting in elevated cytotoxicity in cancerous cell lines.

Figure 5. Viability of (a) HeLa cells and (b) MCF-7 cells incubated with free DOX and DOX loaded NPs for 24 h and 48 h. The data is represented as mean ± SEM (n = 3). * p < 0.05; ** p < 0.01; *** p < 0.001; **** p < 0.0001.
Download figure:
Standard image High-resolution image3.5. Mechanism of apoptosis induction in cancer cells
Generation of ROS plays a crucial role for induction of apoptosis in cancer cells [51–53]. Therefore, to understand the possible underlying mechanism of enhanced cytotoxicity of the DOX loaded NPs, the change in intracellular ROS level of MCF-7 cells after incubation with blank NPs, DOX in free form and DOX loaded NPs were measured via CLSM. For comparison, untreated cells and cells treated with hydrogen peroxide (a standard ROS inducer) were used as negative and positive controls, respectively. A cell-permeable nonfluorescent molecule 2´,7´-dichlorofluorescin diacetate (DCFH-DA) was used as ROS sensor which gets oxidized to 2´,7´-dichlorofluorescin (DCF) expressing green fluorescence once exposed to ROS. As shown in figure 6(a), very weak green fluorescence was observed in cells treated with blank NPs, similar to ROS level in negative control cells. In contrast, the cells treated with free DOX exhibited similar ROS level compared with positive control cells. Interestingly, DOX loaded NPs caused a rapid hike in the fluorescence intensity of DCF suggesting elevated cytotoxicity in comparison to free DOX which is consistent with the MTT data obtained before. The quantitative determination of ROS level followed the same trend as shown in figure 6(b).

Figure 6. ROS formation captured via (a) confocal images and (b) quantification of mean fluorescence intensity (MFI) in MCF-7 cells when incubated for 24 h with blank NPs, DOX in free form and DOX loaded NPs. Here, the incubation with H2O2 (for 30 min) was treated as positive control whereas negative control denotes the untreated cells. The data is represented as mean ± SEM (n = 3). Scale bars: 50 μm. **** p < 0.0001. (c) Gel electrophoresis of DNA and (d) expression of apoptosis-associated genes as evaluated by RT-PCR analysis. The measurements were carried out after various treatments of MCF-7 cells for 24 h. Lane M: 1 kb ladder, Lane 1: control cells, Lane 2: blank PDMS NPs, Lane 3: DOX in free form, Lane 4: DOX loaded PDMS NPs.
Download figure:
Standard image High-resolution imageThe apoptosis mechanism of DOX loaded NPs was further investigated by DNA laddering assay. During apoptosis induced by DOX, DNA gets fragmented due to double or single stranded breaks, giving rise to a series of bands known as 'DNA ladders'. As shown in figure 6(c), DNA fragmentation (bright band) was not observed when MCF-7 cells were treated with blank NPs (Lane 2) or in control cells (Lane 1). This suggests that blank PDMS NPs possess good cyto-compatibility and hence they did not damage the DNA of cancer cells. The DNA fragmentation became visible only when the cells were treated with free DOX (Lane 3) or DOX loaded NPs (Lane 4). The strongest DNA fragmentation was observed for DOX loaded NPs which again confirms the superior cell killing efficiency of DOX delivered via PDMS NPs than in free form.
Cellular apoptosis through excessive generation of ROS and DNA damage is usually associated with modulation of various apoptotic signaling pathways [54]. Therefore, RT-PCR was employed to analyze the expression levels of various apoptosis-associated genes e.g. caspase-3, caspase-9, survivin, p53 and bcl-2 in MCF-7 cells after treatment with blank PDMS NPs, free DOX and DOX loaded PDMS NPs (figure 6(d)). Caspase-3 gets activated in the early phases of apoptosis by caspase-9 and is responsible for cellular DNA fragmentation [55, 56]. Caspase-9 activation causes change in morphology of mitochondria and production of ROS [57]. P53 is a tumor suppressor gene that causes cell cycle arrest leading to apoptosis by regulating expression of apoptotic and anti-apoptotic genes [58]. Survivin belongs to the apoptosis inhibitor family and suppresses cell death signaling in tumor cells [56]. Bcl-2 induces tumorigenesis and resists cellular apoptosis [56]. The housekeeping gene GAPDH was utilized as internal control. The expression levels of apoptotic genes (caspase-3 and caspase-9) and tumor suppression gene (p53) were considerably up-regulated in MCF-7 cells treated with DOX loaded NPs as compared to other controls. Although the expression of survivin was comparable in both type of DOX treated cells, significant down-regulation of bcl-2 (anti-apoptotic gene) expression was observed in cells treated with DOX loaded NPs than that of free DOX. No significant change in the expression level of the above genes were observed in blank NPs treated cells in comparison to untreated control. The expression changes of the above apoptosis-associated genes collectively present a strong evidence on the role of PDMS NPs in improving the anti-tumor efficiency of DOX by strengthening apoptosis in cancer cells.
3.6. In vitro antitumor efficacy in multicellular 3D spheroid model
Conventional 2D cell culture model has been explored intensively in the past for investigating the antitumor efficacy of anticancer drugs. However, the 2D environment raises serious concern on the reliability to mimic the 3D tumor environment in vivo [59]. As an alternative, various 3D cancer models have been developed recently to mimic the innate morphology and microenvironment of actual solid tumors [60]. Among these models, 3D multicellular spheroid models are the most common owing to their ease of preparation [60]. Therefore, we have evaluated the antitumor efficiency of DOX loaded PDMS NPs in 3D multicellular tumor spheroids of MCF-7 cells, prepared using a modified hanging drop methodology. The viability of MCF-7 cells within the spheroids was determined through live/dead cell assay using FDA and PI for staining live cells in green and dead cells in red, respectively (figure S14(a)). The presence of green fluorescence throughout the spheroid without significant red signal indicates that most of the cells within the spheroid remained viable after culturing for 7 d. In order to compare the penetration ability of blank NPs, free DOX and DOX loaded NPs, the spheroids were incubated with them for 4 h and then observed via CLSM. The blank NPs were labelled with RITC to visualize the localization of NPs. A gradually diffused red fluorescence pattern was observed from the periphery to the center of the spheroids for RITC-labelled NPs (figure S14(b)). Free DOX was visible in the form of clusters indicating interrupted penetration through the spheroids (figure S14(c)). However, DOX loaded NPs penetrated uniformly throughout the spheroid expressing intense red fluorescence signal occupying the entire spheroid (figure S14(d)). The faster penetration and homogeneous intratumoral distribution of DOX loaded NPs is expected to inhibit tumor growth more effectively than that of free DOX.
To study the inhibition efficiency of DOX loaded NPs, the MCF-7 spheroids were exposed to culture medium, blank NPs, free DOX and DOX loaded NPs for 10 d and the representative optical images are shown in figure 7. The spheroids became very compact might be due to the intracellular interaction and secretion of the native extracellular matrix [61]. The spheroids treated with fresh culture medium or blank NPs did not show any significant inhibition to the growth of spheroids during 10 d. On the contrary, treatment with free DOX and DOX loaded NPs exhibited significant inhibition on the spheroid growth. After 7 d, free DOX treated spheroid started disintegrating whereas complete collapse of the spheroid was observed for treatment with DOX loaded NPs. Free DOX treated spheroid entirely collapsed after 10 d. The deep penetration ability of free DOX and DOX loaded NPs into the core of the spheroids might be responsible for the complete collapse of the spheroids after 10 d and 7 d, respectively. This result also suggests that DOX loaded NPs can more effectively inhibit the growth of solid tumor than DOX in free form which is quite consistent with the cytotoxicity results obtained before.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. In vitro antitumor efficacy tests in MCF-7 spheroid model. Representative optical images of spheroids treated with (a) culture medium as control, (b) blank NPs, (c) free DOX and (d) DOX loaded NPs for different number of days. Scale bar: 200 μm.
Download figure:
Standard image High-resolution image{kind=link}
4. Conclusion
In this study, for the first time, we have developed a simple, robust and scalable process to synthesize sub-50 nm soft hydrophobic PDMS NPs at room temperature. The synthesized PDMS NPs demonstrated good aqueous stability, favorable biocompatibility and in vitro pH-responsive drug release properties suitable to be considered as drug delivery nano-vehicles. The NPs possessed intrinsic mitochondria and nuclear dual-targeting capability probably due to their inherent hydrophobicity, soft and deformable nature apart from smaller size. Also, the NPs were able to deliver anticancer drug DOX into the intracellular organelles of cancer cells, inducing apoptosis through ROS generation, DNA fragmentation and expression of apoptosis associated genes. More than four-fold increase in the cytotoxicity of DOX was observed when delivered via PDMS NPs, as determined by IC50 value measurement. The enhanced therapeutic efficiency of DOX loaded PDMS NPs was also evident in destroying artificially grown 3D spheroid based in vitro tumor model just within 7 d (10 d for free DOX). These findings at in vitro level reveal that the synthesized soft PDMS NPs is a promising material for intracellular delivery of anticancer drugs. However, in vivo studies must be performed in future to conclusively affirm the potential of the synthesized PDMS NPs towards developing advanced nanomedicine for cancer therapy.
Acknowledgments
The authors are thankful for the financial support from Tata Consultancy Services (TCS) and Department of Biotechnology, India for this work.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
Supplementary data (1.4 MB PDF)