
Abstract
In this review we will discuss the topic of high order harmonic generation (HHG) from samples of organic and bio-molecules. The possibility to extract useful dynamical and structural information from the measurement of the HHG emission, a technique termed high harmonic generation spectroscopy (HHGS), will be the special focus of our discussions. We will begin by introducing the salient facts of HHG from atoms and simple molecules and explaining the principles behind HHGS. Next the technical difficulties associated with HHG from samples of organic molecules and biomolecules, principally the low sample density and the low ionization potential, will be examined. Then we will present some recent experiments where HHG spectra from samples of these molecules have been measured and discuss what has been learned from these measurements. Finally we will look at the future prospects for HHG spectroscopy of organic molecules, discussing some of the technical and in principle limits of the technique and methods that may ameliorate these limits.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
We begin with a brief overview of the process of high harmonic generation (HHG) and the essential attributes that allow it to be used for structural and ultrafast pump–probe studies. To do this we will try to establish a physical picture of the salient features of HHG by invoking some of the underlying theory, but we will not make any formal derivation of that theory. The conceptual framework we use in this exposition is the so called strong field approximation (SFA) [1] which is a quantum mechanical treatment of the process of HHG based upon a classical intuition that is valid when the laser field is sufficiently strong that for the active electron the Coulomb potential of the molecule can be assumed weak. The reader is advised to consult the original paper by Lewenstein et al [1] and other sources cited below for a more complete explanation.
HHG can be viewed within the classical form of the strong field picture [2, 3], alternatively referred to as the three-step model or the Simple Man's picture, from which the SFA theory was inspired. HHG takes place in an intense laser field and has a sub-cycle temporal structure with the ionisation of the molecule, propagation of the ionised electron in the field and recombination of that electron with the molecule as the three steps. In the three-step model HHG follows from tunnel ionization in the strong field [4]. Ionisation occurs near the peaks of the oscillating electric field amplitude with the tunnelling rate exponentially sensitive to the area under the barrier. This rate is determined by the bound state energy and the laser field strength with additional factors concerning the shape of the electronic wavefunction to determine the absolute rate [5–8]. The electron is then accelerated by the laser field and, if the electron is released after the field peak, the trajectory it takes can lead to it returning to the ion from which it was ionised within an optical cycle. The recolliding electron can then recombine with the molecular cation with a probability amplitude determined by the dipole moment for the recombination of an electron of the appropriate momentum. The strong field semi-classical picture is summarised in figure 1.

Figure 1. In HHG the strong laser field clocks and controls the process by (step 1) causing tunnel ionization that is confined to the vicinity of the electric field peaks, accelerating and returning the electron liberated by ionization (step 2) and causing a possible recollision that can lead to recombination of the electron into the hole it left in the molecule (step 3). (Reproduced from [7] with kind permission from Springer Science and Business Media).
Download figure:
Standard image High-resolution imageA crucial feature of this strong field picture is the large amplitude motion of the active electron, resulting in a substantial separation from the parent molecular ion at the zenith of the trajectory. This separation is related to the classical wiggle amplitude x0 for a free electron of mass m in a strong laser field of peak field strength E0 and angular frequency ω,
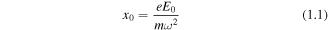
this may become large enough that for a significant portion of the electron's trajectory the Coulomb interaction with the ionic core can be regarded as a weak perturbation or ignored. The classical return energies of the electron can be up to 3Up, where Up is the ponderomotive energy (cycle average kinetic energy) of the electron given by
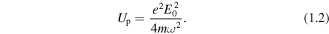
This return energy in a typical HHG experiment can range from a ∼10 eV to a 1 keV, and this plays the crucial role in determining the high energy cut-off of the HHG photon energy spectrum,
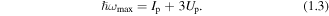
The corresponding de Broglie wavelength of the returning electron will be in the range 10−9–10−11 m. For the typical conditions of a molecular HHG experiment (λ = 1800 nm and I = 5 × 1013 W cm−2) the maximum excursion distance is >3 nm and at this location the Coulomb potential is <1/50th of the electron ponderomotive energy. The assumptions of SFA, that the interaction with the laser field dominates over the Coulomb field whilst the electron is in the continuum, is thus reasonably valid in this example.
The consequence of this strong field limit for HHG is that it has the potential to be a high temporal resolution structural tool [9] with evidence in small molecules that the molecular wavefunction can be retrieved [10] and even temporal evolution of the electronic wavefunction of the molecular cation tracked [11–13]. Furthermore the HHG signal, through the dependence on the recombination dipole matrix element, is highly sensitive to the rotational, vibrational and electronic state of the molecule and so it has been suggested to use HHG in the probe step of femtosecond pump–probe studies with a number of demonstrations of this for simple molecules [14, 15].
This is not a general review of HHG spectroscopy of molecules but will concentrate specifically on organic and bio-molecules. The molecules we will be considering in the context of high harmonic generation spectroscopy (HHGS) include simple hydrocarbons (e.g. alkanes [16, 17] and alkenes [18, 19]), halo-substituted alkanes [20] and more complex molecules like substituted benzenes [21] and nucleobases [22]. These are the focus of the review in part as small biomolecules are at the limit of what can currently be studied through HHG. Further it is anticipated that these molecular systems will display non-trivial coupled electronic and nuclear dynamics following sudden ionization.
A brief comment will now be given regarding some of the current objectives of attosecond/few femtosecond ultrafast structural dynamics studies in organic and bio-molecules, and how some of the measurement challenges associated with these objectives might be met by HHG spectroscopy. Tracking charge and energy flow in a molecule in the first few femtoseconds following photoionization or photoexcitation is a central challenge of attosecond science. As photoionization and photoexcitation are the primary event in photochemistry including those processes that are of key importance to; e.g. photosynthesis, light driven catalysis and biological damage mechanisms, this problem is of importance not only because it addresses fundamental dynamical questions of chemical physics but also because of the potential practical benefits for future technology. Moreover the study of photo-initiated processes may lead to a deeper understanding of the chemical reaction pathways of a wide range of non-photo initiated reactions. The possible existence of charge migration occurring on a timescale much shorter than any nuclear motion is much discussed in the literature [23, 24]. In fact it is now anticipated that the role of the nuclear state is critical in these processes, in both the initiation of the electron wavepacket [25] and in changing the electronic evolution as the nuclear states evolve [26]. To unravel this we need advanced theoretical tools (i.e. beyond the Born–Oppenheimer approximation) and measurements that can not only resolve few-femtosecond electronic dynamics but also structural changes on barely longer timescales. It is posited that due to the temporal structure of the generation mechanism HHG may have a role in making such measurements for molecules immersed in a strong field from which some general principles may be deduced.
The scope of this review is confined to HHG spectroscopy, i.e. those measurements involving HHG from a molecular sample that tell us something about the initial state of the molecule, about a chemical process that it undergoes or about the state of the molecule in a strong field. There are other applications of HHG from molecules, for example as a possible useful source of circularly polarised XUV pulses with short duration [27] that are beyond the scope of the current work.
To be clear from the outset HHG is a method that relies on a strong field. That the presence of the strong field has a profound effect on the processes we want to study at the few femtosecond timescale cannot be overstated. On the other hand the strong field gives unique opportunities for control and interaction with the cation that may reveal new information about the molecule e.g. ultrafast field driven population changes, time-dependence of chiral activity etc. We will argue that the most important application of HHG spectroscopy is that it is a powerful tool that is uniquely sensitive to the response of a molecule to a strong field.
This review is arranged in the following way. In section 2 we will highlight the capabilities of HHG spectroscopy for accessing attosecond and femtosecond domain structural and electronic information. We will then in section 3 turn to the technical requirements for the measurement of HHG spectra from a broad class of organic molecules and bio-molecules. In section 4 we will review recent measurements of the HHG spectra from these molecules and draw some conclusions regarding what may be learned in these first measurements. Finally in section 5 we consider the future prospects for this research and try to draw some conclusions as to where it may prove of utility and how new techniques may make an impact.
2. The capabilities of HHG spectroscopy for attosecond and femtosecond domain measurements
Although this section is not intended as a comprehensive review of the subject of HHG spectroscopy (this topic is covered elsewhere [28, 29]) it will set the scene for the work on organic and biomolecules. We will cover the salient aspect of what has been achieved in the HHG studies of mostly small inorganic molecules, such as H2, N2, CO2, SF6. We treat first experiments that have looked primarily at the harmonic field amplitude i.e. HHG spectral intensity. Then we will go on to consider more complete measurements where the harmonic field phase and polarisation state are also measured that leads to a complete determination of the HHG harmonic field information.
The most straight forward and direct channel of information from the HHG emission is the measurement of the intensity spectrum. The measurement requires a spectrally dispersive instrument that records the intensity (electric field amplitude squared) of the emission at a distant detector. Typically if the spectrometer/detector is spatially resolving in the direction perpendicular to the spectral dispersion then we have a far field spectrum that has also information of the far field spatial divergence. Already a great deal can be learned from the information in the spectrally dispersed and divergence resolved intensity spectrum. To illustrate this we show an example of such spectra recorded for a series of molecules (CO2, N2, O2, H2 and D2) [30] is shown in figure 2.

Figure 2. The on axis contribution to the observed HHG recorded in various molecules with an 800 nm drive field spectra comes mostly from the short trajectories. These are emitted at shorter times (1–1.7 fs) after ionisation. The more diffuse halo at higher divergences arises from the long trajectories (emitted 1.7–2.7 fs) after ionisation. We see that for CO2, N2 and O2 there is appreciable long trajectory intensity recorded. In contrast the long trajectory intensity is weaker for D2 and absent for H2, a consequence of the faster nuclear dynamics in these species. (Reproduced from [30] with permission from Elsevier.)
Download figure:
Standard image High-resolution imageThe divergence allows us to distinguish for instance short and long trajectory [31] (more and less divergent respectively) components and to even observe the interferences between them [32]. It has been established that the short trajectories encode return time into the spectrum [33] and so can be used for temporal resolution of dynamics. The temporal form of this is close to what has been predicted for the SFA and an empirical formula for the return time as a function of harmonic photon energy has been extracted by Lein [34] for the short trajectories.
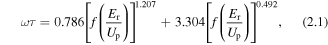
where the function f(x) is defined as  with Er and Up the harmonic photon energy and ponderomotive energy respectively. Similarly there is a well defined chirp for the long trajectories that carries the opposite sign as has been confirmed by measurement [35]. A consideration of these opposites chirps leads to an important fact that for the lowest order harmonics emitted from a 800 nm field there is a ∼1 fs difference in emission times between short and long trajectories. The differences in the short and long trajectory emission in the spectra in figure 2 can be qualitatively explained by this time difference. So whilst the short trajectories (observed on axis) are emitted at shorter times 1–1.7 fs after ionisation, the more diffuse halo at higher divergences arises from the long trajectories emitted 1.7–2.7 fs after ionisation. We see that for CO2, N2 and O2 there is appreciable long trajectory intensity recorded. In contrast the long trajectory intensity is weaker for D2 and absent for H2, a consequence of the faster nuclear dynamics in these species that leads to a much reduced harmonic amplitude (see below). Moreover to an extent harmonic dipole phase information can be extracted from the interferences [32] between the short and long trajectory components.
with Er and Up the harmonic photon energy and ponderomotive energy respectively. Similarly there is a well defined chirp for the long trajectories that carries the opposite sign as has been confirmed by measurement [35]. A consideration of these opposites chirps leads to an important fact that for the lowest order harmonics emitted from a 800 nm field there is a ∼1 fs difference in emission times between short and long trajectories. The differences in the short and long trajectory emission in the spectra in figure 2 can be qualitatively explained by this time difference. So whilst the short trajectories (observed on axis) are emitted at shorter times 1–1.7 fs after ionisation, the more diffuse halo at higher divergences arises from the long trajectories emitted 1.7–2.7 fs after ionisation. We see that for CO2, N2 and O2 there is appreciable long trajectory intensity recorded. In contrast the long trajectory intensity is weaker for D2 and absent for H2, a consequence of the faster nuclear dynamics in these species that leads to a much reduced harmonic amplitude (see below). Moreover to an extent harmonic dipole phase information can be extracted from the interferences [32] between the short and long trajectory components.
The harmonic relative phase, both between neighbouring harmonic orders and between harmonics emitted from molecules aligned at different angles, contains information about molecular structure, intra-molecular dynamics and spectral resonances in the continuum. Resonances are expected to cause a jump in the phase between harmonics that lie above and below the energy of the resonance. The phase between neighbouring harmonics can be measured via the resolution of attosecond streaking by interfering two-photon transitions (RABBIT) method. Here the photoelectron spectrum induced by the HHG field is measured in the presence of a delayed component of the fundamental field. This leads to satellite features (side-bands) arising from absorption or emission of an additional photon. Interference between the quantum paths arising from these two channels shows up in the intensity of the photoelectron side-bands and the delay dependence in this interference can be used to extract the harmonic phase [12, 33]. An all optical technique that utilises the addition of a weak second harmonic field in the HHG generation process has also been used to measure the relative phase of harmonics. Again a variable delay for the second harmonic field is introduced and the differential effect it has on the even harmonic intensities used to extract the phase. In this way the chirp in both short and long trajectories was successfully retrieved [35]. Similarly interference of the light from two adjacent harmonic sources containing molecules in different alignment states has been used to measure phase changes associated with interference features in molecular HHG [11].
The polarisation state of the harmonics from a molecule and their dependence upon the laser polarisation is of great interest in molecules as they are anticipated to display a tensor non-linear response to the applied strong field. Components both parallel and perpendicular to a linear drive field polarisation were predicted [36] and observed [37]. The situation is more complex for an applied elliptical laser polarisation and in this case a high degree of circular polarisation may be imparted on the harmonics in the proximity of a continuum resonance since this can impart differential phase shifts to the components of the harmonic polarisation [27]. The polarisation state of the harmonics can be measured by use of an appropriate XUV polarisation analyser. For instance the polarisation state can be measured through the reflection (at 45° incidence) from an appropriate metal mirror as the XUV reflectance shows a large variance between s and p polarisation. This can determine the s and p components of the polarisation. Since the HHG emission may be only partially polarised a full determination of Stokes parameters using more advanced methods may be required and efforts towards this goal are being made.
The majority of experiments performed so far that determine phase and polarisation of the harmonic field have been carried out on the harmonics generated by a 800 nm laser field. In part this is because that is the wavelength of the standard femtosecond titanium sapphire CPA laser widely used in the laboratory. It is also true that in contrast to HHG generation with longer wavelength fields (see below) the efficiency of HHG at 800 nm is far higher due to the strongly disadvantageous efficiency scaling with λ (for the single molecule response the scaling is ∼λ−6) as is discussed below. This is especially restrictive when molecular gases of limited density (see below) are employed as is typically the case for organic molecules.
We now delve deeper into why HHG spectroscopy can retrieve information on the temporal evolution and structure of a molecule. The HHG emission amplitude for a single molecule in the strong field limit can be interpreted in terms of quantum paths as was implicit in the earlier SFA theory [1] and explicit in the work of Salieres et al [38]. This idea leads to a great deal of insight that allows the harmonic amplitude to be expressed in a physically interpretable form [39].
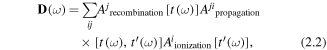
where, reading from right to left, are the amplitudes for ionisation at t', propagation between t' and t, and recombination at t; the summation is over different 'quantum trajectories'(i.e. including different possible cation channels) that result in the same return energy. The recombination amplitude is governed by the recombination dipole matrix element:
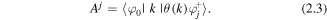
This is the transition matrix element between the continuum state (θ(k) with returning electron momentum k) and the bound state for a particular molecular ion intermediate state j. This quantity is therefore closely related to the photoionisation amplitude connecting the neutral molecule and the final state of the cation and electron continuum. The form of equation (2.2) is common to the SFA based theory where the continuum states have the form of plane waves (with no Coulomb interaction) and also to other theories that go beyond the standard strong field assumptions such as quantitative rescattering theory [40] where a physically more accurate form for Aj is used.
Given that the HHG spectrum reflects the recombination dipole matrix element and the angular dependence of the tunnel ionisation step it is not surprising that details of the molecular geometry are encoded into the harmonic field. Early on it was recognised that structural data can be extracted from molecular HHG [41, 42]. The possibility for experiments to test and exploit this idea stems from the emergence of methods for laser alignment of the molecular axis in the last two decades. These methods result in an ensemble of molecules that can be partially aligned in a controllable way. This alignment occurs in the presence of a strong non-resonant laser field through the second order induced dipole interaction leading to a torque on the molecular axis during the interaction. Laser alignment can be carried out either in the impulsive (laser pulse shorter than molecular rotational period) or adiabatic (laser pulse longer than the molecular rotational period) regimes, with laser fields ranging in intensity from ∼1010 to 1013 W cm−2 [43, 44]. It was first shown that it was possible to search for evidence of the role of molecular structure in HHG by using laser aligned samples of CS2 and CO2 [36, 45–47].
Two centre interference was first identified theoretically in 2002 as an effect likely to be present in the HHG spectra from an aligned ensemble of H2 molecules [36, 47] and the first experimental reports were found in the rotational revivals signatures of CO2 in 2005 [48, 49]. Since then there have been numerous observations of two centre interference signatures for example in H2 [50], N2O [51] and acetylene C2H2 [52]. The condition in a simple diatomic molecule in a gerade (even) state (e.g. H2) for two-centre interference leading to destructive interference between the contributions from the two atomic centres can be expressed as:
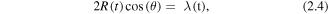
where R(t) is the instantaneous internuclear spacing, θ the alignment angle of the molecular axis with respect to the laser polarisation and λ(t) is the instantaneous de Broglie wavelength of the returning electron that will recombine to one or the other of the centres. Although this expression requires SFA (plane continuum waves) to be rigorously correct it provides a qualitative basis from which to develop a more sophisticated picture where the Coulomb effect on the electron propagation can also be considered. We illustrate these ideas for a time varying internuclear separation [50] in figure 3.

Figure 3. As the inter-nuclear separation increases in time in the few femtoseconds following ionization the two-centre interference condition will also change. This model was used to interpret the HHG spectrum for an H2 sample driven by a high intensity 800 nm field where there was inferred to be a significant degree of laser induced molecular alignment. (Reprinted with permission from [50] by the American Physical Society.)
Download figure:
Standard image High-resolution imageIn principle, if the alignment angle is known the position of a minimum or maximum in the harmonic spectrum can be used to determine the internuclear distance. In this way it is possible for HHG spectroscopy to follow the change of the internuclear coordinate either on a sub-femtosecond timescale during the HHG process [50] or on a femtosecond timescale during a photochemical reaction triggered by a separate laser event.
Subsequent to the theoretical identification of two centre interference there was the first demonstration of the more general method of orbital tomography in N2 molecules [10, 53]. The possibility to implement tomography comes from the Fourier transform relationship between the molecular wavefunction and the harmonic spectrum (see equation (2.3)). This is strictly valid if the returning electron is truly a plane wave as is assumed in the standard SFA [1] analysis of the three step model. A modification beyond the plane wave is feasible via QRS [40] and other more accurate theoretical treatments and the possibility for tomographic reconstruction is retained although the analysis becomes considerably more complicated. In the first implementations of tomographic reconstruction the harmonic spectrum alone of N2 was used, with the phases required to reconstruct the wavefunction being inserted 'by hand'. Going beyond this assumption other workers have used direct phase measurements in addition to the amplitude spectrum [12] or a reconstruction method that employs the full information available through the rotational revival [54]. Tomographic methods require a high degree of molecular alignment and/or orientation.
A high degree of molecular alignment is only possible for a restricted set of molecules, and is precluded in most large organic molecules (see below). One possible route forward in molecules that cannot be aligned is to use the spatial steering of returning trajectories that is made possible in a two-colour field composed of orthogonally polarised components (typically ω/2ω). Varying the phase between the fields has been shown to control the angle of return of the electron [55] and to select the relative probability of return of short or long trajectories [56]. In an unaligned molecular sample of ethane (C2H6) it has been shown that the resulting shaping of the recollision angle can probe the orbital structure of the molecule [57].
Dynamical information can be extracted from the HHG spectrum of molecules due to the attosecond timescale chirp of the emission that encodes the recollision time into the photon energy. In a molecular system, in contrast to an atom, it is expected that both electronic and nuclear dynamics will occur during the short interval between ionization and recombination (i.e. on the order of an optical cycle time). The use of the attochirp property of HHG (see equation (2.1)) to measure such fast dynamics was first proposed by Lein for applications to measuring the nuclear dynamics of H2, D2 and T2 [34]. The measurement of the relative magnitude of the nuclear autocorrelation function, that reflects the nuclear motion, follows naturally from the measurement of the ratio of the HHG intensity spectrum in H2 and D2 as it can be shown that within the SFA the harmonic amplitude spectrum is given by:
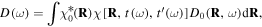
where the electronic dipole (in the absence of nuclear motion) is given by D0(R, ω) and the nuclear wavefunction at the initial time t is χ0 and at the time of recombination t' has evolved to χ. Hence the intensity of a given harmonic is given by the square of the nuclear autcorrelation function between the birth and the recombination times t and t'.
An experimental demonstration was soon after made for H2 and D2 with a good agreement with predicted nuclear autocorrelation function [9], and the retrieval of the proton dynamics in the  cation in the first few femtoseconds following ionization was demonstrated. Further the nuclear dynamics of the CH4 and CD4 molecules were revealed in the same study through the measurement of the nuclear autocorrelation functions. The proton dynamics in the methane cation as the C–H bonds undergo rotation from the equilibrium tetrahedral geometry of CH4 to the C2v geometry of the
cation in the first few femtoseconds following ionization was demonstrated. Further the nuclear dynamics of the CH4 and CD4 molecules were revealed in the same study through the measurement of the nuclear autocorrelation functions. The proton dynamics in the methane cation as the C–H bonds undergo rotation from the equilibrium tetrahedral geometry of CH4 to the C2v geometry of the  cation is evidenced to be very rapid evolution of the nuclear autocorrelation function in these measurements. This result is illustrated in figure 4.
cation is evidenced to be very rapid evolution of the nuclear autocorrelation function in these measurements. This result is illustrated in figure 4.

Figure 4. (a) The measured ratio of harmonic emission form CD4 and CH4 as a function of harmonic order (black solid squares). Also plotted on the horizontal axis is the recollision time obtained from equation (2.1). The red squares are a control measurement that measures the ratio of CH4 to CH4 runs. (b) The structural change in  cation that gives rise to the modulation of the HHG ratio in (a). (From [9] reprinted with permission from AAAS.)
cation that gives rise to the modulation of the HHG ratio in (a). (From [9] reprinted with permission from AAAS.)
Download figure:
Standard image High-resolution imageSubsequent theoretical analysis has shown the important role the Jahn–Teller effect plays in the steepness of the cation potential energy surface in methane [58]. Calculations by Patchkovskii [58] and others [59] have found that the predicted field free dynamics are close to what is measured in this HHG experiment. Other applications of the method to H2O [60] have revealed the important role played by additional states of the cation, each with a different potential energy surface governing the nuclear dynamics. The observed HHG signal is dominated by those potential energy surfaces that give rise to the slowest dynamics [61], the other surfaces with steeper gradients lead to an insignificant contribution due to rapid damping of the amplitude due to the nuclear motion. With the use of longer wavelength fields, with correspondingly longer laser cycle times, it was possible to employ this method to follow the motion in the ammonia molecule over nearly 4 fs [62]. In this last paper examining the ammonia molecule the possibility to predict the dynamics from the single photon ionisation spectrum of the molecule was discussed. It was found that the population of the cation vibrational states established by strong field ionisation differs qualitatively from the single photon ionisation limit.
The strong field response of a multi-electron molecule can also be revealed through the intrinsic temporal mapping of the return time into the HHG spectrum. It was demonstrated that multiple cation channels contribute to HHG in N2, both HOMO and HOMO-1 proving important, as they can both be efficiently accessed by tunnel ionisation in a sufficiently strong laser field [63]. Electron correlations effects in the tunnelling process are found theoretically to lead to new pathways for exciting multi-cation states in addition to direct tunnel ionisation [64]. With two or more cation states contributing to the total HHG amplitude one can anticipate interferences as the contributing amplitudes come in and out of phase due to the different energies of the states Ei (phases evolving as ![$\mathrm{exp}[-{E}_{i}t/\hslash ]).$](https://content.cld.iop.org/journals/0953-4075/49/13/132001/revision1/jpbaa1b6fieqn5.gif) This two (or multiple) channel interference can lead to a local spectral minimum/maximum in the emitted HHG intensity. Control on the dominant tunnel ionisation channels can be gained by aligning the molecule with respect to the strong field polarisation.
This two (or multiple) channel interference can lead to a local spectral minimum/maximum in the emitted HHG intensity. Control on the dominant tunnel ionisation channels can be gained by aligning the molecule with respect to the strong field polarisation.
This effect was first measured in an aligned sample of CO2 molecules by Smirnova et al (2009) using an 800 nm laser [11]. The dynamical interference was observed in the cut-off region where the two channels involved, the HOMO and HOMO-2 states of the cation, were of balanced amplitude due to the location in the cut-off and the choice of molecular alignment direction so that the destructive interference had a contrast deep enough to be observed. Subsequently the interplay between dynamical minima and structural minima (due to two-centre interference) was elucidated [65, 66]. In these experiments a longer wavelength source (e.g. 1350 nm) was used to investigate HHG from CO2. The wavelength scaling of the ponderomotive energy led to the HHG plateau to extend over the entire region where the structural interference occurs (∼50–60 eV) in the HOMO state of CO2. As a consequence of the destructive interference in the amplitude of the HOMO emission the additional dynamical interference between the HOMO and otherwise weaker HOMO-2 channels was apparent (see figure 5).

Figure 5. The dependence of the ratio of the HHG intensity for aligned versus unaligned samples of CO2 as a function of the angle between the alignment axis and the drive laser field measured at four different intensities of the 1350 nm drive field is shown in the first four panels. In the lowest panels are (left) ratio plotted for the angle of alignment θ = 0° for the four intensities, this shows how the position of the minimum depends upon the intensity which is interpreted as arising from the interplay of the contributions from structural minimum of the HOMO and the changing dynamical interference signature due to the relative phases of the HOMO and HOMO-1 amplitudes. In the right hand lowest panel the measured minimum position (black) is compared to the calculation based upon a model that incorporates these two contributions. (Reprinted with permission from [65] by the American Physical Society.)
Download figure:
Standard image High-resolution imageThe concept of studying the cation state amplitudes in a strong field ionized molecule and their dynamics through HHG has now been extended to a number of molecules including SF6 [67]. It is found that additional signatures may arise due to the laser driven transitions occurring between the cation channels of the molecule. The contribution of the different cation channels depends sensitively on their geometry (determining the initial tunnel ionization probability) and the dipole moments between different states.
Since HHG only occurs in the presence of a strong field the laser driven transitions may be a large effect. Laser field induced couplings between the cation states manifests itself through the population changes of these states between the times of ionization and recombination. This appears to be a general feature of molecular HHG. Whereas in atoms the ionic energy level structure does not generally lead to any states in resonance with the laser field in contrast a typical molecular cation may have a number of states separated from the HOMO by 0.0–4.0 eV. The sensitivity of HHG to dipole allowed couplings between cation states driven by the laser field between ionization and recombination has been established in experiments for a number of molecules [68]. The possibility of other orders of light field coupling, e.g. magnetic dipole couplings, has also been identified. This has been used recently to understand the observation of harmonic order dependent polarisation sensitivity in chiral molecules [69].
We have so far concentrated upon the capabilities of HHG spectroscopy for resolving sub-femtosecond dynamics, i.e. the dynamics of the molecular ion that occur in the time between ionization and recombination. It is also true that the properties of the emitted HHG field, including the amplitude, phase and polarisation, are uniquely sensitive to the initial state of the molecule. In principle any variation of the inter-nuclear separation or existence of rotational, vibrational or electronic excitations will manifest in details of the HHG field [70]. As a consequence using the HHG process as a highly sensitive probe in a femtosecond pump–probe experiment, where the pump field is a separate laser excitation, has been explored by a number of groups [14, 15]. Developments of this technique include the demonstration of transient grating methods in the excitation to increase the signal contrast [71, 72] and exploitation of the amplitude beating with the unexcited sample to increase sensitivity [73]. This promising technique has yet to be fully tested against conventional methods that also give femtosecond domain resolution, e.g. multi-dimensional visible/UV spectroscopy, femtosecond photoelectron spectroscopy, time-resolved x-ray spectroscopy, but it seems likely that it will offer some distinct advantages in certain cases. The main drawback is that the strong field may perturb the information available from the probe through significant field induced modifications to the electronic states [74] and distortion of the potential energy surfaces near any conical intersection [75].
3. HHG from organic and biomolecules technical challenges
In the next two sections of this review we will deal with progress so far investigating HHG in organic molecules and biomolecules. First we will discuss the technical issues and challenges associated with measuring the HHG spectrum from these molecules. In the next section the results obtained so far on this topic will be presented. The range of molecules in this category is very wide; ranging from hydrocarbons of different chain length and bonding type, through cyclic hydrocarbons, arenes to a wide range of amino acids and nucleobases. From the point of view of HHG studies all of these molecules present us with two major technical challenges. These are: (a) the low ionization potential (in the range from 8 to 10 eV) common to almost all these molecules (with the exception of a few of the alkanes), and (b) the fact that the vast majority of these compounds are solids or liquids at room temperature.
The low ionization potential results in a commensurately low strong field ionization intensity threshold and saturation intensity. Empirical data for a selection of small organic molecules [76] suggests ionisation saturation to occur at intensities around 2–5 × 1013 W cm−2 for molecules with 8–10 eV ionization potentials. In fact the ionisation saturation intensities for these molecules were found to be significantly higher than for atomic species of the same ionisation potential (when there is a suitable comparison available). It is postulated that this empirical observation is due to the more complex electronic orbital structure of molecules with additional nodal planes that can supress the tunnel ionisation amplitude. This ionisation saturation range is indeed borne out by recent HHG studies where the observed cut-offs are in good agreement with these ionisation studies.
The ionisation saturation intensity sets the limit for the maximum exposure intensity of a neutral molecule. In HHG the limit to the photon energy cut-off (equation (1.3)) is therefore set by the maximum value that the ponderomotive energy Up can reach at the limit set by ionization saturation. The scaling of ponderomotive energy and through this the harmonic cut-off with wavelength  from equations (1.2) and (1.3) shows that the use of a longer wavelength field is advantageous. For example a 800 nm laser field with an intensity 4 × 1013 W cm−2 (i.e. a field strength near the saturation intensity of the molecules of interest) has a ponderomotive energy of ∼5 eV, so for a molecule of Ip = 10 eV the maximum harmonic possible is about 25 eV. In practice HHG studies of organic molecules with 800 nm fields, and those of shorter wavelength, are compromised by these limits. The approach of extending the cut-off by using a longer wavelength laser has successfully been employed using high power femtosecond optical parametric laser. So with a 1600 nm laser field the cut-off high harmonic energy in the example above can be extended to beyond 60 eV.
from equations (1.2) and (1.3) shows that the use of a longer wavelength field is advantageous. For example a 800 nm laser field with an intensity 4 × 1013 W cm−2 (i.e. a field strength near the saturation intensity of the molecules of interest) has a ponderomotive energy of ∼5 eV, so for a molecule of Ip = 10 eV the maximum harmonic possible is about 25 eV. In practice HHG studies of organic molecules with 800 nm fields, and those of shorter wavelength, are compromised by these limits. The approach of extending the cut-off by using a longer wavelength laser has successfully been employed using high power femtosecond optical parametric laser. So with a 1600 nm laser field the cut-off high harmonic energy in the example above can be extended to beyond 60 eV.
Long wavelength radiation from an optical parametric amplifier OPA [77] or an optical parametric chirped pulse amplifier OPCPA [78] laser system is available over the wavelength range 1100–4000 nm. Commercial systems are available that that are pumped by titanium sapphire CPA lasers and can provide ∼1 mJ energy per pulse at a 1 kHz repetition rate with signal wavelengths from 1200 to 1500 nm and idler wavelengths from 1600 to 2000 nm. Typical pulse durations from these systems are ∼40 fs. Further pulse shortening can be achieved using hollow fiber pulse compression [79] or filamentation methods [80] and have been shown to permit the generation of CEP stable few cycle pulses at the idler wavelength with >0.5 mJ per pulse. HHG using longer drive wavelengths has been studied for a number of years, for example in uncovering the wavelength scaling of atomic HHG [81]. These laser wavelengths are well suited for HHG spectroscopy studies of organic molecules, and here we will concentrate on those applications to the quantitative study of the HHG spectrum of organic molecules.
There is, however, an important limit placed on the single molecule efficiency scaling by the use of longer wavelength drives due to the quantum mechanical nature of the process. The continuum electron wavepacket spatial spreading between ionisation and recombination has a severe effect on the probability of recombination. The wavepacket spreading increases with the time spent by the electron in the continuum, this time for the cut-off harmonic is proportional to the laser wavelength. This accounts for a λ−3 scaling when the 3-d spreading is considered. Moreover because of the spectral extension of the harmonic bandwidth the intensity per harmonic is further reduced by a factor scaling as λ−2. It is therefore predicted that the HHG efficiency scaling to be approximately λ−5 (or faster). This scaling is confirmed from recent empirical studies [82] suggesting it is closer to λ−6. Thus we anticipate that the emission per harmonic driven by an OPA idler at 1850 nm compared to an 800 nm laser under otherwise identical conditions is reduced by a factor of ∼6 × 10−3. In some circumstances optimised phase matching can be used to ameliorate this large drop in the single molecule harmonic efficiency but this typically requires the use of elevated sample densities and extended geometry which are not in general feasible for HHGS of organic molecules (see below).
Longer wavelengths (to >4 μm) can be generated at high power using difference frequency mixing, but the adverse wavelength scaling is even more severe. As a compromise most experiments on HHGS of organic molecules are being carried out currently with wavelengths 1000–2000 nm and with a repetition rate of 1 kHz (or greater). All the same the reduced power available from the primary laser source (compared to multi-milli-joules available from a Ti:S CPA) and the adverse wavelength scaling make the photon yield a serious limit from the point of view of accumulating data with a good signal to noise.
It is important to appreciate that the macroscopic HHG signals that are measured in an experiment are not from a single emitter but come from an extended ensemble. The topic of phase matching and propagation (including reabsorption of the emitted radiation in the gas) is crucial to understanding the formation of the measured HHG field in an experiment. This is a topic where the details are well outside the scope of this review, and the reader is advised to look elsewhere for a more complete account [31, 83–86]. An essential requirement for observable HHG signals is to have a dense enough gas sample. For HHG experiments in atomic and molecular gases typical working number densities are in the range from 1015 to 1018 cm−3 [87–89]. For gas phase molecules that are available in standard compressed gas format reaching the required sample pressure (∼10−4–10−1 bar) is usually straightforward. For a 50 μm spot radius and an interaction length 2 mm this density corresponds to ∼107–1010 molecules in the interaction region that take part in the HHG process. It is therefore easy to appreciate that the observed HHG field does not always directly reflect the single molecule response that as was argued above encodes structural and dynamical information.
Special precautions are needed to be able to reliably relate what is measured in an HHG experiment to the single molecule response. Longer interaction lengths may complicate formation of HHG spectrum by introducing; (a) more complex and photon energy dependent phase matching effects (when the interaction length exceeds a coherence length), (b) strong reabsorption also photon energy dependent (length longer than XUV absorption length) [88], and (c) large variation of the field intensity along the interaction region. An ideal experiment should employ a thin target that is much shorter than the laser focus Rayleigh range. A high density should be used to optimise the conversion efficiency whilst confining the generation to a region of constant intensity (along the propagation direction). Expansion through a small nozzle of 50–500 μm diameter is an appropriate solution to ensure a high local density in a spatially confined region whilst preserving a good enough vacuum for the propagation and detection of XUV harmonics once they are formed. Given that the density drops rapidly after expansion through a nozzle typically backing partial pressures of ∼100 times the desired working pressure need to be supplied (i.e. 0.1–10 bar). Whilst for gas phase molecules this will not be hard to achieve most organic molecules are initially liquids or solids and so appropriate sample handling arrangements must be introduced (see below).
Cooling may occur if a supersonic expansion from a nozzle is used. This approach has advantages as it may reduce the rotational temperature of the sample [90] in some cases this may be of utility if the molecule can be laser aligned [43, 44]. This alignment capability has been widely used for HHG spectroscopy of small molecules as was discussed above. Generally for organic and bio-molecules there are complications associated with the sample cooling and attempts to impose laser alignment. One must be wary of cluster formation that accompanies cooling at high density and is very likely for molecules with larger intramolecular interactions (often the case in the organic molecules) [91, 92]. Of course it must be recognised that the majority of organic/biomolecules are not suitable for alignment in any case as they are; (a) not sufficiently rigid, (b) occur in multiple conformers, this means the strategy of using a well-defined molecular alignment or orientation to interrogate the HHG spectroscopy properties is only an available option in special cases.
Small organic molecules that are available in the gas phase are typically stiff and relatively easy to cool so alignment effects can be studied. Examples include acetylene (C2H2) which has been extensively studied [19, 52, 93] and butadiene [94] (C4H6) and in both cases the alignment dependence of the HHG signal has been measured and signatures of two-centre interference observed. For most other organic molecules, including the alkanes, the structure undergoes internal rotation (torsion) and bending that make alignment challenging under the density conditions needed for HHG (although control of torsional modes has been achieved in low density cold molecular beams [95]).
A large number of organic molecules occur in the liquid phase at room temperature. Depending upon the temperature dependence of the vapour pressure it is possible to achieve an acceptable gas phase pressure even at room temperature for some molecules, or to apply a degree of heating. In this category also are those substances that are solid at room temperature but that have melting points low enough that they can be brought into the liquid state relatively easily by modest heating. Typically for these molecules vapour pressures can be obtained in the 100 mbar–1 bar range for a temperature below 400 K. Early efforts to do this were reported by Hay and co-workers where a heated and temperature controlled reservoir containing the sample and vapour feed directly a solenoid actuated pulse gas valve [46]. The vapour sample can be delivered to the nozzle as a pure vapour or in the presence of a higher pressure carrier gas.
Precautions must be taken to ensure that the resulting molecular vapour is accurately maintained at a precise temperature as this is critical to ensure quantitatively reliable HHG spectra. Further any 'cold' spot in the gas handling system can lead to condensation that can results in blocking of the vapour flow and other problems. To overcome this we have recently constructed an apparatus for the quantitative comparison of the harmonic spectrum from a number of substituted benzene systems. The fact that a number of molecular vapours can be prepared and switched quickly facilitates the comparisons between molecules. This apparatus employs a continuous flow of the gas jet into the vacuum via a 100 μm orifice (figure 6), this avoids any difficulties associated with operating a kHz repetition rate pulsed valve reliably over an extended period.

Figure 6. Schematic of apparatus used at Imperial College. Liquid molecular samples are loaded in stainless steel vessels and held at the required temperature in a heated water or oil bath with high temperature stability. The pipes and valves of the vapour delivery manifold are maintained at a slightly higher temperature using heating tape. The nozzle is also heated and injects a continuous jet of gas through a 100 μm diameter orifice located close to the laser focus. (Courtesy of F McGrath, private communication.)
Download figure:
Standard image High-resolution imageThe apparatus shown in figure 6 has recently been used to record an extensive series of HHG spectra using an OPG operating at 1850 nm in vapours of benzene, methyl substituted benzenes (toluene and xylene) and halo-substituted benzenes. The aim was to achieve harmonic spectra that could be compared between the molecules under identical conditions, including identical sample densities. The 100 μm orifice meant that the interaction length was less than 0.5 mm compared to a focus Rayleigh range of >10 mm. In this way the harmonic spectra come close to the ideal of the single molecule response. The density could be controlled accurately by varying the temperature. We can see in figure 7 that even though the backing pressure was varied over a factor of three the normalized HHG spectrum from benzene (when corrected for the n2 scaling where n is the sample density) is essentially identical. This indicates that under these conditions deleterious effects such as cluster formation, reabsorption of the harmonic radiation and reshaping of the spectrum due to phase matching effects play a minor role. It seems that we can thus be confident that with the correct precautions we can record HHG spectra from organic molecule vapours that can be quantitatively related to the single molecule response.
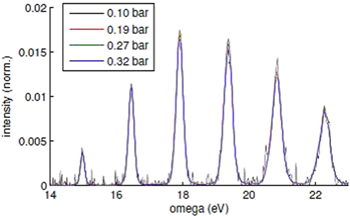
Figure 7. Pressure scaling of the normalised spectrum demonstrating the constancy of the spectral shape of the HHG over the range of pressures investigate. (Courtesy of D Austin, private communication.)
Download figure:
Standard image High-resolution imageMany biomolecules occur in a state that is initially liquid or in solid phase and have typically a very low vapour pressure at room temperature. For instance the nucleobase thymine has a vapour pressure of only 4 × 10−7 bar (density ∼1012 cm−3) at a temperature of 408 K [96]. Molecules such as this are a serious challenge for HHG spectroscopy techniques since the sample density required for HHG needs to be ≫1014 cm−3 to record a good quality HHG spectrum. It is known that excessive heating will lead to thermal dissociation and other deleterious effects so simply increasing the temperature is not an option.
Heating samples with low vapour pressure such as biomolecules or fullerenes in an oven is feasible, but the densities will typically be too low for HHG experiments although may be sufficient for strong field fragmentation experiments [97]. An alternative method for obtaining vapour phase samples of sufficient density for HHG is to use laser ablation. This technique was reported to have been used for HHG from fullerenes [98] and from carbon ablation plumes [99] Motivated by these successes with fullerenes attempts have been recently made to perform HHG from ablation plumes of uracil and thymine. In the uracil case good quality HHG spectra were recorded (figure 8(a)), but in contrast no HHG was observed from a plume formed from thymine under otherwise identical conditions [22].

Figure 8. (a) Set-ip for obtaining stable HHG spectra from an ablation plume that employed a rotating target coated in thymine/uracil powder. (b) The HHG spectrum from the uracil plume can be clearly seen, but under nominally identical conditions not HHG spectrum was recorded from a thymine plume. (c) An ion TOF measurement of the mass fragments in the uracil plume under the same ablation conditions reveal even in this molecule the vapour has a complex composition. (Reproduced from [22] with permission of PCCP Owner Societies.)
Download figure:
Standard image High-resolution imageMoreover even in the case of uracil it was not clear from which species in the plume the HHG emission originated. A study of the composition of these plumes using ion mass spectroscopy techniques reveals that in fact multiple species are formed in the plume (figure 8(b)).
It therefore remains uncertain as to whether ablation plume techniques will ever give sufficiently precise control over the vapour composition to permit their use in quantitative HHG spectroscopy.
4. Status of HHG spectroscopy with organic and biomolecules
In this section we will focus on the work so far reported on measuring quantitative HHG spectra from organic molecules and biomolecules. We will note the advances towards the retrieval of time-resolved and structural information from HHG of these molecules where they have been made and what might be possible in the near future and where the limits may lie. So far the main emphasis is on the easiest to study small organics, where some progress can be made using the full tools of HHG spectroscopy. We will look first at these cases. The alignment dependence of HHG and even phase measurements of the HHG emission are still feasible for some molecules. This is because these molecules are in many cases stiff enough to contemplate molecular alignment.
The first experiments setting out to measure HHG from organic molecules compared HHG spectra with other molecules and atoms [17, 18]. An early study by Fraser et al aiming to investigate the role of electron delocalisation on the HHG process compared the HHG yield from butadiene with that from butane [18]. Evidence was found that the lower order harmonics were relatively enhanced in butadiene, supporting the hypothesis that delocalisation can enhance HHG. A relatively long (∼200 fs) 800 nm laser pulse was used and interpretation of these early studies should be treated with some caution due in part to the complication arising due to the contribution to the HHG from various fragments of the parent molecule as fragmentation may occur earlier in the laser pulse. Recently investigations using a laser at 1850 nm Alharbi et al [100] have investigated the role of delocalisation (aromaticity) in benzene C6H6 (aromatic) and furan C4H4O (non-aromatic) and found higher yield for the benzene which they argue is due to the contribution from the π orbitals that contribute to the delocalisation corroborating the earlier finding of Fraser et al.
Early work on cyclic hydrocarbons (benzene, naphthalene, cyclopropane, cyclohexane and hexane) using similar laser parameters to those of Fraser et al followed and some systematics in the comparison between molecular species were observed [101] finding evidence again for enhanced efficiency due to aromaticity, and the ellipticity dependence of HHG yield was measured [102]. Nevertheless the long pulse was found to lead to deleterious effects as was confirmed when results with a 70 fs laser pulse were compared with those from a 200 fs pulse [103]. These problems were partly circumvented by adopting significantly shorter pulses. This approach was successfully applied to a study of HHG from a series of small molecules where the output of a 800 nm laser was compressed using the technique of hollow fiber compression to durations of 6 fs [104]. A series of alkanes was studied using a 25 fs laser pulse and the spectra quantitatively compared with SFA calculation [16]. The results of these measurements were interpreted by the aid of SFA theory based upon the component atomic orbitals that suggested the p-orbitals played an important role in the HHG spectrum of these species.
To advance this work it was realised that more complete studies that could access the geometrical properties of the molecule would be valuable. First measurements on HHG from laser aligned inorganic molecules were conducted using a 800 nm laser wavelength and CS2 and CO2 were studied (see above) [45, 46]. First steps aimed at retrieving structural information from the HHG spectrum in organic molecules were taken by Torres and co-workers, where impulsive alignment methods were applied to molecules in a beam emitted from a small (100 μm) nozzle and used to study the HHG yield as a function of molecular alignment [19]. The alignment dependence revealed an insight into the strong field orientation dependence of the HHG signal in acetylene (C2H2) and allene (C3H4) and ethylene [93]. These studies revealed aspects of the electronic structure of the HOMO of these molecules, indicating the presence of nodal planes in the orbital that affect the alignment dependent HHG yield through the influence of the ionisation and recombination amplitudes. The harmonic orders studied were, however, restricted to relatively low harmonic photon energies (to orders below <H25) due to the use of an 800 nm laser field. This limitation on the photon energy, and therefore the recolliding electron momentum, prohibited the observation of any structural interference signatures in the HHG.
As discussed in section 3 ionisation saturation limits the intensity that can be used for most organic molecules to <5 × 1013 W cm−2 and for 800 nm this severely restricts the width of the HHG spectrum. By employing an OPA Torres et al showed how the advantageous wavelength scaling of HHG enabled clear observation of an extend cut-off in molecules of low ionisation potential [52]. Using a 1300 nm field HHG spectra from N2O (Ip = 12.9 eV) and C2H2 (Ip = 11.4 eV) were generated with cut-offs exceeding 50 eV. A further advance was made when longer wavelength fields were applied along with molecular alignment. In a study of butadiene using a 1450 nm few-cycle field generated directly from an OPA [105] operating at 1450 nm it was possible to record the angular dependence in acetylene, allene, ethylene and 1,3-butadiene [94]. In the case of the latter molecule the ability to measure an extended harmonic spectrum (to photon energies ∼50 eV) and to study the angular properties of harmonic yield was especially notable as the Ip < 10 eV (see figure 9). The latter observation can be seen as opening the door to HHG spectroscopy of biomolecules that typically have an ionization potential between 8 and 10 eV.

Figure 9. HHG spectrum of 1,3 butadiene recorded using a 17 fs laser pulse with a wavelength of 1750 nm from an OPA system. The HHG variation with molecular alignment verifies that the HHG emission is from an intact molecule even though the molecules ionisation potential is only 9.1 eV. (Courtesy of C Vozzi, private communication.)
Download figure:
Standard image High-resolution imageGoing beyond the use of molecules available as compressed gases is challenging and requires the use of special preparation methods as described above if the single molecule response is to be uncovered. One method adopted by the group in Ottawa University was to construct semi-infinite and finite length vapour cells in which vapour pressures of >1017 cm−3 could be attained over an extended path length. In this way high efficiency HHG from water vapour with optimised phase matching was observed using both 800 nm and 1300 nm lasers [106]. The extended cut-off compared to atomic gases under similar conditions was attributed to ionization suppression resulting from the shape of the water outer orbital that includes nodal planes. The finite cell approach was used by the same group to study HHG from chloromethane molecules (the series CCl4, CHCl3 and CH2Cl2) with a 1300 nm and 1500 nm laser wavelength. A Cooper minimum was observed to appear close to the value appropriate for atomic chlorine and additionally two-centre interference structures were reported indicative of the electronic structure of the molecules [20] (see figure 10). In a similar experimental configuration the role of differing strong field ionization rates on the HHG from randomly aligned cis- and trans-isomers of the same molecules (stereoisomers of 1,2 dichloroethylene C2H2Cl2 and butene C4H8) were reported [107]. The connection of the observed spectra to the single molecule response still needs to be confirmed for these experiments.

Figure 10. Harmonic spectra measured from Xe, CH2Cl2, CHCl3 and CCl4 displaying Cooper minima and structural interference signatures. (Reprinted with permission from [20] by the American Physical Society.)
Download figure:
Standard image High-resolution imageAn interesting series of recent experiments has been carried out at the University of Ottawa and CELIA in Bordeaux with theory from the Max Born Institute in Berlin. This work has investigated the HHG response of the chiral organic molecules, epoxypropane and fenchone, to the ellipticity and helicity of the laser field. These experiments were performed in vapour cells using 1770 nm laser fields. The ellipticity was varied from linear towards circular polarisation with both helicities measured. Although the behaviour is somewhat different in these two molecules it was clear that the left and right handed entaniomers display a different, and opposite, asymmetric ellipticity response in the HHG yield [69] (figure 11). Analysis has indicated that the dominant contribution to the helicity dependence arises from magnetic dipole allowed transitions driven in the molecular cation by the laser field. The remarkably high degree of sensitivity of the HHG signal to chirality is posited as a new opportunity to determine the stereometric mix of chiral molecules and perhaps this will help with various schemes of entaniomer enrichment [108]. The result also indicate another example where the strong laser field drives transitions in the cation and the high significance this has in HHG. In this case magnetic dipole transitions are the critical ones, but the principle is similar to the effects inferred for N2 [68] where it was the electric dipole allowed transitions that play the dominant role.

Figure 11. Dependence of harmonic yield upon laser ellipticity/helicity in the two entaniomers of the epoxypropane molecule. Signatures of helicity dependence in some harmonics (around 35 eV) are believed to reflect the chiral response of the cation undergoing HHG. (Reprinted by permission from Macmillan Publishers Ltd [69].)
Download figure:
Standard image High-resolution imageRecently a retrieval of the hole dynamics induced by strong field ionisation in HC2I (iodoacetylene) using HHG spectroscopy was reported [13]. Here the linear molecule was laser aligned and orientated and the harmonic amplitude and phase compared to that of an unaligned sample. This comparison was made for alignment of the molecular axis both parallel and perpendicular to the strong field polarisation. Measurements were performed at 800 nm and 1300 nm in order to separate dynamical from structural effects. For the parallel aligned case the laser field coupling was found to significantly modify the dynamics of the superposition of hole states formed by strong field ionisation whereas for the perpendicularly aligned molecules (for which the laser coupling matrix amplitudes are much smaller) the hole dynamics in the absence of significant subsequent field driven dynamics was retrieved.
So far then there has been good progress towards understanding HHG from small organic molecules. Considerable challenges remain to extend the methods to larger organics and biomolecules. Moreover the use of vapour cells may prohibit the fidelity of measuring the single molecule response due to masking by reabsorption and phase matching effects. This has motivated the work by our group to study larger organic molecules using a small diameter (100 μm) continuous gas jet. In this work we have demonstrated the ability to systematically compare the HHG yield and spectrum of a series of methyl- and halo-substituted benzenes. Use of the gas jet geometry has enabled the thin sample limit to be achieved as evidenced by quadratic density scaling of the HHG yield (see figure 8). In these experiments it was important to confirm that the HHG spectra were reproducible, were not recorded in a strong saturation regime and did not show obvious evidence of distortion due to high density effects. In that regard these measurements come close to the uncovering of the single molecule HHG response. The motivation for this work is that we expect the interplay between; (a) structural effects in the dipole forming the HHG spectrum, (b) possible nuclear dynamic effects (of the kind earlier studied in CH4 and NH3), and (c) multi-electronic states of the cation introducing additional channels of harmonic amplitude that may display dynamic interplay and possibly laser driving effects. By use of changing the substitution chemically and geometrically it is intended to unravel these effects. Although this work is at the time of writing at a preliminary stage in the data analysis we note interesting recent theoretical work. Predictions have been made of the manifestation of short time dynamics at a conical intersection in C6H6 and C6H5F with distinct differences reported between these molecules [109].
5. Current limitations and future prospects for the HHG spectroscopy of organic and biomolecules
We will now consider some of the limits that need to be overcome in the near and medium term to make progress in understanding HHG from molecules. First we must reiterate the limitations due to lack of molecular state alignment. Hot vapour cells can provide no significant cooling and continuous nozzles lead to only limited rotational cooling. This is because unless there is a very fast pumping of the vacuum chamber the use of a continuous flow nozzle leads to raised background pressure and so cooling is not efficiently achieved. In future work use of heated pulse gas valves to achieve lower base pressures may lead to a significant improvement to the degree of rotational cooling. This objective should be pursued. Nevertheless these approaches will not, in general, be as effective in achieving aligned molecule samples as in the simple linear molecules (e.g. N2, CO2 and HC2I) so far studied. This is for several physical reasons, including; (i) a higher propensity of many of the organic molecules to cluster when rotationally cooled at high density [110], (ii) the complex geometry of many organic molecules (typically asymmetric tops) requiring rotation around independent multiple axes to be controlled which has only been achieved in low density molecular beams by either adiabatic alignment [111] or by adiabatic switching which requires a femtosecond timescale laser shutter [112], (iii) the fact that many of the molecules of interest occur as multiple conformers and/or are not rigid and so alignment concepts are of limited validity.
Given the difficulties associated with achieving molecular alignment it is very important in parallel to understand what can be learned from unaligned samples of these molecules. A promising technique (mentioned in section 2) that uses a field synthesized from orthogonally polarised fundamental (800 nm) and second harmonic (400 nm) components has been shown to reveal dynamical information from HHG in C2H6 [113] In this technique the propensity of the molecule to tunnel ionise only when aligned at a specific angle with respect to the field is used to produce an aligned ensemble of molecules that participate in HHG [114]. The phase delay between the field components is then used to control the direction of the recollision whilst the direction of the recombination dipole is determined from the ratio of odd and even harmonics [55]. From these measurements evidence of the dynamics in the valence wavepacket formed by the strong field ionisation is found. Such a method may be extendable to some larger molecules, although such measurements will be more challenging to implement with the longer wavelength fields required and the more complex molecular geometry.
An in principle limitation to all HHG spectroscopy implementations is the fact that the valence states that are ionized are tied to the tunnel ionization process. In effect this means the electron dynamics that can be probed is inherently limited only to those processes that can be triggered by strong field ionisation. As the sensitivity to electronic dynamics in HHG is primarily confined to the superposition formed by the ionization process, it is not yet clear that any correlation driven dynamics beyond those that can be deduced by simple photoelectron spectroscopy can be uncovered. Moreover the further effects of the strong field on a molecular cation are profound and include the possibility of coupling of cation states by radiative transitions driven in the strong field [68, 69] and the dynamic polarisation of the remaining electrons in the system as was found to play a role in the strong field ionisation of metal atoms and clusters [115–117]. Invariably in the cation of an organic or bio-molecule there exists a high density of electronic states [118]. It is thus inevitable that in many cases there will be dipole allowed transitions near to resonance with the strong field. For a dipole allowed transition with a dipole moment of 1 atomic unit in a field of 2 × 1010 V m−1 we can expect coherent population transfer timescales of just a few femtoseconds, i.e. complete population transfer between cation states within the time between ionization and recombination.
The role of the strong field in the modification of the electronic structure in HHG is explicitly ignored in SFA models of HHG. A recent experiment on orientated molecules CH3F and CH3Br has shown that the measured intensity ratio between odd and even harmonic orders and aligned/unaligned samples requires the strong field modification of the electronic structure to be taken into account to explain the experimental observations [74]. The magnitude of this effect is dependent upon the size of the static polarisability of the molecule. It is clear that the influence of the strong field on the HHG will complicate the interpretation of molecular HHG as structural or dynamical probes and these complications will grow for more complex organic molecules.
We will now attempt a critical analysis of the prospects for the HHG spectroscopy technique for unravelling structure and dynamics when applied to organic and biomolecules. It will be informative to draw a comparison to alternative approaches. Some speculations as to how some of the limits may be overcome using new concepts and higher repetition rate and/or higher power laser sources will be included. We will try to identify the best prospects for where HHG spectroscopy of more complex molecules will impact future science and also where HHG spectroscopy may not be the ideal technique.
First we consider what may be possible using conventional HHG spectroscopy and make a brief critical analysis of alternative approaches. The central limitation is that the presence of the strong field is likely to 'break' the attosecond timescale electronic dynamics except those explicitly caused by the strong field. This is especially true of the case of electron–electron correlation driven electron dynamics i.e. the case of electron dynamics most relevant to native photochemical response that is posited to drive charge migration in valence states. Essentially all the valence shell electrons will interact with the strong field laser field that is applied. This may be through quasi-resonant interactions, see the examples for laser driven transitions in the cations cited above. Even when there is no single resonant interaction that dominates it has been speculated for a long time [115] and recently shown explicitly in experiment [74] that the strong field influences the basic physics of the HHG process well beyond what is possible to accurately describe in naïve SFA treatments. It is possible to apply more sophisticated theoretical treatments [119, 120] to account for these multi-electron effects. It remains to be seen as to whether this will permit HHG spectroscopy to become a routine measurement method for electron dynamics of molecular valence states in more complex organic and biomolecules that is free from the need to make demanding theoretical calculations specific to every molecular species investigated.
There are of course alternative attosecond pump–probe methods to investigate ultrafast valence dynamics that have been the focus of much research over the last decade, the reader is advised to consult several recent reviews on this subject [121–123]. The majority of the techniques demonstrated use HHG sources either for the probe [124] or the pump steps [118] with a strong laser field employed for the other step. This has several drawbacks, unless special precautions are employed to shorten the laser pulse to below a single cycle [125], since the field used in ionisation does not provide sub-femtosecond resolution. The use of a strong field as a probe [118] causes severe interpretational complications similar to those discussed above in the context of HHG spectroscopy. Work progresses towards true attosecond pump–attosecond probe experiments and generating the two-colour pulse capability required was recently demonstrated [126]. Time-resolved photoelectron spectroscopy, angularly resolved photoelectron spectroscopy and transient absorption methods are all looking to be viable alternatives where if HHG based sources are employed the high temporal resolution needed can be achieved. These are perhaps more appropriate solutions to measuring the full dynamics of charge migration and the nuclear response to sudden photoionisation because they in principle do not require either of the pump or probe fields to be of a strength that is non-perturbative. Nevertheless for the HHG sources presently available the fields may be too weak to permit a pump–probe measurement with adequate signal/noise to be accomplished. Alternative detection schemes that allow for observation of the probe signal against no background are being postulated to circumvent this [127]. In the meantime work on developing brighter HHG based attosecond sources continues.
A future alternative, where source brightness is not thought to be a limit, is to use a soft-x-ray FEL to carry out pump–probe measurements. Although as yet x-ray FELs are not operating in the sub-femtosecond regime the large potential source bandwidth is sufficient to generate pulses of a few hundred attoseconds and schemes to do this are being actively theoretically investigated [128]. Recently two-pulse two-colour operation was demonstrated at LCLS with pulses as short as a few-femtoseconds [129]. A number of strategies have been proposed to use these multi-pulse x-ray FEL capabilities to measure ultrafast valence electron dynamics [130–132] but these are yet to be demonstrated.
It is perhaps the remaining difficulties associated with alternative attosecond methods that continues to motivate the development of HHG spectroscopy for the attosecond measurement of molecular dynamics associated with photoionisation. The use of HHG spectroscopy to deduce nuclear dynamics within the field cycle has been shown for small molecules. These dynamics appear not to have been greatly modified by the presence of the strong field and so there is some prospects that the use of the method for larger organic molecules will be viable. Here more systematic experimental and theoretical work, along the lines of [58], is needed to extend the interpretation and quantitative comparison to a wider range of molecules. For following structural changes on a longer (femtosecond) timescale in a molecule undergoing a photochemical change the prospect seems also quite promising for HHG spectroscopy.
In contrast to the problems just highlighted regarding using HHG spectroscopy to retrieve the native photochemical behaviour of molecules under perturbative conditions it must be recognised that the HHG signal carries the most detailed insight into the response of a molecule in a strong field. Indeed there is maybe no better tool available to understanding what goes on in a molecule exposed to a strong field at the detailed quantum level and on the inherent sub-femtosecond timescale than through the light emitted in the HHG process. This may prove the greatest utility for HHG spectroscopy of organic and biomolecules. So for instance the need to understand strong field induced fragmentation dynamics, not only in terms of the final fragment states but also in terms of the short lived intermediate states and mechanisms leading to fragmentation, may lead to breakthroughs that underpin future technologies in for example femto-second laser induced fragmentation mass spectroscopy analysis of proteins [133]. and the strong field coherent control of molecules [134, 135]. Likewise femtosecond pump–probe measurements, with sensitivity to the instantaneous state of the molecule at the start of the HHG process, are a promising area for developing better strong field control methods.
So given that there may be a unique scientific benefit of applying HHG spectroscopy to organic and biomolecules how do we overcome the main technical limitations? First, of course, there is the not inconsiderable difficulty of getting a single component gas/vapour of the molecule of interest at sufficient density to record a HHG spectrum. As we have recently shown a wide range of organic molecules, i.e. those that are in the liquid phase at around room temperature, can be handled using heated systems for vapour handling (see section 3). The generation of HHG from dense confined jets of these vapours is the best way to ensure that we can come closest to the single molecule HHG response. For molecules starting as solids at room temperature, and for which a great deal of heating is needed to get to the vapour pressures required for a successful HHG measurement, the required state may be harder to achieve. This makes handling biomolecules much harder than it is for low density techniques, e.g. compared to the electrospray method widely used in mass spectroscopic analysis. Application of too much heating can cause the molecule to appear in additional conformers or to dissociate so that multiple molecular fragments are present in the sample to be investigated. The problem of multiple fragments and the degree of sample heating that take place in an ablation plume has so far prevented laser ablation plumes being used in quantitative HHG spectroscopy work. More work is needed to try to resolve these issues, but there is no guarantee of a satisfactory outcome as the ablation process is known to be very complex with little precise control available over the thermally driven channels that occur within the plume and in the initial heating of the solid.
An inherent problem already encountered in our work on HHG from jets of organic molecules is the relatively low signal level caused by both the low density of the sample and the need to use a longer wavelength laser. A possible future solution is the use of high repetition rate MIR laser sources providing high average power. Currently we employ systems that run at 1 kHz repetition rate and deliver ∼1 W average power, a similar laser of 100 kHz repletion rate will need to deliver a 100 W power or greater. Similarly per shot low signal levels might be overcome by using higher power MIR sources so that a larger focal volume can be employed, but again this demands an increase in the average power. Combining higher rep-rate and higher peak power sources is clearly something that should be investigated as our technology for high power ultrafast lasers in the 1–4 μm wavelength range develops. The former capabilities are being developed through OPA and OPCPA technologies [136–138]. There is already proven the potential to go to the few-cycle limit with sources at 1 kHz repetition rate.
As stated above a fundamental limit of conventional HHG spectroscopy is that it is tied to the strong field ionisation process. The consequence of this is that the electronic states of the molecular cation that it can probe are restricted to the lowest outer valence states, and these can only be looked at in the presence of a strong laser field. An alternative approach that overcomes this limit to an extent is x-ray initiated HHG where single photon ionisation is used instead of strong field ionisation as the initial step in the HHG. This idea was first proposed for HHG in atomic systems as a way of selecting specific quantum paths [139, 140]. In atoms the idea has been demonstrated in several recent experiments [141, 142]. As the photon that initiates ionisation can in principle be of sufficiently high energy to excite a hole in an inner valence or inner shell state (see figure 12) it overcomes one of the limits of conventional HHG. Advantages include: (a) it can in principle access deeper lying states and those for which tunnel ionization is harder/not feasible, (b) because the IR field doesn't need to be strong enough to cause ionisation the intensity can be dropped by a little (maybe a factor of 2), but is still limited to fields strong enough to return the electron. In recent analysis it was shown that by using this method and the return time to photon energy encoding properties of HHG the hole evolution of in an inner shell in Kr and in an inner valence shell in 1,3-butadiene could be reconstructed from this measurement technique [143] (see figure 12).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 12. The concept of XiHHG illustrated for an inner shell hole formed by single photon ionisation. HHG resulting from the IR field driven electron return will compete with the fast decay rate of the hole. In the left panel the use of the wavelength and order scaling of the return times for retrieving fast exponential and non-exponential hole decay is illustrated (green—krypton, blue—butadiene, purple—propanal). (Reprinted with permission from [143] by the American Physical Society.)
Download figure:
Standard image High-resolution image{kind=link}
The implementation of the method requires a primary HHG generation region that generates either an attosecond pulse train or more ideally an isolated attosecond pulse that is then delivered to the secondary HHG source containing the molecule in synchronization with an IR laser field. Sufficient intensity must be delivered from the primary source in the photon energy range of interest to excite a sufficient density of molecules. In practice this is no easy situation to achieve, but it will widen the scope of molecular HHG spectroscopy if it can be implemented.
6. Summary and conclusions
We briefly reviewed the topic of HHG from molecules and the salient background to using HHG for time-resolved and structurally sensitive studies of molecular dynamics via the methods of HHGS. The extension of HHG and HHGS to organic molecules and biomolecules was then discussed and the technical challenges and likely limitations of the technique were identified. An effort was made to review all pertinent experimental work so far reported in this area and to briefly survey the relevant literature on the theoretical treatment. The value of the use of longer wavelength drive fields (in particular in the range 1200–2000 nm) was made clear as these drive wavelengths have been used in the majority of investigations so far reported on HHG from organic molecules. Critical issues of sample handling to deliver a sufficient gas density (1014–1018 cm−3) for HHG whilst not masking the single molecule harmonic response were also discussed.
As a consequence of the efforts of a number of groups the progress in the field has been encouraging over the past decade. The ability to control and fully characterize the harmonic response from a range of small molecules, including small organic molecules, has led to the possibility to study the internal dynamics of molecular cations in a strong field. Nevertheless we caution that because of the presence of the strong field the method is not well suited to reveal the dynamics following sudden photo-ionization of molecules in the absence of a strong external field. As the HHG emission is tied to the sub-cycle strong field evolution and exquisitely sensitive to the quantum state of the molecule, both the initial state and the evolution of the state during the process, the signal carries unprecedentedly detailed information about the strong field interaction with the molecule. To extend this capability to a wider range of molecules the development of advanced sample handling methods, especially for solid compounds, is urgently required. Likewise extension of molecular alignment methods where possible is an important direction for future work. Probing molecules ionized to inner valence cation states may extend the sensitivity of technique, and may allow the interrogation of dynamics less affected by the strong field. This will demand higher intensity HHG sources to ensure a significant ionization fraction following the initial single photon ionization step. So far this method has only been successfully demonstrated in atoms and extension to molecules would be a timely development. Finally with advances in high power laser technology towards higher repetition rate high average power sources in the 1–4 μm spectral range we may be able to overcome the intrinsic limits so far encountered with the HHG signal.
Regardless of progress in HHGS of organic and bio-molecules the ability to use a wider variety of molecules will access potentially new opportunities in manipulating the high order non-linear optical response for the generation of soft-x-ray fields with tailored properties.
Acknowledgments
I must acknowledge discussion and advice from a large number of researchers both at Imperial College; John Tisch, Misha Ivanov, Vitali Averbukh, Amelle Zair, Dane Austin, Ricardo Torres, Thomas Siegel, Nathaniel Kajumba, Felicity McGrath, Allan Johnson, Zsolt Diveki, Emma Simpson, Peter Hawkins, David Wood, Tobias Witting and elsewhere Paul Corkum, David Villeneuve, Manfred Lein, Raffaele Velotta, Carlo Altucci, Ravi Bhardwaj, Olga Smirnova, Yann Mairesse, Nirit Dudovich, Hans-Jakob Worner, Caterina Vozzi, Sergei Patchkovskii, Chii-Dong Lin and Andrei Bandrauk. There are numerous others to whom I am also indebted for discussions and insights over the last decade or more. Funding for this work was provided by EPSRC (EP/I032517/1) and the ERC (ASTEX project 290467).