
Abstract
X-ray free-electron lasers (XFELs) and table-top sources of x-rays based upon high harmonic generation (HHG) have revolutionized the field of ultrafast x-ray atomic and molecular physics, largely due to an explosive growth in capabilities in the past decade. XFELs now provide unprecedented intensity (1020 W cm−2) of x-rays at wavelengths down to ∼1 Ångstrom, and HHG provides unprecedented time resolution (∼50 attoseconds) and a correspondingly large coherent bandwidth at longer wavelengths. For context, timescales can be referenced to the Bohr orbital period in hydrogen atom of 150 attoseconds and the hydrogen-molecule vibrational period of 8 femtoseconds; wavelength scales can be referenced to the chemically significant carbon K-edge at a photon energy of ∼280 eV (44 Ångstroms) and the bond length in methane of ∼1 Ångstrom. With these modern x-ray sources one now has the ability to focus on individual atoms, even when embedded in a complex molecule, and view electronic and nuclear motion on their intrinsic scales (attoseconds and Ångstroms). These sources have enabled coherent diffractive imaging, where one can image non-crystalline objects in three dimensions on ultrafast timescales, potentially with atomic resolution. The unprecedented intensity available with XFELs has opened new fields of multiphoton and nonlinear x-ray physics where behavior of matter under extreme conditions can be explored. The unprecedented time resolution and pulse synchronization provided by HHG sources has kindled fundamental investigations of time delays in photoionization, charge migration in molecules, and dynamics near conical intersections that are foundational to AMO physics and chemistry. This roadmap coincides with the year when three new XFEL facilities, operating at Ångstrom wavelengths, opened for users (European XFEL, Swiss-FEL and PAL-FEL in Korea) almost doubling the present worldwide number of XFELs, and documents the remarkable progress in HHG capabilities since its discovery roughly 30 years ago, showcasing experiments in AMO physics and other applications. Here we capture the perspectives of 17 leading groups and organize the contributions into four categories: ultrafast molecular dynamics, multidimensional x-ray spectroscopies; high-intensity x-ray phenomena; attosecond x-ray science.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Linda Young
Argonne National Laboratory and University of Chicago
The roadmap starts with topics generally familiar to the AMO community: femtochemistry viewed with an x-ray probe, multidimensional spectroscopy extended to the x-ray regime, and then winds toward the less commonly encountered areas of high-intensity x-ray phenomena and attosecond science. The first category describes how accelerator-based x-ray sources are used to probe ultrafast molecular dynamics: understanding ultra-intense x-ray pulse interactions with matter as a prelude to x-ray free-electron laser (XFEL) probes of femtosecond molecular dynamics (Ueda, section 2), optically induced molecular dynamics probed with XFELs (Gühr and Bucksbaum, section 2), and the novel use of long-pulse, monochromatic x-rays from a synchrotron source to induce and probe ultrafast inner-shell molecular processes (Simon, section 2). The second category describes multidimensional x-ray spectroscopies enabled by XFELs: a theoretical perspective where analogy to NMR, infrared and optical realizations is used to highlight the potential of x-rays to monitor the phase and dynamics of non-equilibrium valence wavepackets (Mukamel, section 3), a discussion of routes from an atomic x-ray laser to control of stimulated Raman processes with XFELs (Rohringer, section 3), and an account of the realization of coherent control and four-wave mixing in the XUV regime using only the fully coherent, seeded XFEL, FERMI (Prince and Masciovecchio, section 3). The third category deals with high-intensity x-ray phenomena created in XFELs in systems of increasing complexity: nonlinear multiphoton processes and polarization control in atoms (Meyer, section 4), charge and nuclear dynamics after inner-shell absorption in molecules (Rudenko and Rolles, section 4), imaging and scattering in nanoscale clusters (Bostedt, section 4), hard x-ray nonlinear optics (Fuchs and Reis, section 4), and, to describe quantitatively these phenomena, the theory of electronic structure under extreme conditions of x-ray irradiation (Santra, section 4).
The final category addresses table-top and attosecond x-ray science as enabled by high harmonic generation (HHG) sources. Technical frontiers in HHG include extension to x-ray wavelengths, enhancement of the single pulse energy and increase of the average power for short-wavelength radiation. We start with a general perspective on table-top-scale ultrafast coherent x-ray science that leads toward a future that can be 'smaller, cheaper and (ultra)faster' (Kapteyn and Murnane, section 5), followed by a description of a route to high-average-power soft x-ray ultrashort pulses via mid-infrared drive lasers (Ibrahim and Légaré, section 5), a discussion of attosecond and femtosecond XUV science (Vrakking, section 5), quantitative studies of photoionization time delays in atoms (Isinger, Kroon, Gisselbrecht and L'Huillier, section 5), evolution of attosecond spectroscopies from the XUV to the x-ray regime and from isolated molecules to the liquid phase (Wörner, section 5), and finally a description of soft x-ray transient absorption and the first multidimensional spectroscopies in the attosecond domain (Leone, section 5).
To guide the reader we have sketched the ultrafast photon source capabilities, dynamical phenomena and experimental techniques discussed in this roadmap. Source capabilities and phenomena that can be studied are inextricably linked. In figure 1, the performance of accelerator-based XFEL and HHG sources are shown. Both are labeled with photons/pulse/1% bandwidth for individual pulses. The complementarity of the two classes of sources is clear. Most XFELs are based upon self-amplified spontaneous emission (SASE) radiation, lack longitudinal coherence, struggle to obtain pulse lengths shorter than a few femtoseconds and struggle to synchronize with external sources. HHG sources, on the other hand, have exquisite temporal coherence and pulse duration, but are challenged to obtain large photon numbers per pulse, to extend their reach to short wavelengths and to obtain high average power. At the carbon K-edge XFELs produce many orders of magnitude more photons/pulse/1% bandwidth than HHG sources. Figure 2 maps the fundamental dynamical phenomena observed in isolated atoms and molecules, as well as the collective phenomena occurring in condensed phases, onto their respective time and length scales. Figure 3 maps the experimental techniques enabled versus photon number per pulse and photon energy. Overlaid are some research areas that are beneficiaries of these studies in atomic and molecular physics and the accompanying source and methodology development. The incredible progress of the past few years and the logical paths for source improvement augur a very exciting future for ultrafast x-ray science.

Figure 1. Phase space covered by the XFEL and HHG sources discussed in this roadmap. For XFELs, the pulse duration represents that of a single pulse, whereas for HHG, the range spans both single pulses and pulse trains spaced by the period of the driver laser. The numbers in each island indicate the number of photons/pulse/1% bandwidth. Research to extend the limits of all represented quantities, photon energy, time scale and photon number per pulse, is pursued for both XFEL and HHG sources. The emphasis for XFELs is to extend the time scale to the attosecond regime and photon energy above 20 keV; the emphasis for HHG is to extend the photon energy range to hard x-ray and photon number per pulse. For properties not represented by these basic quantities, XFELs seek enhanced temporal coherence and synchronization with external sources, and both sources seek increased average power and controlled polarization.
Download figure:
Standard image High-resolution image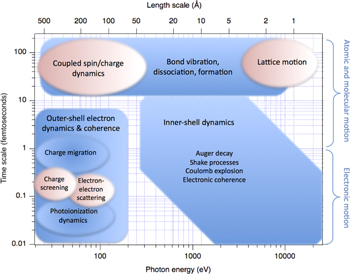
Figure 2. Fundamental atomic, molecular and electronic phenomena probed on ultrafast timescales (blue). Fundamental collective phenomena in the condensed phase probed on ultrafast timescales (pink).
Download figure:
Standard image High-resolution image
Figure 3. Experimental techniques used in ultrafast x-ray science mapped onto photon number per pulse and photon energy typically used (blue). The high-fluence regime enables nonlinear x-ray spectroscopies and single-shot imaging, potentially with atomistic resolution. Low fluences are employed to remain in the linear x-ray absorption regime to probe ultrafast transient processes. (Saturation fluence for a carbon atom at 290 eV, just above the K-edge, is ∼1010 photons/microns2.) Overlaid are research areas addressed with ultrafast x-ray methodologies that stem from understanding fundamental atomic and molecular physics processes (pink).
Download figure:
Standard image High-resolution imageAcknowledgment
The submitted manuscript has been created by UChicago Argonne, LLC, Operator of Argonne National Laboratory ("Argonne"). Argonne, a U.S. Department of Energy Office of Science laboratory, is operated under Contract No. DE-AC02-06CH11357. The U.S. Government retains for itself, and others acting on its behalf, a paid-up nonexclusive, irrevocable worldwide license in said article to reproduce, prepare derivative works, distribute copies to the public, and perform publicly and display publicly, by or on behalf of the Government. The Department of Energy will provide public access to these results of federally sponsored research in accordance with the DOE Public Access Plan. http://energy.gov/downloads/doe-public-access-plan
2. Ultrafast molecular dynamics
2.1. Probing ultrafast structural and electronic dynamics with XFELs
Kiyoshi Ueda
Tohoku University
Status
Currently, two hard XFELs, LCLS in USA and SACLA in Japan, are in operation for users, and a few more will open for operation this year. The ultrashort and intense x-ray pulses of these XFELs are revolutionizing the field of ultrafast imaging, allowing determination of so far unknown structures of transient species, proteins and any matter undergoing reactions. Also, since XFEL pulses are giving access to a new regime of x-ray intensities, they are opening up new avenues in studying the interaction between intense x-rays and various forms of matter. Understanding ultrafast reactions induced by XFEL pulses is of fundamental interest, as well as of crucial importance for structural determination with XFELs.
AMO science with XFELs may also be classified into the two groups described above. The first group is the investigation of the interaction of the intense XFEL pulse with atoms, molecules and clusters. In the early experiments at XFEL facilities, the target samples were just irradiated by a single XFEL pulse and the products (ions, electrons and fluorescent photons) were detected. Such experiments revealed new phenomena whenever the photon energy of the XFEL pulse and/or its pulse energy entered into a new regime. The findings may be summarized as follows. X-ray absorption initially creates an inner-shell hole in a specific atomic site. Then electronic relaxations, or Auger cascades, follow. An XFEL pulse is so intense that it can cause multiple overlapping cycles of deep inner-shell photoemission and Auger cascades. Competitions between sequential photoemission and Auger cascades have been studied extensively using rare-gas atoms as a target [1–3]. To study the coupled motion of electrons and ions induced by an intense XFEL pulse, on the other hand, a single molecule composed of a small number of atoms is an ideal target since various levels of theoretical modeling and experimental methods are available or can be developed. Indeed, to study competition between Auger cascades, charge redistribution and Coulomb explosions, a series of studies have been carried out for molecules that contain up to twelve atoms with one heavy atom as an x-ray absorber [4–6]. To study XFEL-induced reactions beyond these molecular model systems, atomic clusters are ideal objects because their size can be varied in a controlled way from a single atom to a bulk-like macroscopic object. When an atomic cluster is exposed to an intense x-ray pulse, many free electrons are created by sequential inner-shell photoionization of many individual atoms followed by Auger cascades. Because a number of electrons escape from the cluster, the cluster becomes highly charged and starts trapping electrons that are emitted from the individual atoms. A nanoplasma is thus formed. These nanoplasma formation processes have been studied by a combination of experiments and simulations [7, 8]. The nanoplasma formation is expected to be a general phenomenon, as it is expected to occur whenever nanometer-size particles are irradiated by an intense XFEL pulse. In connection to structural studies using XFELs, the above described investigations for atoms, molecules and clusters may be regarded as a fundamental study of radiation damage at the atomic level (see figure 4).
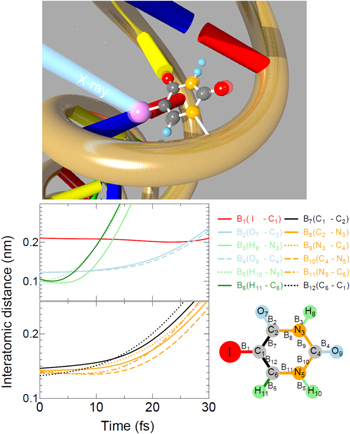
Figure 4. Upper panel, a schematic view of the radio-sensitizing effect of 5-iodouracil (5IU), a nucleobase analog of biological relevance. The work described in [6] illustrates how the molecule breaks apart and what ionic fragments are formed via the breakage of the molecular edifice shortly after the inner-shell ionization, shedding light on the role of energetic ions in the initiation of damaging reactions. Lower panel, time evolution of interatomic distances in 5IU after XFEL irradiation, obtained by model simulations; upper left, interatomic distances of I–C (red line), O–C (sky-blue lines), H–C (dark-green line) and H–N (light-green lines) pairs; lower left, interatomic distances of C–C (black lines) and C–N (orange lines) pairs, illustrating that most atoms other than hydrogen remain intact during the XFEL pulse duration of 10 fs. Reprinted figure with permission from [6], Copyright 2016 by the American Physical Society.
Download figure:
Standard image High-resolution imageNowadays, a combination of optical laser and XFEL pulses is available at XFEL facilities. Consequently, one can probe the XFEL-induced reactions described in the previous paragraph, employing an optical laser pulse as a probe. Another way to probe XFEL-induced reactions is to use XFEL pulses for both pump and probe. A split-and-delay assembly was introduced to XFEL facilities for such pump–probe experiments. Recently, XFEL facilities started to provide double pulses at two different x-ray photon energies with variable time delay. This operation mode has some advantages over the use of the split-delay assembly since, e.g., one can set the two photon energies above and below a certain edge of a certain atom. Such time-resolved studies on XFEL-induced reactions have just started at XFEL facilities [9] and are in progress.
The availability of optical lasers at XFEL facilities opened a route to the second group of AMO science at XFELs, i.e. investigations of ultrafast reactions induced by light, or an optical laser, employing an XFEL pulse as a probe. For such studies on gas-phase samples, ions or electrons are usually detected because of small cross sections of x-ray scattering. X-ray diffraction experiments of isolated molecules in photoreaction have, however, become feasible in the last few years [10], demonstrating that watching atomic motion in an isolated molecule is no longer a dream. X-ray imaging experiments for rare-gas clusters have also been combined with optical laser-pump techniques and have just started to probe the structural evolution of clusters heated by an optical laser [11].
Current and future challenges
As noted above, most experiments with XFELs are nowadays based on a combination of optical laser and XFEL pulses or two XFEL pulses. Thanks to the development of an arrival timing monitor for XFEL and optical laser pulses, which allows us to correct temporal jitters between the optical laser and XFEL pulses, one can investigate light-induced ultrafast reactions in real time, at a time resolution of tens of femtoseconds, which is comparable to that for the XFEL-pump and XFEL-probe experiments. To fully explore electronic dynamics or interplay between the electronic and structural dynamics, however, it is desirable to have better time resolution. Various kinds of electronic decays (Auger and interatomic Coulombic decays as well as laser-enabled Auger decay) may occur on a time scale of femtoseconds to tens of femtoseconds. Nuclear dynamics, especially involving the motion of hydrogen atoms, may also take place on a similar time scale and the system may undergo reactions passing through conical intersections. All these processes contribute to the charge redistribution in the molecule or cluster. The electronic wavepacket may also be created when more than one electronic state is populated via ultrafast electronic decay or irradiation by an ultrashort optical laser pulse. Then the wavepacket motion may be even faster. To see all of these dynamics, a pump–probe scheme with time resolution of a few femtoseconds or less than a femtosecond would be desirable.
To fully extract information about ultrafast structural and electronic dynamics with the ultimate time resolution discussed above, the signal detection scheme also needs to be improved. For molecules, if the number of events are ideally less than one per single XFEL pulse, then one can record electrons and ions in coincidence, in a momentum-resolved manner. Such kinematically complete measurements should be a challenge for the aforementioned time-resolved study. For clusters, single-shot imaging combined with ion and electron spectroscopy at ultimate time resolution should be a challenge.
Advances in science and technology to meet challenges
The XFEL facilities can provide x-ray pulses with durations down to a few femtoseconds. This is also the case for double-pulse operations. Employing these ultrashort double pulses one can achieve a time resolution of a few femtoseconds. Producing pulses of duration below a femtosecond is also technically feasible. So far, the time resolution achieved for the combination of optical laser and XFEL pulses is often limited by the duration of the optical laser, say a few tens of femtoseconds. In principle, the technology of producing an ultrashort pulse down to a few femtoseconds (a few optical cycles) is available and thus the time resolution in a few femtoseconds should be within reach. To record ions together with electrons in coincidence in a momentum-resolved manner is also technically feasible, as has been demonstrated in experiments with synchrotron sources and high-repetition optical lasers. The reason why such measurements were limited for the XFEL experiments (see, e.g. [4–6] for multiple ion coincidence) is their low repetition rates. This situation will dramatically improve when European XFEL, the first high repetition rate XFEL, and LCLS-II will be in operation for users. X-ray detectors and sample injectors that can accommodate in these high repetition rate XFELs will also become available in time.
Concluding remarks
To probe electrons and atoms in action is no more a dream thanks to recent advances in technology related to time-resolved measurements with XFELs and detection techniques. Solving interplay between electronic and atomic motions, which governs photochemistry, is now within reach.
Acknowledgments
2.2. Methods for probing molecular dynamics with XFELs
Markus Gühr1,2 and Philip H Bucksbaum2
1Potsdam University
2Stanford PULSE Institute
Status
Photoexcited molecular dynamics is at the heart of many processes in nature, from light harvesting and atmospheric chemistry to photoprotection of living organisms. Light interacts with molecular electrons, which couple to nuclei to initiate ultrafast and concerted motion of both the electrons and the atomic geometry. In only femtoseconds to picoseconds the subsequent steps for light-energy conversion are determined by this motion. Ultrashort hard and soft x-ray pulses provide valuable insights to understand the connections between the initial motion and the ultimate chemical changes in excited molecules [12]. Time-resolved hard x-ray scattering experiments have revealed light-induced changes in the geometry, while soft ultrafast x-ray spectroscopy is most sensitive to the electronic degrees of freedom. Large inner-shell binding energy differences make x-ray spectra sensitive to both the type and location of atoms in a molecule. Complementary information from ultrafast scattering and spectroscopy can elucidate fundamental processes such as charge transfer, light harvesting and photo-induced molecular damage. Many well-developed x-ray spectroscopy and scattering techniques have been extended to measurements in the femtosecond time domain, and the first decade of ultrafast molecular experiments in XFELs has had several science successes, some of which are highlighted here.
Early science success at XFELs
Fundamental charge transfer has been studied using pulsed x-rays to selectively excite the iodine in ionized methyl iodide while monitoring the fragment charge state [13]. Excited state isomerization of acetylene has been investigated with coincidence methods [14, 15]. More complex nonadiabatic dynamics were also investigated with larger molecules. A longstanding controversy over the detailed photoprotection mechanisms of nucleobases was resolved through a combination of femtosecond Auger spectroscopy and time-resolved x-ray absorption [16]. Figure 5 highlights the extreme sensitivity of these methods to the non-radiative ππ*–nπ* relaxation mechanism [17]. Due to the high lone-pair localization, a strong oxygen 1s–n absorption feature results in ππ*–nπ* relaxation. In liquids, nonadiabatic processes in metallo-organic complexes have been investigated. In the case of Fe(CO)5, for example, resonant inelastic x-ray scattering (RIXS) [18], revealed the singlet spin nature of the photoproduct Fe(CO)4.
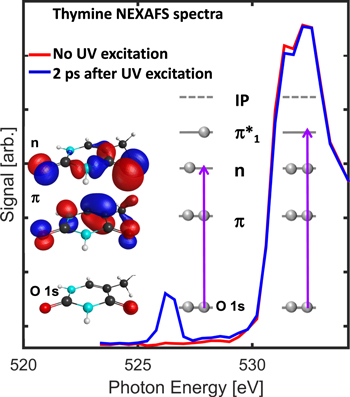
Figure 5. Transient near-edge absorption spectrum of thymine. Excitation of the molecule with ultraviolet light leads to strong oxygen 1s–n absorption due to the half-full oxygen lone-pair orbital n. Reprinted by permission from Macmillan Publishers Ltd: Nature Communications [17], Copyright 2017.
Download figure:
Standard image High-resolution imageHard x-ray scattering from optically excited or aligned molecules gives insight into the transient geometry. Rovibronic wavepackets in iodine, metal compounds in liquids and organic ring openings have all been observed with unprecedented time resolution [10, 19, 20]. Simultaneously, ultrafast electron diffraction has also made considerable progress in gas-phase scattering with atomic resolution [21].
Source parameters
The success of the first XFEL experiments relied on microjoules to millijoules of pulse energy over wavelengths from 0.1 to 100 nm. Pulse lengths can be as short as a few femtoseconds. Pulse timing jitter for laser—x-ray pump–probe experiments can be well under 50 fs. Focused intensities up to 1020 W cm−2 or more permit nonlinear studies, and the high peak fluence enables single-shot diffraction based on the principle of 'diffract before destroy.' The repetition rate in copper-waveguide FELs (LCLS, SACLA, FERMI) can exceed 100 Hz, while superconducting accelerators (FLASH) can provide 103 to 104 higher repetition rates. New sources will soon be available (European XFEL, Swiss-FEL, PAL-FEL, LCLS-II), expanding the capacity for femtosecond x-ray science.
Current and future challenges
Ultrafast photoabsorption in molecules creates an electronic wavepacket. Probing its dynamics with x-rays could reveal both the location of the excitation within the molecule and its local chemical environment. Nonlinear x-ray optics protocols and their early success are described in section 3. Ultimately, multiple attosecond pulses are required. Accelerator-based schemes have already demonstrated x-ray pulse pairs with tunable wavelengths and delays. The full scientific potential of these schemes will be explored with sub-femtosecond x-ray pulses.
Charged particle coincidence detection in combination with light excitation is another emerging method highlighted in sections 2.1 and 4.2. Angle-resolved photoelectron spectroscopy suffers poor spectral stability inherent to self-amplified stimulated emission sources. Spectrally stable seeded sources at high photon energies are desirable. Seeding has been implemented the FERMI VUV FEL (see section 2.3).
An increased repetition rate, reduced pulse duration, and spectral stability would improve most existing ultrafast x-ray probe schemes. In FELs, a higher repetition rate accompanies increased average x-ray flux since the lasing dynamics generally requires that the energy per pulse be kept approximately at the μJ to mJ level. Higher average power will benefit x-ray absorption experiments as well as x-ray emission and facilitate emission and RIXS in dilute liquid samples as well as in the gas phase.
Shorter pulses in conjunction with high pulse energy at wavelengths from the carbon edge near 300 eV to the hard x-ray range above 4 keV will broaden the applicability of ultrafast x-ray methods not only for electronic wavepackets but also to study fast nuclear dynamics. This development must be accompanied by advanced optical x-ray cross-correlation methods to utilize the time resolution available in ultrafast pulses. Improved spectral stability from self-seeding or even high gain harmonic generation will improve the quality of data in absorption and RIXS experiments. Those have been hampered by the attenuation of x-ray monochromators in SASE FELs. With seeding, those experiments could utilize the full-spectrum 'pink' beam, or have much higher throughput when using a monochromator. This in turn would allow the use of more dilute samples or enable more systematic studies, which are beyond the scope of current beamtimes.
Raising the hard x-ray cut-off energy of x-ray FELs will increase the spatial resolution of scattering measurements, opening a path to measure both motion and transient geometries in excited states such as molecules approaching conical intersections.
An important new challenge is in the area of calculation, modeling, and simulation, and how they can help us understand delay-dependent experimental observables such as charged-particle correlations or x-ray scattering patterns. Core-ionized states must be incorporated in electronic-structure codes that work together with valence excited molecules. The field of nonlinear x-ray optics is a fertile ground for theory (sections 3.1 and 4.5). Our conceptual understanding of electron wavepackets is just beginning, and we have much to learn about the manifestation of electron correlation in the time domain as well as how to interpret experimental observables and display the resulting molecular movies in attosecond experiments.
An equally important challenge for the new field of ultrafast x-ray science in FELs is how to broaden participation in the molecular physics and chemistry communities. Currently, experiments at FEL facilities require a high level of sophistication with experienced teams. The knowledge and manpower needs are high compared to synchrotrons, which can discourage investigations by small groups interested in specialized or non-traditional scientific questions. This barrier to use must shrink in the future. Furthermore, the new generation of superconducting high average power x-ray SASE FELs will add the challenges of massive data rates because each laser shot must be stored for later sorting, binning, and further analysis.
If these social and technical barriers can be lowered, higher stability FELs could attract a large community to use more fully the potential of these revolutionary machines in all aspects of science.
Advances in science and technology to meet challenges
FEL sources are improving as the community gains experience with their characteristics. In addition, new sources are coming on line with higher photon energies, higher repetition rates, and higher stability of many crucial variables. The PAL-FEL and Swiss-FEL will begin operating within the next year. Both employ a Cu LINAC and thus are limited to low repetition rates. The European XFEL and LCLS-II use superconducting accelerators, and will be in full operation within the next few years. At the European XFEL, the full wavelength range up to 25 keV will be delivered at the full repetition rate of 2700 pulses spaced by 220 ns within 10 macrobunches per second. At LCLS-II, the high repetition rate accelerator will be limited to 5 keV in the fundamental; the highest photon energy will be derived from the warm LINAC at 100 Hz repetition rate.
The stability should improve with repetition rate. If timing, spectrum and pulse energy can be controlled better, single-shot data collection may be unnecessary for many experiments. Many of the challenges identified above could be resolved in next-generation machines.
For example, several methods are under development to produce sub-femtosecond x-ray pulses. The XLEAP project at SLAC is based on laser-manipulation of the electron bunch phase space to produce isolated sub-femtosecond x-rays precisely timed to an external laser. XLEAP will begin commissioning this year and, if successful, might become a standard operation at LCLS-II [22].
In addition, coincidence techniques will be useful over an enlarged spectral range at the European XFEL and LCLS-II. Emission and RIXS experiments will benefit from the flux increase due to the higher repetition rates, easing constraints on the sample concentration and systematic studies.
Concluding remarks
The new field of femtosecond x-ray probes of molecules is beginning to have an impact in molecular physics, chemistry, and biology. New sources that will be available to the research community in the next decade will expand access to these unique probes and allow new transformative methods.
Acknowledgments
MG is funded by the Volkswagen foundation via a Lichtenberg professorship. PHB is supported through the Stanford PULSE Institute, SLAC National Accelerator Laboratory by the US Department of Energy, Office of Basic Energy Sciences, Atomic, Molecular, and Optical Science Program.
2.3. Ultrafast dynamics with tender x-rays
Marc Simon
CNRS and Pierre and Marie Curie University
Status
There is a revival of studies on the processes occurring after absorption of a tender x-ray photon (2–10 keV) by isolated atoms or molecules. Historically, this energy domain has been intensively studied before the soft x-ray domain, mainly because it was technically easier to deal with x-ray tubes than discharge lamps. Later on, the field was declining in terms of number of scientists in favor of the soft x-ray domain, which was taking advantage of technical improvements offered by synchrotron radiation facilities. Using synchrotron radiation, there are, for the moment, only five groups in this research field around the world with their own experimental setup: one group in Ljubljana (Slovenia), one group in Argonne (US), one group in RIKEN-SPring 8 (Japan), one group in PETRA III (Germany) and our group in Paris. This research field on single x-ray photon absorption spectroscopy is also boosted by the huge interest in the community on the results of multiphoton absorption in this x-ray domain obtained at different XFELs: LCLS (US), SACLA (Japan) and soon at European XFEL in Hamburg (Germany). Recent technical developments in high-brilliance third-generation synchrotron radiation facilities, electron or x-ray spectrometers and COLTRIMS (cold target recoil ion momentum spectroscopy) now allow investigations with tremendously higher performance than before. This field is attracting more and more scientists and leading to interesting discoveries. A review of our recent results can be found in [23].
One of the main interests of the tender x-ray domain comes from the short core-hole lifetimes (one femtosecond or less) used as an internal clock for the studies of ultrafast processes. After resonant inner-shell excitation, when the intermediate state is dissociative, ultrafast nuclear motion occurring in a time shorter than the core-hole lifetime has been observed in different chlorinated compounds. Because of the cascade Auger effect, the ultrafast nuclear motion occurring in the first intermediate state is amplified through the different dissociative intermediate states and leads to the ultrafast dissociation as illustrated in figure 6: the Auger electron is emitted by the fragment and not by the molecule [24, 25]. Because of the short core-hole lifetimes and large kinetic energies of the emitted Auger electrons, just above threshold large post collision interactions (Coulomb interactions between the photoelectron, Auger and the ion) as well as recapture of the photoelectron have been observed [23].
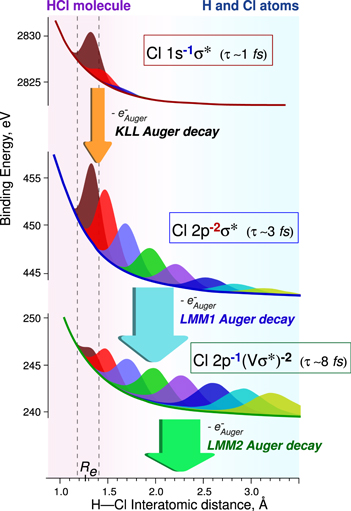
Figure 6. Potential energy curves for different steps of the KLL Auger cascade following Cl 1s → σ* excitation. Wave-function distributions are shown in different colors for up to 8 fs after x-ray photon absorption with 1 fs increments.
Download figure:
Standard image High-resolution imageBelow the ionization threshold, large lifetime broadening induces overlap in energy of the different discrete resonances located below the inner-shell ionization threshold, corresponding to the promotion of the inner-shell electron to an empty molecular orbital. Photon absorption leads to the coherent resonant excitation of different electronic states decaying to the same final states, giving rise to pronounced electronic lifetime interferences, as recently studied [26].
Because Auger cascades occur after deep inner-shell ionization, multiply charged ions are produced that then explode by Coulomb repulsion. The vector momenta of the ions and the photoelectrons have been recorded in a multi-coincidence COLTRIMS setup revealing interesting features such as fragmentation dependence of the localization/delocalization of a deep inner-shell hole in a symmetric molecule [27].
At high photon energy, the large kinetic energy of the escaping photoelectron induces large translational recoil observed via the atomic Doppler Auger effect [28], illustrated in figure 7 as well as vibrational recoil, which we have started to study in collaboration with Turku University (Finland).

Figure 7. Schematic diagram of the atomic Doppler Auger physical phenomenon. Reprinted by permission from Macmillan Publishers Ltd: Nature Communications [28], Copyright 2014.
Download figure:
Standard image High-resolution imageDouble core hole (DCH) states have recently received growing interest, mainly because larger chemical shifts than with single core hole states are expected. We have shown that conventional electron spectroscopy in the tender x-ray domain can be used to study DCH states: one core electron is promoted into an empty orbital and another core electron is simultaneously ejected into the continuum and detected [29, 30]. We can then have access to the different states converging toward DCH states and determine their core-hole lifetimes. These studies on molecules have started in collaboration with the University of Gothenburg (Sweden).
On the CS2 molecule, a strong evolution of the RIXS spectral profile with the excitation energy much detuned below the lowest discrete resonance has been observed. With theoretical support, we understood this evolution as the onset of electron dynamics triggered by a coherent excitation of multiple electronic states [31].
Current and future challenges
One of the challenges is to control ultrafast fragmentation in the sub-femtosecond time scale; the effect of the nuclear wavepacket propagation on the first excited states in a time shorter than one femtosecond plays a crucial role in the ultrafast fragmentation observed. Measurements on van der Waals clusters should exhibit nice surprises. Interatomic Coulomb decays, charge transfer dynamics, the solvation effect, etc, will probably be an important field in the future. X-ray emission or hard x-ray photoelectron spectroscopy (HAXPES) from organometallic molecules, isolated or in solution, should soon be a field of interest.
Access to higher photon energies will allow reaching deeper inner shells with shorter core-hole lifetimes. The core-hole clock method applied to tens of attoseconds lifetimes should allow us to reach dynamics occurring in a time shorter than 10 attoseconds. Another alternative would be to use large photon detunings; electron dynamics in the tens of attosecond time scale will become measurable, as already observed in the hundred of attoseconds time scale [31]. Compton scattering should be possible to study with hard x-rays. Using a high flux photon beamline, it has been already possible to elucidate the role played by Compton scattering in the He++ formation in the 8–28 keV photon energy range [32, 33]. Non-dipole effects are becoming strong in the hard x-ray regime. Angular distribution measurements of the photoelectrons are mandatory to extract non-dipole parameters [34].
Photon–ion and Auger electron–ion coincidences are, for the moment, impossible to measure in this energy domain because the luminosity of the x-ray or electron spectrometers are not high enough. These kinds of coincidences would give important information such as the fragmentation dependence on the electronic states and the possibility to record fixed-in-space photon x-ray emission. Photon–ion coincidences could be decisive for the understanding of the radiative Auger process; an x-ray photon and a slow Auger electron are emitted simultaneously and share the excess of energy.
Probing the non-radiative electronic relaxation of doubly excited states as has been performed for radiative relaxation [35] would be very interesting.
Advances in science and technology to meet challenges
The future of research in this field very much depends on optical issues, synchrotron facilities upgrades and technical improvements of end-stations. Sophisticated high-resolution monochromators on a large photon energy range should allow significant improvement of the measurement's resolution. The low emittance of modern synchrotron radiation facilities using multi-bend-achromat magnets is very promising; a large increase of the photon flux even in the hard x-ray regime up to 60 keV or more could be achieved. An increase in this limit is becoming more and more desirable for new beamlines extending toward harder x-rays. HAXPES spectrometers are for the moment limited to the detection of maximum 15 keV of electron kinetic energies. An increase in this limit will be needed for HAXPES measurements at high photon energy. Large increases in the collection efficiencies of electron and x-ray spectrometers are needed to perform photon–ion and Auger electron–ion coincidences. Such crucial developments have already started for x-ray spectrometers.
Concluding remarks
This research domain should continue to rapidly grow in the future. The recent availability of intense XFELs in the hard x-ray domain is additionally stimulating any type of spectroscopies in this energy domain in order to understand and to predict interesting processes.
Acknowledgments
Maria Novella Piancastelli, Renaud Guillemin, Loïc Journel, Oksana Travnikova, Tatiana Marchenko, Iyas Ismail, Gildas Goldsztejn, Ralph Püttner, Denis Céolin and Jean-Pascal Rueff are warmly acknowledged.
3. Multidimensional x-ray spectroscopies
3.1. Multidimensional nonlinear x-ray spectroscopy of molecules
Shaul Mukamel
University of California, Irvine
Status
Multidimensional spectroscopic techniques, first developed in nuclear magnetic resonance [36], and gradually extended to higher frequency (infrared, and optical) regimes [37, 38], probe the electronic structure and nuclear dynamics of molecules through their response to sequences of short pulses with variable, carefully timed delays. By using femtosecond to attosecond x-ray pulses resonant with core transitions in selected atoms, these techniques can be extended to probing core electronic states and couplings, the real-time tracking of impulsively created valence electronic wavepackets and electronic coherences. Nonlinear experiments that combine x-ray and optical beams have been reported in atoms and crystals, and all x-ray wave mixing measurements in molecules are on the horizon [39].
Thanks to their broad bandwidth (10 eV for a 100 attosecond pulse), x-ray pulses can create coherent superpositions of a large number of electronic and vibrational states that are localized at a target atom, and monitor their evolution on a very short time scale. 2D x-ray spectroscopy may be used to investigate the interactions between core excitations [40]. Since core energy levels are highly element specific, this technique can provide structural and dynamical information with high spatial, temporal, and spectral resolutions not feasible with optical pulses.
Apart from the direct study of core excitations, core resonant excitations offer a fast and versatile way to trigger valence electronic excitations impulsively at selected positions and times and monitor their subsequent dynamics via stimulated Raman processes. Notable advantages of Raman signals are that they do not require phase control of the pulses and their ability to probe valence excitations that are more chemically relevant than core excitations. Sequences of coherent broadband x-ray pulses thus offer new windows into the dynamics of nuclei and electrons in molecules. Resonant core transitions provide a short time window, limited by the core lifetime. Valence excitations prepared by a Raman process provide a much longer observation window.
Ultrafast processes such as intersystem crossing and radiationless decay monitoring are ubiquitous in complex molecules but have evaded complete understanding due to the extreme temporal and spectral parameter regimes necessary for their observation. Tunable, intense attosecond x-ray pulses can probe such ultrafast processes and reveal the nature of elementary photophysical and photochemical events [41]. Multidimensional x-ray techniques may be used to study charge and energy transfer in photosynthetic complexes, donor–acceptor aggregates and the coherent control of long-range electron transfer.
The nonlinear response of valence electrons to multiple cores excited at variable delays provides a unique window into electron structure and correlations. The complex nature of excited-state dynamics leads to characteristic patterns in nonlinear 2D correlation plots, which provide signatures of strongly coupled electron and nuclear dynamics. γ-ray pulses could open up time domain nuclear spectroscopy.
Current and future challenges
Time-resolved, off-resonant scattering (diffraction) provides movie-like snapshots of the charge density. By tuning the x-ray beam to be resonant with core excitations, these experiments reveal a qualitatively higher level of many-body information related to the coupling of various core excitations connected by delocalized valence states. Diffraction can be extended to multiple dimensions by photon coincidence measurements obtained by subjecting the molecule to sequences of pulses [42]. These require single-molecule photon counting and provide information on charge fluctuations through multi-point correlation functions of the charge density. Note an important qualitative difference: coherent multidimensional spectroscopy involves several perturbations followed by a single measurement, whereas multidimensional diffraction consists of a series of measurements. Off-resonant x-ray pulses interact with matter through the charge density while resonant x-ray pulses interact with the current density; hybrid combinations of off and on resonance multipulse experiments are possible.
The breakdown of the adiabatic (Born Oppenheimer) approximation in strongly coupled electron-nuclear dynamics may be monitored through electronic coherences generated at conical intersections (CoIns). Nonlinear x-ray spectroscopies can track the ultrafast passage through conical intersections and reveal the time evolution and couplings of electronic coherences. At conical intersections, the energy splitting of the electronic states involved in the coherence can be read from the Raman shift and the coherent oscillation period reveals this time-dependent level splitting averaged over the nuclear wavepacket [43]. These offer a novel window into the electronic and nuclear dynamics as well as the coupling between various core and valence level excitations. This technique could further detect direct signatures of geometric (Berry) phase effects in molecular dynamics near CoIns.
Diffraction from a molecule prepared in a coherent superposition state can arise from diagonal as well as off-diagonal elements of the charge-density operator. The former, known as charge densities, are obtained in conventional diffraction and used to probe the structure of molecules in a given electronic state. The latter, known as transition charge densities, are associated with electronic and vibrational coherence and carry additional dynamical information about electronic excitations and their delocalization [44]. They are naturally created during the passage through conical intersections. Nonlinear techniques such as sum frequency generation and time-resolved diffraction can be used for imaging the transition charge densities.
Advances in science and technology to meet challenges
X-ray pulses produced via high-harmonic generation are easier to create and of much higher quality than XFEL pulses, but they are of significantly lower intensity, making it harder to use them in higher-order nonlinear spectroscopies. They are further limited to XUV and soft x-ray <1 keV. Improving existing sources to combine the FEL photon flux with the high quality of HHG will be an important development. Future investigations could also include incoherent detection of the coherent signals induced by pulse sequences through fluorescence, currents, photoelectron or Auger electron emissions.
A complete control over the phase and amplitude of intense x-ray pulses would allow the extension of sophisticated shaping techniques used in optical and IR spectroscopy [45] to the x-ray regime. Coherent control techniques should allow us to conveniently create multiple pulse sequences out of a single pulse. A combination of broadband (femtosecond) and narrowband (picosecond) pulses has proven to provide an optimal temporal and spectral resolution in Raman spectroscopy in the optical regime. The same idea can be extended by combining attosecond (HHG) and femtosecond (FEL) x-ray pulses. Many experiments use the HHG field itself as the signal [41]. Such signals are robust but harder to interpret than, e.g., a pump–probe, since the spectroscopic information is convoluted with the HHG multistep mechanism. Selecting a few HHG harmonics for spectroscopy will be a major step forward. High quality circular and linear polarized beams should provide novel probes of time-resolved chirality [46, 47].
The design and interpretation of nonlinear spectroscopy of core and valence excitations pose enormous theoretical and simulation challenges. Handling the nonlinear response of electrons and nuclei to attosecond broadband x-ray radiation calls for the development of new computational tools. These signals should provide sensitive direct tests of many-body theories of electron correlations.
The short correlation time of noisy light sources has been exploited in the optical regime to perform femtosecond experiments with stochastic nanosecond light, see chapter 10 in [37]. Similar strategies could be applied to the XFEL. Conventional nonlinear spectroscopy uses classical light to detect matter properties through the variation of its response with frequencies or time delays. Quantum light opens up new avenues for spectroscopy by utilizing parameters of the quantum state of light as novel control knobs and through the variation of photon statistics by coupling to quantum light. Quantum optics techniques involving interferometric detection can be imported to the x-ray regime. Ultrafast spectroscopy signals generated by quantum light, focusing on applications involving entangled photons with nonclassical bandwidth properties will be an interesting new frontier of x-ray science [48]. Parametric down conversion (PDC), which generates entangled photon pairs in the hard x-ray-regime [49, 50], has been demonstrated recently with XFELs [51]. One notable advantage for spectroscopy is that entangled photon pairs are not subjected to the classical Fourier limitations on the joint temporal and spectral resolution. Ghost imaging [52] and spectroscopy, which generate a signal by one member of an entangled pair and detect it in coincidence with its twin that does not interact with matter, could improve the detection sensitivity as well as the temporal and spectral resolutions. Photon statistics [53] and coincidence detection such as Hanbury-Brown–Twiss measurements detect higher-order light intensity correlations. Employing them in diffraction to reveal additional matter information should be an exciting development.
Concluding remarks
Historically, coherent nonlinear spectroscopy have gradually evolved towards higher frequency regimes as phase control of the relevant light sources became feasible. The key nonlinear optical spectroscopy experiments were based on earlier ideas developed and implemented in radio waves (NMR). Similarly, x-ray techniques can be designed by learning lessons from the visible and infrared regimes. Historically, stimulated Raman has been the most robust nonlinear optical technique. By extending it to core transitions, it could impulsively excite and detect valence excitations at selected locations and times. Optical pulses can excite vibrations impulsively. Broadband x-ray pulses can do the same to valence electronic excitations. The rich variety and sophistication of vibrational spectroscopies could be thus imported to the x-ray regime. A resonant Raman process can create valence excitations localized in the vicinity of a selected atom and probe them near another selected atom at variable delays. These signals offer new powerful windows into many-body effects in electron and nuclear dynamics.
Acknowledgment
The support of the Chemical Sciences, Geosciences, and Biosciences division, Office of Basic Energy Sciences, Office of Science, US Department of Energy through Award No. DE-FG02-04ER15571 and the National Science Foundation award CHE-1663822 is gratefully acknowledged.
3.2. Nonlinear x-ray pump–probe spectroscopy with XFELs
Nina Rohringer
CFEL/DESY and University of Hamburg
Status
Nonlinear x-ray pump–probe spectroscopy has been identified as a potentially powerful spectroscopic x-ray technique. Even before the advent of XFELs, Mukamel and colleagues discussed several different x-ray pump–probe schemes [54, 55] based on sequences of attosecond x-ray pulses in order to unravel coherent electron dynamics of processes such as long-distance coherent charge and energy transfer [56], or the passage of vibronic wavepackets through conical intersections [57]. Similar to optical nonlinear spectroscopy, by carefully choosing the time order, frequencies and propagation direction of a series of x-ray pulses, different x-ray nonlinear response diagrams contributing to higher-order susceptibility can be addressed that probe electron dynamics with high temporal and spatial resolution, and give novel insight into dynamical properties of electronic coherence. The basic building block of these nonlinear x-ray techniques is stimulated resonant Raman scattering (see figure 8). Despite the ultrahigh intensities necessary for realizing stimulated x-ray emission (1017–1018 W cm−2 in the soft and 1019–1020 W cm−2 in the hard x-ray range), amplified spontaneous x-ray emission [58] and stimulated resonant inelastic x-ray Raman scattering (sRIXS) [59] have been demonstrated in atomic Ne in the soft x-ray spectral range. Moreover, stimulated emission was demonstrated in the hard x-ray range in Cu [60] as well as Mn salts in solution [61], where chemical shifts of the K-α emission were demonstrated in highly amplified x-ray emission. More importantly, similarly to the 'diffract before destroy' approach of femtosecond serial crystallography, the chemical information of the oxidation state of the different Mn compounds was demonstrated to be preserved. These studies, demonstrating coherent amplification and Raman gains of several orders of magnitude, are encouraging to realize nonlinear optical x-ray pump–probe spectroscopy of optically dense samples in the liquid or solid phase in future XFEL sources.
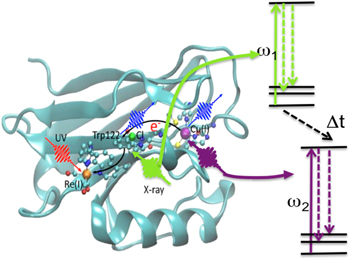
Figure 8. Schematic diagram of UV x-ray nonlinear pump–probe spectroscopy to observe long-range charge transport in biologically relevant samples. A UV pulse initiates charge migration on a specific site in the molecule. Two attosecond x-ray pulses that are tuned to specific inner-shell absorption resonances of metallic centers in the molecule serve as a probe by inducing two stimulated electronic Raman scattering events at two different sites in the molecule. The nonlinear x-ray spectroscopic signal is obtained by varying the delay between the two x-ray probe pulses and monitoring the Raman gain. Representation of Re-modified Azurin Molecule with courtesy to S Mukamel and Yu Zhang. Adapted with permission from [56]. Copyright 2014 American Chemical Society.
Download figure:
Standard image High-resolution imageCurrent and future challenges
The current feasibility of nonlinear x-ray Raman spectroscopy is limited by the attainable peak brilliance, limited temporal coherence, control of pulse duration, and realizable experimental geometries in present-day XFEL sources. Current proof-of-principle experiments of sRIXS, the building block of nonlinear optical spectroscopy, focus on optically thick samples that are specifically chosen to maximize the amplification gain. As opposed to the studies in neon, where a Raman gain of up to eight orders of magnitude has been demonstrated [59], unaligned molecular targets at currently obtainable peak brilliances will give rise to soft x-ray Raman amplification of only a few per cent [62, 63]. In contrast to the more sophisticated nonlinear spectroscopies involving higher-order x-ray susceptibilities, the direct signal of stimulated Raman scattering is not background free and measured in homodyne detection with the transmitted x-ray probe beam that stimulates the Stokes transition. Therefore, it is particularly challenging to measure these small Raman gains with spectrally highly fluctuating SASE pulses.
In the soft x-ray range in gas-phase targets the Raman gain could be strongly enhanced by several orders of magnitude by pre-aligning the molecular target [62]. Although impulsive rotational laser alignment of a molecular gas target was recently attempted in the sRIXS setup, the technical challenge is to find the temporal overlap of the rotational revivals and the x-ray pulse. Current timing techniques that allow for an a posteriori characterization of the relative arrival times of an external optical laser and the XFEL pulses rely on an x-ray optical cross-correlation measurement on a solid target, which typically alters the x-ray spectrum. This destructive technique is therefore not suited for sRIXS, where pump and signal x-ray beams are co-propagating along the same direction. Moreover, in the course of the sRIXS process in optically thick samples, the x-ray pulses are generally heavily attenuated. The x-ray optical cross-correlation measurement relies on a change of index of refraction of an insulator by the x-ray pulse and typically requires an unattenuated x-ray beam. Hence, nondestructive cross-correlation and timing techniques have to be developed that are suitable for the sRIXS setup. Moreover, experimental beamlines that allow measuring the incoming FEL spectrum for every single pulse are required for detection of small Raman gain.
Recent proof-of-principle studies to demonstrate sRIXS in CO gas were performed with two temporally overlapping XFEL SASE pulses, one tuned to the O π* pump transition, the other to the Stokes-shifted transition. The ∼50 fs long SASE pulses, in addition to driving resonant transitions, also resulted in the creation of higher charged molecular and atomic ions, with absorption bands overlapping with the spectral area of the outgoing sRIXS signal, thereby producing a strong background (absorption dips in the spectral region of sRIXS), that precluded the unambiguous demonstration of Raman gain. In a second experiment the SASE-pump pulse was substituted with a self-seeded beam with overall less pulse energy but comparable photon flux on the pump transition. Despite the self-seeding, the pulses showed large shot-to-shot spectral variations. We, however, demonstrated that post-sorting the pair of pulses according to their total pulse energy and the electron-beam energy resulted in very stable, reproducible spectral averages, so that relative differences of >5% are measurable using this pulse combination [63]. A conclusive demonstration of sRIXS was, however, not demonstrated in this experiment. A comparison to our comprehensive theoretical model showed that the achieved experimental conditions were only at the onset of an observable Raman gain.
Advances in science and technology to meet challenges
Similarly to the beginnings of optical nonlinear spectroscopy, we are currently facing the dilemma of having high-intensity, high-brilliance x-ray sources available that in principle can drive nonlinear effects, but their spectral coherence, control of time duration and shot-to-shot reproducibility is poor due to the intrinsic SASE character, so that inherently small nonlinear x-ray signals are difficult to observe in homodyne detection. Necessary advancements in XFEL technology will be new machine setups or seeding techniques [64, 65] that allow for the production of several isolated, transform-limited x-ray pulses of variable and controllable delay and pulse duration. Depending on the different types of nonlinear x-ray spectroscopy, different pulse sequences are necessary. Two promising nonlinear x-ray probe techniques are depicted in figure 9. The general idea is to apply a pair of x-ray pulses as a nonlinear probe following, for example, an activating UV pump pulse that starts a reaction of interest. Nonlinear spectra can be either obtained by applying two sub-fs pulses tuned to specific absorption edges of the molecule of interest and measuring the integrated Raman gain of the second probe pulse as a function of the sample delay time. The spectrum is then obtained by Fourier transform with respect to Δt, which is varied in sub-fs steps to up to ∼100 fs to obtain high spectral resolution. A second method, an extension of the common coherent vibrational nonlinear Raman spectroscopy, applies a long probe pulse that pumps a well-defined core excitation, overlapping with a sub-fs Stokes-shifted probe pulse, that stimulates to a variety of valence excited final states. In this scheme the spectrum of the transmitted broadband probe pulse has to be dispersed and monitored as a function of the pump–probe delay τ.
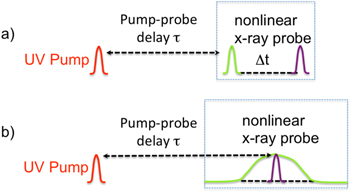
Figure 9. Two different x-ray pulse sequences for coherent attosecond x-ray Raman pump–probe spectroscopy. (a) Application of two attosecond x-ray pulses of controllable time delay Δt. The first pulse prepares a coherent, localized electronic valence wavepacket by an impulsive stimulated Raman process that is mediated by a resonant core to valence transition. After time Δt the electronic wavepacket is probed with a second x-ray pulse that induces a second impulsive x-ray Raman scattering process. The nonlinear signal consists of measuring the Raman gain of the second pulse (relative difference of pulse energies of x-ray pulse with and without UV pump) to obtain a spectrum by Fourier transform with respect to Δt. (b) Combination of a narrow-band and a Stokes-shifted sub-fs probe pulse. A narrow-band, ∼100 fs long x-ray pulse tuned to a specific core to valence transition transiently prepares the system in a core-excited state. An attosecond probe pulse creates a coherent valence wavepacket by stimulated emission of the core-excited system to several valence excited final states. The nonlinear spectroscopic signal is given by the dispersed spectral differences of the transmitted attosecond pulse with and without UV pump.
Download figure:
Standard image High-resolution imageConcluding remarks
Nonlinear x-ray pump–probe spectroscopy would open the pathway towards studying coherent electronic wavepackets with unprecedented spatial and temporal resolution, thereby allowing the study of processes such as coherent charge and energy transport in macromolecular complexes of biological relevance, coupled coherent rovibrational wavepackets crossing conical intersections or photocatalysis. The development of these various spectroscopic pump–probe techniques requires a concerted effort involving in-depth theoretical signal predictions on a quantitative level, development of new configurations of the FELs that allow for the production of several high-intensity attosecond pulses of adjustable photon frequency, pulse duration and pulse separation, as well as the development of a dedicated beamline that provides the necessary optics to focus and distribute the various pulses on the sample with a large flexibility of incidence angles, so that the nonlinear x-ray signals can be detected background free. Nonlinear x-ray spectroscopy could develop into a powerful probe technique, revolutionizing our understanding of coherent electron dynamics in a broad variety of systems of chemical and biological relevance, but could also be applied to understand the build-up of correlated electronic states in solids.
3.3. Ultrafast x-ray atomic and molecular physics—nonlinear processes and coherent control
Kevin C Prince and Claudio Masciovecchio
Elettra Sincrotrone Trieste
Status
Free-electron lasers (FELs) are extending laboratory laser experiments to shorter wavelengths, and adding element and chemical state specificity by exciting and probing electronic transitions from core levels. The high pulse energies available ensure that nonlinear optics can cross the frontier into this new wavelength range. The transition from masers to lasers brought about a scientific revolution, and a similar transformation in physics may be expected from these sources.
Since the first short-wavelength FEL FLASH began operation in Hamburg [66], development has been very rapid. The LCLS was specified to produce 200 fs pulses, but soon produced few fs pulses [67]. Schemes have been developed to create multipulse and polychromatic radiation, and FEL light has been used to pump atomic lasers in the x-ray region [58, 68]. The XUV/soft x-ray laser FERMI is the first fully coherent FEL, as the light possesses the full longitudinal coherence missing from SASE FELs [69].
In this article, we will discuss how advances in the performance of FELs, especially in multicolor pulse production and exploitation of coherence, may drive the development of new experimental strategies to study non-equilibrium behavior of matter at the femtosecond–nanometer time-length scales. This would have a tremendous impact as an experimental tool to investigate a large array of phenomena ranging from nanodynamics in complex materials to phenomena that are at the heart of conversion of light into other forms of energy.
Current and future challenges
With the development of FEL radiation sources, a new era of x-ray spectroscopy commenced, which may have a comparable impact to that of lasers in optics and spectroscopy. In nonlinear ultrafast time-resolved techniques, state-specific information is often provided through multiphoton resonances with combinations of sequential photons. Theoretically, combinations of multiple x-ray photons resonant with core transitions can also characterize different excitation processes due to specific sequences of light–matter interaction.
Thus particular sub-processes can be enhanced by matching the pulse frequencies to transitions between molecular eigenstates. This provides high selectivity and flexibility due to momentum and energy conservation of the interacting photons with the material.
The different nonlinear processes can typically be ordered by the number of incident photons: sum frequency generation, two-photon absorption, and stimulated emission use two photons; other nonlinear x-ray phenomena, such as time-resolved transient gratings (XUV-TG) or four-wave mixing (XUV-FWM) spectroscopy, use three incoming photons.
Fluorescent decays in the soft x-ray region allow unique access to the structure of the occupied valence states, while keeping the element selectivity and chemical state specificity of soft x-ray spectroscopies. Unfortunately, the probability for fluorescent decay in the soft x-ray range is below 1%, and this represents a challenge for signal detection. Spectrometers are also inefficient (typical acceptance <10−5 of the full solid angle), so soft x-ray emission spectroscopy (XES) or RIXS are experiments where single photons have to be counted for hours in order to obtain useful information. This challenge can be met by using stimulated emission spectroscopy with high-intensity, small-bandwidth, tunable soft x-ray beams of the appropriate color. A stimulated beam containing the same information as a fluorescence spectrum can be formed—both mitigating the low acceptance angle of the spectrometers, and suppressing the dominating Auger decays that also create electronic damage to the sample. With such techniques, two to three orders of magnitude in the signal levels can be gained through the suppression of Auger processes, while the beam directed into the spectrometer promises to gain about five orders of magnitude in detected signal levels—clearly having the potential to revolutionize how we can study matter with XES or RIXS based spectroscopies.
Among wave mixing processes, four-wave mixing (FWM) has a special place, since it is at the basis of most experimental methods, besides being the lowest order nonlinear process that does not vanish by reason of sample symmetry considerations.
FWM with optical lasers is an important technique, which can be used in fields as diverse as combustion science [70] and molecular spectroscopy [71]. Extending FWM to the XUV and soft x-ray regime would open up the possibility to probe the sample with chemical selectivity exploiting electronic resonances. This would allow monitoring charge and energy transfer and understanding photodynamics.
Not only do we want to understand molecular processes, but we also wish to manipulate them. In the quantum mechanical world this can be done by the established methods of coherent control (figure 10). When this approach is developed for FELs, the new frontiers opened up include control of inner valence and core levels, and consequently chemical sensitivity. Optical coherent control is temporally limited to pulses that are several times the duration of an optical cycle, which is several femtoseconds. In the XUV to x-ray range, the periods are from hundreds to a few attoseconds. Thus extremely fast processes can be controlled. First steps have been taken to establish coherent control [69] and the challenge now is to develop the technique.
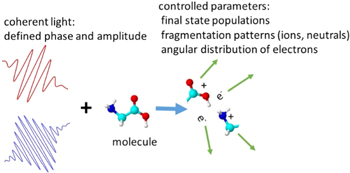
Figure 10. Schematic view of coherent control. Phase coherent light of one or more colors interacts with a target. The outcome of the interaction is determined by the phase and amplitude of the light. Ion yield, direction of emission of ions and electrons, etc can be controlled.
Download figure:
Standard image High-resolution imageMuch effort is being invested to develop ever shorter pulses in FELs. The arrival of phase control of multicolor pulses now means that they can be designed to produce trains of very short pulses, since a train of attosecond pulses can be constructed from a coherent superposition of coherent femtosecond pulses with commensurate wavelengths. This ability to tailor pulses may eventually lead to 'designer' single pulses of attosecond duration.
Although already proposed in the literature [72], only recently Bencivenga et al, [39] demonstrated the feasibility of degenerate FWM in the XUV regime. Two identical FEL pulses were used to create transient grating and an optical pulse was used to probe it. The natural evolution is to create coherent excitations, i.e. excitons, polarons, in both condensed matter and molecular targets (see figure 11).

Figure 11. Range of excitation energies (eV) and wavevectors (nm−1) potentially accessible by XUV and soft x-ray FWM experiments.
Download figure:
Standard image High-resolution imageFor example, this can be done by using two different FEL pulses resonant with a core hole and probing the delocalization with a third FEL pulse resonant with a core hole of a different atom of the system.
Advances in science and technology to meet challenges
The critical developments needed to further the field are increased longitudinal coherence, shorter pulses and multiple pulse production. SASE machines are making progress in the first of these using self-seeding, single spike generation and other innovative approaches. External seeding confers a high degree of coherence (and control), but at the price of longer temporal duration of the pulses, as a sufficient number of (long wavelength) optical cycles has to be contained in the seeding pulse. The natural evolution would be to develop FELs in order to allow few-fs emission through the exploitation of innovative methodologies, based for example on echo enhanced microbunching [73]. This is necessary in order to have a pulse duration shorter than the typical core hole lifetimes (4–8 fs for O, N and C). Moreover, the requirement of different pump and probe pulses calls for frequency tripling for the few-fs FEL emission scheme.
Concluding remarks
The XUV/soft x-ray FWM technique is a powerful tool for investigating many systems, which include: (i) spin, charge and structural dynamics of macrocycles, dyes, and metal-based molecular complexes, which are important in solar energy applications; (ii) in condensed matter, charge carrier and spin dynamics of metal oxides; (iii) field-driven phase transitions. These studies are the first steps to exploit the novel possibilities offered by XUV/soft x-ray FWM. In addition, the powerful methods of coherent control allow us to manipulate the outcome of photon–matter interaction, maximizing or minimizing cross sections for a particular process. In addition, control of the phase and amplitude of short-wavelength radiation will allow pulses to be designed to order. In the long term, these first applications of nonlinear XUV/soft x-ray interactions will trigger the development of further experimental tools and applications, in analogy to what happened in the last few decades in the field of optical laser-based research, which has witnessed enormous growth of diverse nonlinear applications.
Acknowledgments
KCP thanks all of the authors of [39] for useful discussion.
4. High-intensity x-ray phenomena
4.1. Multiphoton processes and polarization control at short-wavelength FELs
Michael Meyer
European XFEL GmbH
Status
Since the advent of FELs producing intense photon pulses in the VUV, XUV and x-ray wavelength regime, many new and exciting applications using the unique properties of the radiation have been introduced. In particular, the high number of photons comprising the ultrashort femtosecond pulses gives rise to the production of high charge states, which can only be explained by the absorption of many photons from the same pulse (e.g [1, 74]). Contrary to excitations with optical lasers reaching only the outer electronic shell in an atom or molecule, the short-wavelength radiation interacts preferentially with strongly bound core electrons and leads to competitions between relaxation via Auger decay and further ionization steps. However, as the main mechanism, stepwise sequential ionization was identified (figure 12). Only for higher charges, where the energy of one photon is not sufficient to induce the next ionization step, do two (or more) photon processes have to be included for a consistent interpretation. In general, except for some special cases, good agreement between theory and experiment, mainly based on ion spectroscopy, has been obtained. These studies serve as an important model system to investigate fundamental processes in the interaction between matter and highly intense radiation pulses. The importance and influence of multiphoton processes have to be understood as they provide a firm basis for the reliable interpretation of processes in larger systems.

Figure 12. Example for the sequential ionization process upon core excitation. Reprinted by permission from Macmillan Publishers Ltd: Nature [1], Copyright 2010.
Download figure:
Standard image High-resolution imageMore detailed investigations of the underlying dynamics requiring, e.g., the analysis of the electron spectra have also been undertaken. Direct two-photon processes have been observed in the manifestation of above threshold ionization [75] and formation of double core hole states [76]. These studies represent the starting point for a new class of investigations uniquely possible at FEL sources and demonstrate that the intense short-wavelength pulses open access to new phenomena and to additional information, which cannot be obtained in the one-photon regime [75]. Similarly, a different class of resonances inaccessible by one-photon excitation were studied [77]. Changes in the dynamics of autoionizing resonances induced by the presence of a strong XUV or external optical field [78] highlighted the possibilities and the richness of these studies enabled by the use of the x-ray FEL sources, especially if the polarization of the FEL pulses can be varied and controlled [79].
Furthermore the short time duration of XUV pulses provides an ideal ground for time-resolved pump–probe studies, using either two parts of the same XUV pulse, which can be controlled independently by a split-and-delay unit, or in combination with a pulsed synchronized optical laser. Since the electron dynamics takes place on a sub-femtosecond time scale, time-resolved information has been obtained mainly on molecular targets and competition between electronic and nuclear movement was in the focus of these studies. Pulses of some femtosecond durations enabled access to processes such as isomerization or charge transfer processes during dissociation [13], i.e. processes where the movement of the atomic components could be traced by measuring the electronic response (see section 4.2).
The attosecond regime comes into reach through techniques like THz streaking allowing setting a very precise time marker and identifying pulses with sub-femtosecond durations [80]. Proof-of-principle experiments have been demonstrated, but application was achieved due to the seeding scheme for generation of FEL pulses [69] (see section 6). The controlled production of the FEL pulses also enables the control of the spectral phase of the pulses and few attosecond temporal resolution was realized in an experiment using the interference between ionization processes induced by two-photon absorption of the fundamental radiation and one-photon absorption of the second harmonics.
Current and future challenges
To fully explore the FEL sources the perfect characterization of the XUV pulses has to be realized. Generally, the production of the intense pulses is based on SASE. As a consequence, the temporal, spectral and intensity distribution of the pulses show strong variations and are characterized by the presence of many spikes. For a detailed interpretation of the observed phenomena and for a meaningful comparison with theoretical calculations the precise characterization of all these parameters for each individual pulse is required and has to be taken into account in the analysis.
Scientifically one of the major interests relies on the investigation and application of nonlinear spectroscopy in the short-wavelength regime. For optical lasers multiphoton spectroscopy is established as an extremely powerful tool; at shorter wavelength the general application of the direct multiphoton process is still a challenge, although the feasibility and potential has been demonstrated [75–77]. Multiple simultaneous ionizations and the subsequent relaxation processes, multiphoton direct ionization and resonant excitations, will give access to unexplored dynamics and symmetries, which have not been studied so far. In particular, the possibility for atom- or site-specific excitation in molecules, due to the localized core electrons, provide an unprecedented basis for stimulating specific processes and controlling the dynamics of fragmentation and electronic decay. The strong competition with one-photon processes, which are characterized by several orders higher cross sections, is limiting presently the full exploration of these new possibilities. In the experimental arrangement, the multiphoton signal has to be clearly separated from the strong one-photon signal and possible depletion of the target volume has to be taken into account.
In addition to the investigation of nonlinear phenomena, detailed knowledge of the temporal evolution of the system under these extreme conditions is of fundamental interest. For molecules, charge transfer and charge migration can be studied in detail using two pulses of different photon energies. Theoretical analysis predicts these processes happening on sub-femtosecond timescales [81]. Charge rearrangement before, during and after fragmentation of the molecule can be determined by selective excitation of a specific site in the parent molecule or/and the selection of a dedicated fragment. In addition, unexplored regions of the potential curve of the core-excited state can be studied in combination with an optical laser [82]. The optical laser creates a wavepacket in the ground state, which can be probed in a time-delayed way by the XUV pulses. The two-photon process explores, therefore, different Franck–Condon areas and gives access to different molecular geometries and dissociation dynamics. Ultimately the control of photon energy, pulse duration, temporal delay and spectral phase of the two pulses will be possible and the intramolecular processes involving the electrons and ions can be traced in detail on a sub-femtosecond time scale.
The recent advances in producing variable polarization present additional benefits for the above-mentioned studies. In atomic multiphoton processes, symmetries of resonances and of outgoing electrons depend on the polarization state of the linearly and circularly polarized light. Combining the results of both studies enables the 'complete' experiment, i.e. the extraction of all quantum mechanical parameters. In the case of molecules, circular polarization is an especially crucial ingredient to provide new information on chiral molecules. In time-resolved studies the ionization and fragmentation dynamics of these molecules, particularly the possible transfer of chirality to the fragmentation products, can be determined.
Advances in science and technology to meet challenges
In the last few years much knowledge has been accumulated in operating FEL sources and has strongly influenced the design of the upcoming sources, e.g. the European XFEL in Hamburg. The main and, up to now, unique characteristic of the European XFEL is the high number of photon pulses, namely 27 000 pulses per second. This number provides a drastic improvement for all applications based on coincidence techniques. In consequence, the SQS (Small Quantum System) scientific instrument at the European XFEL, which is dedicated to gas-phase experiments, will fully exploit this possibility, including the installation of a dedicated reaction microscope. Separation of different ionization processes caused at different stages of a sequential ionization or arising in the course of molecular fragmentation can be realized using these techniques. Specially designed detectors for coherent diffraction imaging experiments will make use of this high repetition rate improving dramatically the statistics in the related studies on clusters, nanoparticles or large biomolecules.
In addition, high-resolution electron spectroscopy and the determination of the electron angular distributions for high kinetic energy electrons have been successfully developed. In particular, the polarization dependent studies rely strongly on the more detailed analysis of the electrons. Specially designed 1D imaging fluorescence spectrometers, enabling probing of the interaction of a dense molecular target with the intense XUV radiation in the direction of the FEL propagation, open up new and unique access to studies of nonlinear phenomena and their temporal evolution.
The quality of focusing mirrors has also strongly increased and it is possible to fabricate extremely large mirrors (80 cm length) with ultrahigh surface flatness (50 nrad). These are necessary requirements for producing intensities of about 1018 W cm−2 in the interaction volume by achieving a focus of 1 micron diameter. Higher intensities will be possible, but space restrictions will limit the general use of such devices for a broad range of applications.
Concluding remarks
Investigations using FELs operating in the short-wavelength regime have already demonstrated their strong potential and scientific interest in various areas of research. New developments at these sources, including seeding and application of dedicated devices for producing variable polarization and generating two (or more) FEL pulses of largely different wavelengths and durations at variable time delays as well as pulses with durations in the attosecond regime, are foreseen and will tremendously increase the possibilities for research. New results in nonlinear spectroscopy and time-resolved pump–probe spectroscopy are within reach, taking advantage specifically of the localized excitation of core electrons at short wavelength.
Acknowledgments
4.2. Probing charge and nuclear dynamics after inner-shell photoabsorption
Artem Rudenko and Daniel Rolles
J R Macdonald Laboratory, Kansas State University
Status
One of the key features that make x-rays a very efficient tool for imaging structure and dynamics of matter is their element specificity. It makes it possible to probe a specific site or atomic species inside a larger molecule, crystal, or nanoparticle. Since x-rays predominantly interact with inner-shell electrons, which are highly localized, x-ray photoabsorption in an extended system often occurs locally on one particular atom or atomic species. Following this initial photoabsorption step, the induced charge and deposited energy can be efficiently redistributed to the neighboring atoms. The time scale of such a charge rearrangement can vary from sub-femtosecond to a few femtoseconds, depending on the particular mechanism. Because of the high degree of initial charge localization, ultrafast dynamics after inner-shell photoabsorption have attracted considerable attention in the context of charge transfer studies. However, until recently, these studies were limited to measurements in the energy domain [83] because of the lack of short x-ray pulses. The situation has changed with the development of XFELs and high harmonic based sources capable of delivering femtosecond or even sub-femtosecond x-ray pulses.
The local nature of x-ray–matter interactions makes the role of elements with high atomic numbers particularly important. In the soft x-ray regime, such a heavy atom can have a photoabsorption cross section 10 to 50 times higher than that of carbon or oxygen. In the hard x-ray domain, the same cross section ratio is typically in the range between 100 and 1000. Under these conditions, even a single heavy atom constituent can significantly alter the total energy deposition in a large macromolecule and cause increased local radiation damage. This is observable in the x-ray diffraction patterns from biological objects containing, e.g., sulfur, selenium, and iodine atoms [84], or heavy metal centers, and can be exploited for phasing of the acquired diffraction data [85]. Therefore, the behavior of heavy atoms embedded in an environment consisting of lighter elements under intense, short-pulsed x-rays has recently become a focus of a series of dedicated experiments at FLASH [86], LCLS [4, 13, 87], and SACLA [5, 6], as sketched in figure 13. In particular, it has been shown that at extreme x-ray intensities (exceeding 1019 W cm−2), the ionization of a small molecule can be significantly enhanced via a recurrent charge redistribution resulting in the emission of more than 50 electrons within few tens of fs [87].
Figure 13. Experiments at FLASH, LCLS, and SACLA have studied x-ray interaction with molecules containing one heavy element such as Se or I, contrasting it with the response of an isolated atom with similar absorption cross section (Kr and Xe, respectively). Understanding the local molecular response in the vicinity of embedded high-Z constituents is important for x-ray imaging of many biological objects.
Download figure:
Standard image High-resolution imageCurrent and future challenges
The behavior of molecules irradiated by intense x-ray pulses represents a serious challenge for theory and for the interpretation of the experimental results. While recent theoretical studies have considered examples of pure charge migration [81] and charge transfer [88] following inner-shell ionization of a particular atom, in general, there can be a variety of pathways driving charge dynamics in such a core-ionized state. The mechanisms that unfold on a time scale slower than ∼1 fs are necessarily interwoven with (and often driven by) nuclear motion. Furthermore, for two- or multiphoton processes, the absorption of each additional photon triggers similar dynamics now unfolding on a modified set of ionic potential surfaces. The characteristic time between the absorption of two photons is defined by the x-ray pulse intensity and the target cross section. For typical XFEL conditions, it is of the order of a few fs, which matches the timescales of both electronic relaxation and nuclear motion.
The presence of a heavy atom, which is attractive for experimentalists because it significantly enhances the degree of initial charge localization, represents an additional challenge for theory since a large number of possible electronic configurations need to be considered [2]. Nevertheless, recent theoretical efforts have yielded a quantitative understanding of ionization and fragmentation dynamics for CH3I molecules under extremely intense hard x-ray light [87]. Besides complementing the theoretical description to include resonant and relativistic effects, extending this kind of simulation to larger systems is currently one of the main challenges for theory (see section 4.5). It also remains one of the key experimental challenges. While several experiments have addressed nanoscale systems either consisting of high-Z elements (e.g., Xe clusters [89]) or containing a few heavy atoms in the environment of low-Z elements (Xe atoms embedded into He microdroplets [90]), as illustrated in figure 13, no ionization or fragmentation studies have been reported for large systems of biological relevance containing heavy atom centers. Performing such experiments and linking the results to features observed in the corresponding diffraction patterns is therefore an important goal for the future. Technically, the main challenge is to bring biological objects into the gas phase at high enough densities.
Despite considerable interest in the behavior of heavy atoms under intense x-ray pulses, most of the typical XFEL targets, in particular, organic molecules, mainly consist of light elements. Since most of them have comparable x-ray absorption cross sections, the determination of the initial absorption site is experimentally challenging. In principle, it can be done by measuring the kinetic energy of the emitted photoelectron. However, to efficiently exploit it for revealing site-specific features of x-ray–molecule interaction, one often needs to detect the photoelectron in coincidence with ionic fragments and/or other (e.g. Auger) electrons, which is hardly feasible in the current low-repetition-rate XFELs. Finally, for many interesting dynamics unfolding upon inner-shell photoabsorption, the pulse duration of the present XFEL facilities still remains too long. For example, while the few-fs transfer dynamics of a double valence hole [88] created by core-shell ionization and subsequent Auger decay might be barely accessible with the few-fs pulses available at the LCLS, sub-fs charge migration discussed in [81] is currently beyond reach. Following such ultrafast motions would require even shorter, attosecond pulses.
Advances in science and technology to meet challenges
By the end of 2017, the European XFEL is expected to become operational. With a repetition rate of 27 kHz, it will be the first high-repetition-rate XFEL facility. A few years later, it will be followed by LCLS-II, which will approach MHz repetition rates. With these next-generation XFELs, efficient multi-coincidence measurements will become feasible. These developments will open up new horizons for XFEL experiments in atomic and molecular physics as well as in gas-phase photochemistry, and, in particular, will allow several of the challenges outlined above to be addressed.
A sketch of a generalized coincidence experiment in a high-repetition-rate XFEL is shown in figure 14. Electrons and ions created by a pair of pump and probe pulses, are measured by two time- and position-sensitive detectors. This can be realized, e.g., by a 'reaction microscope' type spectrometer or by a double-sided velocity map imaging setup. Even though efficient coincident detection of rather fast Auger electrons combined with sufficient energy resolution for slower photoelectrons and for ions represents a serious technical challenge; this will, most likely, be resolved at the experimental end-stations of the next-generation XFEL facilities. Coincident detection of a photoelectron will allow filtering the events triggered by the x-ray absorption at different atoms within a molecule even if their absorption cross sections are comparable, while a combination of ion charge state and energy measurements with the coincident Auger spectra will enable reliable experimental identification of the intermediate states involved.
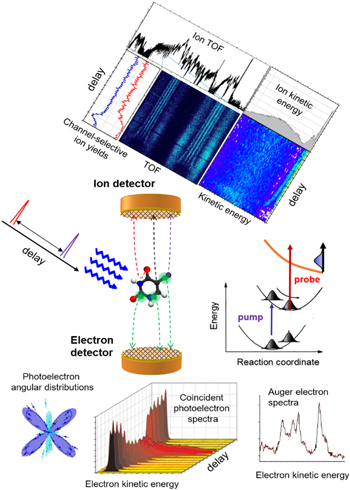
Figure 14. Sketch of a multi-coincidence pump–probe experiment with detection of ions and electrons. Ion time-of-flight spectra allow one to identify the final products of a particular reaction channel, while the kinetic energies and emission angles of ionic fragments and of the photo and Auger electrons measured as a function of pump–probe delay can fully characterize the intermediate dynamics.
Download figure:
Standard image High-resolution imageThe expected high repetition rates of the upcoming XFELs will also help in resolving sample delivery issues since the ionization signal will become observable for considerably lower target densities. Along with dedicated source development, this should enable experimental studies on larger, biologically relevant systems such as small proteins containing a cluster of higher-Z metal atoms, or on radiosensitizer molecules with embedded iodine atoms.
Finally, the current pulse duration and temporal-resolution limitations might be addressed by employing sub-fs HHG sources. Recently, isolated attosecond pulses in the water window have been demonstrated [91] (see sections 5.5–5.6). Since these pulses are not intense enough for x-ray pump–x-ray probe experiments, it remains to be seen to what extent the achievable time resolution for inner-shell dynamics will be limited by the synchronized infrared laser pulse. An alternative route is the development of attosecond capabilities at XFELs (see section 2.2). With rather large photon densities available even in this ultrashort pulse regime, they will enable x-ray pump–x-ray probe schemes with sub-fs temporal resolution and two-color pulses.
Concluding remarks
With the parallel developments of new XFELs and HHG-based x-ray sources, unique opportunities are emerging for studies of ultrafast dynamics triggered or probed by inner-shell interactions. While, in the nearest future, nonlinear x-ray processes will most likely remain accessible only at XFELs, HHG pulses are becoming intense enough to not only initiate the dynamics, but also to efficiently probe the reactions driven by a synchronized laser. The combination of HHG-based and XFEL-based approaches will provide the scientific community with efficient solutions for many fundamental problems in molecular physics and ultrafast photochemistry.
Acknowledgments
The authors acknowledge support by the Chemical Sciences, Geosciences, and Biosciences Division of the Office of Basic Energy Sciences, Office of Science, US Department of Energy under contract No. DE-FG02-86ER1349.
4.3. New opportunities from ultrafast single-particle imaging and scattering in AMO physics
Christoph Bostedt
Argonne National Laboratory
Status
The intense and coherent x-ray pulses from free-electron lasers (XFELs) have opened up new avenues and opportunities for visualizing dynamics and transient states in single, nanometer-sized particles in a single shot. Since the first short-wavelength FELs became operational, this field has rapidly developed and imaging as well as diffraction techniques have been used for various purposes.
The first proof-of-principle experiments for single-shot imaging of single nanometer-sized samples in the gas phase (figure 15) were targeted at determining cluster shapes and sizes [92], the morphologies of cluster assemblies [93], and imaging biological specimens [94]. The experiments rapidly advanced to obtaining three-dimensional information from metallic clusters with sizes of the order of 100 nanometers [95]. Further, single-shot diffractive imaging was used to investigate quantum phenomena in nanometer-sized superfluid 4He droplets [90]. From the shape analysis of the superfluid droplets it could be concluded that they rotate with speeds beyond the stability limit for viscous samples. Doping the droplets with xenon, which acted akin to an x-ray contrast agent, it was possible to show that quantum vortices exist inside the droplets and that they form lattices with densities up to five orders of magnitude higher than in bulk superfluid helium.
Figure 15. Examples of single-shot diffractive images from nanometer-sized samples. (a) Early diffractive single-shot image of a single van der Waals cluster. Reproduced from [92]. © IOP Publishing Ltd. All rights reserved, (b) a cluster dimer. Reproduced from [93]. © IOP Publishing Ltd. CC BY 3.0, (c) a truncated octahedral Ag cluster. Reprinted by permission from Macmillan Publishers Ltd: Nature Communications [95], Copyright 2015, and (d) a helium droplet with Bragg scattering from xenon-decorated quantum vortices. From [90]. Reprinted with permission from AAAS.
Download figure:
Standard image High-resolution imageA new opportunity for single-shot imaging of single particles lays in the investigation of transient states. For investigating electronically excited states one can exploit that the x-ray scattering signal is generated by the electrons in the sample and that strong resonances near the inner-shell ionization thresholds can leave distinct fingerprints in the diffraction signal [96].
Coincident imaging and spectroscopy is a novel experimental strategy providing additional information about the sample or experimental conditions, information that is often lost during ensemble averaging experiments. This was first demonstrated for x-ray ionization of clusters in the high-intensity limit [89]. Here the coherent image of the clusters was used to select single-particle events and to sort the data according to the exposure intensity of the particles based on the brightness of the image, giving unprecedented insight into nonlinear ionization processes.
More recently, single-shot imaging has been combined with external pumps in order to investigate non-equilibrium dynamics on the nanoscale. This can entail external optical drivers or intense double pulses from the XFEL itself. Using an optical laser, clusters were superheated and their expansion was subsequently imaged with intense x-ray pulses [11]. With this method surface softening at the cluster/vacuum interface was identified with nanometer resolution within 100 femtoseconds of the driving laser pulse. In another study, femtosecond hard x-ray pulses were used to isochorically pump xenon clusters into a highly excited state and their lattice response was probed with a second hard x-ray pulse via Bragg scattering [97]. The experiments showed a surprising transient lattice contraction within 80 femtoseconds, which was attributed to the massive x-ray induced electronic excitation and a collective change in the bond character of the nanoparticles.
In parallel to the experimental efforts, theoretical approaches to describe the x-ray diffraction experiments have rapidly evolved. Massively parallel computing models can now describe the complete x-ray induced process from ionization to inner-vacancy decays, to elastic and inelastic scattering processes, and give guidance on optimized imaging parameters [98]. Other theoretical efforts focus on describing the field propagation in clusters in intense optical laser fields, laying the foundation for imaging experiments of electron density distributions in externally driven systems [99].
Current and future challenges
The field of single-shot imaging of single, nanometer-sized clusters and nanoparticles is rapidly maturing from the first proof-of-principle experiments to an approach for tackling scientific challenges and applications. So far, single-shot imaging of gas-phase particles has been a domain of linear-accelerator-based facilities but with the rapid advancement in, and ever increasing output of, table-top systems one can imagine that optical laser driven experiments will start playing a vital role in this field in the near future [100].
The spatial information from single-shot imaging experiments [89, 90, 92, 93–95] can be used to investigate particle nucleation and growth processes, morphologies of larger nanoparticle assemblies, exotic symmetries in reduced-dimensional systems, as well as phase mixes and phase separations on the nanoscale. More intense and shorter x-ray pulses will allow obtaining higher resolution images and even three-dimensional information [95, 98]. Holographic approaches in the gas phase hold the potential for rapid and high-fidelity imaging, circumventing the need for iterative phase retrieval analysis.
Time-resolved diffractive imaging and scattering with external drive lasers [11, 97] opened up opportunities for the investigation of lattice dynamics, phase transitions, and non-thermal processes. Optical light pulses can excite the nanoparticle as a whole and x-ray pulses can be used to induce localized, element-specific excitations [101]. With the current state-of-the-art, coherent imaging can yield information about the particle shapes and thus their phases. Detailed information about the nanocrystal lattice structure and dynamics can be obtained from Bragg diffraction [97]. The efficiency of Bragg experiments from nanoparticles in the gas phase is low, however, as the particles only stochastically satisfy the Bragg condition, and most of the time only incomplete reflections and partial cuts of the Ewald sphere are recorded.
Another possibility for the future is using coherent imaging and diffraction to deduce information about transient electronic information [96]. The scattering signal is generated during the pulse and therefore the obtainable temporal resolution is only limited by the imaging pulse length. Here, single-shot imaging may be a route to obtaining transient electronic information in nanoscale samples, including visualization of charge transfer processes in, e.g., core-shell systems. Using fs or even sub-fs x-ray pulses and combining the imaging approach with optical laser fields will allow following electron density modulations in space and time during an optical cycle with potential applications in nanophotonics and nanoplasmonics [99].
The coincident imaging and spectroscopy approach [89] can be extended to a wide range of applications. One could envision multipulse schemes where a system is first pumped with an optical pulse and subsequently the excitation is characterized spectroscopically before the sample size, phase, or morphology is probed by means of single-shot imaging. This pump–spectroscopy–imaging approach could yield information about the interplay of electronic and geometric structure during phase transitions or charge transfer and migration processes in complex particles or aerosols. It could open up new routes for obtaining information on how excitations develop on the nanoscale.
On a final note it is important to remember that the single-shot imaging process is always a strong probe and will also strongly interfere with the sample itself. Fundamental questions about the detailed contributions of localized, delocalized, and free electrons are still under discussion. Basic questions about the obtainable contrast as well as effects from transient spatial electronic configurations due to charge transfer mechanisms in the strongly ionized samples need to be answered. Therefore, there is a strong need for an advanced theoretical description for the ionization dynamics and scattering response of strongly ionized and also externally driven nanometer-sized samples in intense x-ray laser pulses.
Advances in science and technology to meet challenges
The resolution obtainable in state-of-the-art single-shot imaging experiments is currently limited by the signal levels at large scattering angles, and so is the lower sample size limit. Both aspects can be improved through higher exposure intensities of the sample, i.e. higher photon output of the laser, efficient beam transport to the sample, and tighter foci. In this context it is noted that the figure of merit is not necessarily the highest number of photons but the highest number of photons per Auger lifetime.
Detectors are crucial for recording high quality diffraction images. Compared to the systems available to date the dynamic range needs to be improved to span more than four orders of magnitude from the intense center of the coherent image to the single-photon signal levels at large scattering angles. Further, the detectors need to cover a large solid angle with minimal dead areas.
A crucial aspect for the pump–probe experiments, in particular for pump–probe experiments aimed at electron dynamics, is the imaging-pulse duration and the synchronization of the optical and x-ray pulses. New operational modes of XFELs currently under development promise femtosecond and even sub-femtosecond pulse lengths. Future superconducting accelerator-based XFELs will have greatly reduced timing jitter between the optical and x-ray laser systems. For sub-cycle resolution phase stabilized systems will be needed, which are already available today from table-top systems, although with comparably low photon output.
Concluding remarks
Ultrafast imaging of single nanometer-sized particles with single x-ray pulses has rapidly developed into a very powerful tool in AMO sciences. Combining imaging and spectroscopy approaches with precisely timed optical or x-ray laser pulses will allow following nuclear and electron dynamics on the nanoscale with femtosecond and nanometer resolution.
Acknowledgments
I would like to acknowledge fruitful discussions with Tais Gorkhover, Phay Ho and Linda Young. This work was supported by the US Department of Energy, Office of Science, Basic Energy Sciences, Chemical Sciences, Geosciences, and Biosciences Division.
4.4. Hard x-ray nonlinear optics
Matthias Fuchs1,2 and David A Reis2
1University of Nebraska
2Stanford PULSE Institute
Status
The generation of second harmonic radiation by Franken et al in 1961 [102] shortly after the invention of the laser is widely viewed as the birth of nonlinear optics. The ubiquity of bright optical laser sources coupled with theoretical treatments of light–matter interactions rapidly advanced the field. The field of x-ray nonlinear optics got its start only a decade after Franken's seminal experiment. In 1971, Eisenberger and McCall [49] observed the first nonlinear effect at hard x-ray wavelengths, namely spontaneous parametric down conversion (PDC) of one 17 keV x-ray into two near 8.5 keV photons in a Be crystal using an x-ray tube as the source. In these experiments, down-converted photon pairs were produced at a maximum rate of about one per hour. The development of significantly more brilliant storage-ring based x-ray sources allowed for much higher rates, leading to a better understanding of x-ray PDC [50, 51]. It also led to new applications, such as the visualization of local induced charge densities in a crystal with atomic spatial resolution [103] (see figure 16). However, nonlinear effects in the hard x-ray regime that involve the simultaneous interaction of multiple incident photons have remained elusive even at the brightest synchrotron sources due to vanishingly small susceptibilities at high frequencies. Only with the billion-fold increase in peak brightness of hard XFELs and with focused intensities approaching those of the most intense optical sources has it become possible to observe multiphoton processes at x-ray wavelengths. First experiments demonstrated that the peak x-ray flux of the LCLS XFEL is sufficient to saturate atomic inner-shell absorption at soft x-ray wavelengths within a few femtoseconds (see section 4.1). These experiments were followed by seminal experiments at the LCLS and SACLA XFELs that demonstrated fundamental nonlinear multiphoton interactions at hard x-ray wavelengths. This includes nonsequential two-photon K-shell absorption [104, 105], phase-matched sum frequency generation [106], second harmonic generation [107] and two-photon Compton scattering (2PCS) [108]. While the former three experiments match theoretical predictions reasonably well, an anomaly was observed in 2PCS involving an excessive red-shift in the energy of the nonlinearly generated photons compared to what was expected in the quasi-free-electron (impulse) approximation. This discovery demonstrates that our fundamental understanding of x-ray matter interactions is still incomplete.
Figure 16. Reconstructed atomic-scale view of the XUV susceptibility of diamond at 60 eV from x-ray down conversion ∼11 keV photons. Reprinted by permission from Macmillan Publishers Ltd: Nature Physics [103], Copyright 2011.
Download figure:
Standard image High-resolution imageCurrent and future challenges
The nonlinearities in dielectric materials at optical wavelengths are mainly due to anharmonicity in the atomic potential. These effects scale unfavorable with the radiation frequency ω. In the case of the lowest (second) order nonlinearity, the induced polarization scales as ω−6 for frequencies well above atomic resonances. For interactions with hard x-rays, the electrons in a material can be approximated as quasi-free when the x-ray energy is well above the atomic resonances. The nonlinearities in this wavelength range are predominantly due to anharmonic motion of quasi-free electrons caused by, for example, the  part of the Lorentz force. In this case, the nonlinearly induced current at the second harmonic scales as ω−2 (and thus the polarization as ω−3), which for x-ray wavelengths is considerably more efficient than the anharmonicities in the atomic potential. Still, for the free-electron nonlinearity, the number of scattered photons per unit timescales as ω−3 for a fixed field strength, which leads to a factor of 10−12 smaller efficiencies for 10 keV x-rays compared to 1 eV optical photons. Thus, the observation of nonlinearities that involve the simultaneous interaction of multiple incident x-ray photons requires intensities that only XFELs can generate. Driving higher-order nonlinearities requires even higher intensities.
part of the Lorentz force. In this case, the nonlinearly induced current at the second harmonic scales as ω−2 (and thus the polarization as ω−3), which for x-ray wavelengths is considerably more efficient than the anharmonicities in the atomic potential. Still, for the free-electron nonlinearity, the number of scattered photons per unit timescales as ω−3 for a fixed field strength, which leads to a factor of 10−12 smaller efficiencies for 10 keV x-rays compared to 1 eV optical photons. Thus, the observation of nonlinearities that involve the simultaneous interaction of multiple incident x-ray photons requires intensities that only XFELs can generate. Driving higher-order nonlinearities requires even higher intensities.
While nonlinearities are considerably smaller at x-ray wavelengths than in the optical regime, they offer new possibilities for understanding structure and dynamics at the atomic scale due to their short wavelength. For example, they could be applied as instantaneous plasma diagnostics for materials in extreme conditions, in multidimensional x-ray spectroscopy ([54] and see sections 3.1 and 3.2) in coherent control of photo-induced charge transfer ([109] and see section 3.3), in element-specific atomic imaging and for imaging induced polarization on atomic spatial and femtosecond to attosecond temporal resolution [106]. However, the transition from demonstration experiments to functional tools requires overcoming some challenges, which are mainly consequences from the small cross sections of the nonlinearities and the resulting low signal count rates. One of the main difficulties is the separation of these small signals from the background, which mainly consists of linear scattering of the FEL fundamental and its harmonics. Because of the currently operational XFELs with comparably low repetition rates of ∼100 Hz, nonlinear experiments need to be performed at x-ray intensities near or above the sample damage threshold and typically using solid samples with a sufficiently high electron density. Due to the low repetition rate, these machines are also comparably noisy in terms of shot-to-shot parameters, such as photon number, pulse duration, longitudinal coherence, wavelength, beam pointing, and the resulting focus position and quality.
Advances in science and technology to meet challenges
The challenges outlined above can to a large extent be overcome with advances in x-ray sources, x-ray optics, diagnostics, experimental design and instrumentation. In particular, as the effects increase nonlinearly with the driving field, one way to improve the signal-to-noise ratio is by using high-intensity beams. This can be achieved through a combination of increased x-ray pulse energy, and decreased pulse duration. Specifically, it requires extending the generated peak power from currently ∼100 GW to significantly above the TW level by increasing the pulse energy and simultaneously shortening the pulses to bandwidth-limited attosecond durations. This has the additional benefit of fully longitudinal coherent pulses. The x-ray intensity can be further increased by orders of magnitude through improvements in x-ray optics that would allow one to decrease the currently smallest achievable focus sizes of 50 nm to a sub-nm diffraction-limited spot with high efficiency, which would also be of interest for testing nonlinear quantum electrodynamics (QED) [110].
The signal count rates can also be increased by using sources that operate at a higher repetition rate, such as the European XFEL or LCLS-II, which can deliver beams with thousands to a million pulses per second, so long as backgrounds are kept low. A combination of monochromators and grazing incidence mirrors can be used to effectively suppress the FEL harmonics of the incident beam without dramatically affecting the spectral brightness. These new machines will allow new types of experiments, including studies of higher-order effects and the usage of more complex nonlinear processes for novel diagnostics. It will also be possible to use targets with lower densities, such as gas-phase experiments that would allow a full characterization of interaction dynamics. Such sources would be of advantage for investigations where a small signal per shot but overall high count rate is preferred, such as studies using entangled photons generated by parametric down conversion. Additionally, high-repetition rate sources are also expected to generate beams with smaller fluctuations.
An even more stable source could be achieved by using an XFEL oscillator [111], which uses a cavity to provide feedback, similar to those used by optical lasers. This would allow the generation of stable, fully coherent x-ray pulses at an extremely high repetition rate. Additional possibilities in the study of nonlinear effects can be accessed by XFELs that can generate more complex pulse structures, such as multi-pulse, multi-wavelength operation with a variable temporal delay between the pulses and where the frequencies can be tuned to a wide range and separation.
To take full advantage of such sources also requires the development of new diagnostics. Of critical importance for the observation of nonlinear x-ray effects is instrumentation with good rejection of FEL harmonics and low-noise diagnostics. Some examples include detectors that can operate at such high repetition rates, high-efficiency hard x-ray spectrometers or reaction microscopes, such as COLTRIMS.
Concluding remarks
Recent FEL experiments have demonstrated the possibility of driving nonlinear effects at hard x-ray wavelengths. Although the detailed interactions of x-rays and matter at high intensities is still in the process of being explored, progress in our theoretical understanding and advances in technology will certainly lead to novel tools and diagnostics for many research areas. The rapid developments of x-ray sources and the accompanying improvements in instrumentation will move this field from the proof-of-principle stage into a phase where the effects can be harnessed for many applications in fields where the structure and dynamics of matter on the atomic and electronic scale are of importance.
Acknowledgments
Work by MF is supported by the US Department of Energy, Office of Science, Basic Energy Sciences under Award #DE-SC0016494. Work by DR is supported by the AMOS program within the Chemical Sciences, Geosciences, and Biosciences Division of Basic Energy Sciences, US Department of Energy under Award #DE-AC02-76SF00515.
4.5. Electronic structure at high x-ray intensity
Robin Santra
DESY and University of Hamburg
Status
Until less than two decades ago, x-ray physics seemed incompatible with the concept of ultrafast science, which was driven by the rapid progress in optical (infrared and visible) technology. In particular, it seemed unnecessary to worry about processes where, in a single x-ray pulse, an atom absorbs more than one x-ray photon. However, now that more and more short-wavelength free-electron lasers are becoming available for user experiments, the situation has changed dramatically. Because x-rays interact predominantly with the electronic degrees of freedom of matter (in a way that ultimately affects outer-shell electrons as much as it affects inner-shell electrons), high-intensity x-ray science presents electronic-structure theorists with unprecedented challenges.
The importance of the electronic-structure problem in this context derives from the fact that the x-ray response of interacting many-electron systems governs the structure and dynamics of matter exposed to high-intensity x-rays. This matters for practically relevant applications of x-ray free-electron lasers such as biomolecular imaging. In such experiments, single-shot x-ray scattering patterns are recorded under conditions in which the x-ray absorption per non-hydrogen atom is practically saturated. Because x-ray scattering is scattering from electrons, and because the electrons are severely perturbed by high-intensity x-rays, the interplay of various processes giving rise to, and affecting, image formation is of both fundamental and practical interest.
For atoms, an experimentally well-tested approach to describing the electronic response to high-intensity x-rays is the model implemented within XATOM [2, 112]. XATOM computes the x-ray-driven time evolution of bound-electron configurations by numerically solving a system of coupled rate equations. For every bound-electron configuration arising during the x-ray pulse, the orbitals are re-optimized at the Hartree–Slater level. Using these individually optimized orbitals, XATOM computes, among other things, x-ray photoionization cross sections, Auger rates, and x-ray fluorescence rates. These quantities in turn determine the rates connecting pairs of bound-electron configurations in the system of coupled rate equations. For heavier atoms, millions to billions of distinct configurations are accessible, corresponding to millions to billions of rate equations. XATOM, therefore, solves such problems using a Monte Carlo strategy, calculating all electronic-structure information on the fly (i.e. only when needed).
Current and future challenges
The central challenge for electronic-structure theory in connection with XFELs is the quantitative treatment of the radiation-driven dynamics of polyatomic systems. In the case of bioimaging, 'polyatomic' can mean hundreds of thousands to millions of atoms. This places enormous constraints on the scaling, with system size, of the computational effort of the model adopted. Moreover, in contrast to small gas-phase systems, intramolecular impact ionization by photoelectrons and Auger electrons is particularly important. In addition, because of space-charge effects, only a fraction of all ionized electrons manage to escape from the system. As a consequence, the theoretical framework must be able to treat not only the valence and inner-shell electrons, but also a potentially large number of mutually interacting, ionized electrons.
An experimentally tested approach to addressing these challenges is the model implemented in XMDYN [112, 113]. XMDYN treats polyatomic systems by combining Monte Carlo and molecular-dynamics strategies with classical force fields. Within this framework, neutral atoms, atomic ions, and ionized electrons are treated as classical point particles. The force between a pair of charged particles is simply Coulombic; the force between two neutral atoms, or between a neutral atom and an atomic ion, is parametrized as in classical force-field calculations. The forces combined with Newton's equations of motion allow one to propagate the particles in phase space. Associated with each atom is not only its position and momentum, but also its bound-electron configuration at a given time. The ionization dynamics of the atoms are treated stochastically by using bound-electron-configuration-dependent cross sections and rates computed with XATOM. The main drawback of this approach is that it cannot account for effects related to dynamical changes of the chemical-bond network. XMDYN is part of the start-to-end simulation package for biomolecular imaging at the European XFEL [114].
When it comes to the development of short-wavelength nonlinear spectroscopy [75, 115], the challenges for electronic-structure theory are maybe even more severe. Spectroscopy fundamentally relies on the availability of quantitatively accurate theory. Theoretical single-photon x-ray spectroscopy is already rather difficult; it is obvious that the nonlinear regime has additional challenges in store. It must also be realized that nonlinear spectroscopy in ultrafast AMO physics is not limited to photon(s)-in photon(s)-out schemes; electron detection is equally important. In other words, restricting the theoretical analysis to the computation of the light-induced polarization (electric dipole moment per volume)—the standard in textbook nonlinear spectroscopy—is insufficient.
Advances in science and technology to meet challenges
A first step towards overcoming the molecular electronic-structure challenge in high-intensity x-ray physics is XMOLECULE [87]. XMOLECULE is similar in spirit to XATOM, but it actually treats the molecular electronic-structure problem for every bound-electron configuration accessible at a given x-ray photon energy and fluence. This involves the optimization of molecular orbitals associated with configurations that may contain one or more inner-shell vacancies. Because standard Gaussian basis sets are optimized for neutral species in their electronic ground state, molecular orbitals in XMOLECULE are expanded using a set of numerical atomic basis functions computed with XATOM. By eliminating a key shortcoming of XMDYN, it was possible, utilizing XMOLECULE, to identify a new ionization enhancement mechanism that involves ultrafast electron redistribution through chemical bonds [87].
Various further advances are still needed. Ultimately, in view of the need to push calculations in the direction of increasing molecular size and complexity, each electronic-structure calculations for a given bound-electron configuration must scale only linearly with the number of atoms. Furthermore, because the number of electronic configurations accessible also increases with system size, it may no longer be possible at some point to achieve an adequate sampling of the electronic configuration space. It could then be advantageous to adopt a more coarse-grained description of the electronic configuration space by using superconfigurations. In addition, many experimental applications of XFELs concern dynamical processes in liquids and on surfaces. Therefore, it is worthwhile exploring the inclusion of environmental effects in excited-state electronic-structure calculations.
Even though image formation in single-shot x-ray imaging is temporally gated by the x-ray pulse duration, this does not mean that the motion of atoms can always be neglected. Calculations on small biomolecules indicate that proton dynamics combined with non-Born–Oppenheimer effects can lead to electron redistribution on the time scale of one femtosecond [88]. For this reason, molecular electronic-structure theory must be accompanied by a strategy to treat nonadiabatic nuclear dynamics in an efficient manner. It is not clear that restricting this to mixed quantum-classical approaches is sufficient. Given the importance of proton dynamics in biological systems, there is a need to systematically investigate the role of nuclear quantum effects for the situations considered here.
A further advance required is the inclusion of relativistic effects in molecular calculations. In many molecular species, higher-Z atoms occur naturally or can be included through chemical synthesis. In connection with imaging via x-ray scattering, anomalous-diffraction effects connected to higher-Z atoms can play a particularly useful role. It is expected, based on isolated-atom calculations, that such effects provide a clean solution to the crystallographic phase problem, even at high x-ray intensity [85]. Therefore, a quantitative treatment of relativity is important. Finally, it should be realized that the unusual conditions arising in high-intensity x-ray imaging provide an opportunity to AMO physicists to re-examine the underlying conceptual foundations. It has been demonstrated that, even at low intensity, ultrafast x-ray scattering from electron wavepackets can hold surprises, which derive from the impossibility to separate electronically elastic and inelastic scattering events when using ultrafast x-ray pulses [116]. Hence, molecular electronic-structure theory in this field must go hand in hand with advanced theory for high-intensity x-ray scattering.
Concluding remarks
As may have become obvious based on what I have written above, I am advocating a systematic approach to software development for high-intensity x-ray AMO physics. The idea is to make available efficient multipurpose software that can reliably tackle a wide range of experimentally relevant problems. This type of software development strategy is rather common in quantum chemistry and in solid-state theory, but still not so much in AMO physics.
An important subject I have not touched upon in this contribution is the strong-correlation problem. (What is strong correlation to the condensed-matter physicist is static correlation to the quantum chemist.) In general, strong correlations affect the electronic structure of multiradical species, which are unavoidably formed when molecules are exposed to high-intensity x-ray pulses. This difficult topic provides natural opportunities for collaborations with scientists working in the areas of condensed-matter physics and ultracold AMO physics.
Acknowledgments
The thoughts presented in this contribution were shaped in part by discussions within the Collaborative Research Center SFB 925 and by my collaboration with Nora Berrah, Henry Chapman, Yi-Jen Chen, Gopal Dixit, Daria Gorelova, Kota Hanasaki, Yajiang Hao, Ludger Inhester, Zoltan Jurek, Antonia Karamatskou, Zheng Li, Adrian Mancuso, Michael Meyer, Daniel Rolles, Artem Rudenko, Sang-Kil Son, Koudai Toyota, Kiyoshi Ueda, Oriol Vendrell, Linda Young and Beata Ziaja.
5. Attosecond science with table-top sources
5.1. Table-top-scale coherent soft x-ray light sources based on high harmonic generation: new quantum light science and applications in nanoscience and nanotechnology
Henry Kapteyn and Margaret Murnane
JILA and University of Colorado, Boulder
Status
The HHG process makes it possible to generate light spanning UV to x-ray wavelengths using table-top-scale ultrafast lasers, with unprecedented temporal and spatial coherence. This capability not only makes possible new studies of ultrafast dynamics on the fastest timescales, but also allows for the small-scale implementation of many analytical, metrology, and microscopy techniques that in the past could only be implemented at large-scale light source facilities. As such, HHG light source technology is poised to become an enduring outcome of intense-field and ultrafast x-ray research.
In HHG, an intense femtosecond laser pulse is focused into a gas to create a nanoscale quantum antenna during the process of strong field ionization, thereby upconverting laser light to much shorter wavelengths. HHG for coherent XUV generation was first described in 1987 [117], with subsequent work clearly showing that HHG is a nonperturbative interaction [118]. The basic mechanism for HHG from single atoms was outlined in numerical [119] and then analytic [120] forms as essentially a coherent version of the Roentgen x-ray tube, through a 're-collision' process where the strong electric field of a laser pulls an electron away from an atom, then slams it back into that ionized atom. As such, the high-energy collision process is fully coherent, resulting in coherent dipole emission from each atom. Isolated attosecond pulse generation is a special case where the emission is confined to a single peak in the laser field [121, 122], although all HHG emission occurs in attosecond bursts. The strong field interaction that results in HHG was relatively quickly explained in increasingly elegant fashion [123], and current experimental work exploring the 'single atom' or single molecule physics has further confirmed, refined, and expanded on this.
However, the understanding of HHG as the most extreme nonlinear optical process has proven a grand challenge, unfolding slowly over the past two decades. The importance of phase matching for generating a coherent beam was recognized early, and elegant work revealed the influence of the generation process on phase-matching conditions [124]. However, the key to useful HHG was the realization that the conversion can in general be phase-matched, with phase matching occurring transiently in the time domain, i.e. at some time during ionization of the gas. This conceptual shift from conventional 'frequency-domain' NLO that requires, for example, equalizing static indices of refraction, has proven to be both conceptually and computationally challenging. However, the implications have been profound—the dynamics of the process itself not only represent the inception of attosecond science [125, 126], where sub-optical cycle dynamics drives the physics, but also where the physics intrinsically drives the HHG process toward shorter, rather than longer, pulses.
The key enabler for both practical application and conceptual understanding has been the ability to make intense few-cycle optical pulses, enabled first by Ti:sapphire laser technology [127]. Driving HHG in a highly transient regime revealed the fact, for example, that HHG phase matching is realized through generation in a weakly ionized gas [128]. In the 2010s, the ability to generate tunable mid-IR ultrashort pulses led to the further realization that HHG scaling made it possible to phase-match the process into the soft x-ray region of the spectrum (see figure 17) [129].
Figure 17. Phase matching of HHG. Experiment confirms predictions that, using longer wavelength driving lasers, produces phase-matched emission to shorter wavelengths for fully phase-matched HHG in weakly ionized gasses [129]. Further experiment and theory may reveal new regimes of efficient HHG [130]. Reprinted with permission from AAAS.
Download figure:
Standard image High-resolution imageThe current status of HHG as a light source is that we have good predictive power over optimization of HHG for simple phase matching in weakly ionized gases; optimally, the peak intensity of the focused beam should drive the ionization to the 'critical' (phase-matched) ionization level at the peak of the pulse [128, 131, 132]. The focus region must sustain this (constant) intensity over a propagation length corresponding to several absorption lengths of the generated light, and the gas region must end abruptly before the laser intensity decreases significantly. This sets a narrow optimum operating range that, depending on the gas and laser wavelength, results in conversion efficiency approaching 10−3 for the VUV, decreasing slowly to ∼10−7 for soft x-ray generation using mid-IR lasers.
Current and future challenges
Further advances continue in several areas. First, and likely foremost, the use of mid-IR (1.5–4 μm) ultrafast lasers now makes HHG sources possible for coherent soft x-rays, even to photon energies >1 keV. Second, new regimes of HHG that take advantage of new propagation physics, for example in using intense deep-UV lasers for HHG in more highly ionized gases [130] may reveal novel more efficient methods. Third, the generation of new polarization states represents new physics as well as an enabler for many applications [133, 134]. Fourth, as ultrafast laser technology becomes more flexible and robust, precise control over the spectrum of the emission will become routine. The unique combination of quantum coherence and macroscopic physics has made it possible both to optimize emission to a particular harmonic order using pulse shaping [126], and to generate the smooth supercontinuum spectrum of a single attosecond burst [135, 136]. However, these techniques are still too difficult for truly routine implementation in complex application experiments. Quasi-phase matching by colliding the driving laser with a weak controlling pulse [137], or sculpting the wave front of emission to generate a focusing HHG pulse, are also promising areas for expanding capabilities.
Growing range of applications in materials and molecular nanoscience and nanotechnology
Another significant recent advance is that commercial table-top EUV light sources can now generate a coherent flux comparable to synchrotrons—with much higher peak brightness and intrinsic synchronization to a femtosecond laser [138]. These sources are increasingly being used to drive new scientific understanding in a variety of areas, especially for materials science and nanotechnology. The breadth of applicability ranges from the practical, such as extending photoacoustic techniques to characterize few-nm mechanical and thermal properties of current relevance to industry [139, 140], or making use of the spatial coherence of HHG beams for unprecedented sub-wavelength resolution imaging in the EUV [141] (figure 18). Other new capabilities now allow us to measure fastest intrinsic charge and spin dynamics in materials on attosecond timescales for the first time, and in a band-specific manner. Recently, the first measurement of attosecond lifetimes of highly excited states in solids was made, with a lifetime of τ ∼ 210 as [142]. This technique also allows the fastest charge screening and scattering to be distinguished. XAFS spectroscopy is also just becoming possible with very broad bandwidth soft x-ray HHG.

Figure 18. HHG as a unique light source to uncover new science and technology at the spatio-temporal limits. Coherent diffractive imaging with sub-wavelength resolution [141]. The lifetime of highly excited states in solids can be directly measured, confirming 'final state' effects in photoemission spectroscopy [142].
Download figure:
Standard image High-resolution imageAdvances in science and technology to meet challenges
One primary technology challenge for advancing HHG sources is in laser development; robust, high-average-power ultrafast lasers will advance the field. Such lasers in the mid-IR, for soft x-ray generation, are especially needed. Although we know that HHG can, in principle, provide usable sources into the soft- and even hard x-ray region of the spectrum, actual implementation will be a continued technological challenge. Mid-IR ultrafast lasers are typically implemented through parametric down conversion (PDC) from shorter wavelengths. However, the peak intensity requirement for HHG is approximately constant while the spot size for a given confocal parameter of the laser increases. Furthermore, the density–length product required for optimal conversion rapidly increases. All these parameters push the pulse energy requirements rapidly higher in moving from EUV to x-ray HHG. The technology that can mitigate this issue is to develop low-loss waveguides that can confined the gas and guide the laser. For HHG into the EUV, simple hollow capillary waveguides can extend the confocal parameter of propagation by 5–10×, while optimizing confinement of the nonlinear medium and minimizing gas flow. This makes it easier to obtain optimal conversion efficiency with modest driving pulse energy. For VUV generation, guiding is often not needed and a cell geometry is suitable; however, for soft- and ultimately hard x-ray HHG, hollow waveguides that can confine the intense infrared light with low loss will represent an increasingly critical enabler.
As discussed previously, more-robust technologies for pulse shaping and manipulation, especially in the mid-IR, promise to find increasing utility. However, it has become clear that group-velocity walk-off issues limit the utility of extremely-short, quasi-single-cycle pulses for HHG [143]. Importantly, this does not preclude isolated attosecond bursts, as the physics naturally drives the process in this direction even when using multi-cycle driving pulses [144]. One area where new concepts are needed is for generating narrow-band HHG in the soft x-ray region.
Another area where progress is crucial is in developing precision predictive power for HHG computation. HHG calculations represent an elegant combination of atomic quantum dynamics with macroscopic electromagnetic propagation, with the scale lengths spanning many orders of magnitude. This diversity in scale length rapidly increases for IR-driven HHG. Furthermore, effects such as filamentation will also be increasingly relevant; there is a clear potential for computation to predict 'sweet-spot' regimes where filamentation and phase-matched HHG coincide, or where novel tapered waveguides can increase efficiency. Development of computationally efficient methods to address such a broad range of scale lengths thus deserves to be considered a grand challenge in computational physics.
Finally, the ultimate high-energy limit for the HHG process has not yet been identified. It is fully possible that intense light pulses at >10 μm wavelength, and in the THz region of the spectrum, will generate coherent hard x-rays [145]. Such an eventuality could revolutionize medical x-ray technology, taking it to its ultimate limits of resolution and dose. These sources would also find use for screening applications, making x-ray technology safe to use even in public for security.
Concluding remarks
In conclusion, it has become more and more clear that the science and technology of HHG-based coherent x-rays is open-ended. As we learn more, further opportunities and challenges present themselves. In the broader context, coherent x-ray science and technology can trace its roots to 'big' science projects such as synchrotrons, or the 'Excalibur' project that drove an x-ray laser with a nuclear explosion [131], and more recently to the extraordinary achievement of the LCLS XFEL. At present, the ability to generate coherent x-rays on a table top is producing new science just as compelling as that done at the large facilities, and further with the potential to take a broad role as a technology that can proliferate into hundreds or thousands of instruments, facilities, and capabilities in science and nanotechnology. To date, at least in the US, this potential has yet to be acknowledged in a concerted way by the funding agencies. In short, an exciting future for ultrafast and coherent x-ray science is smaller, cheaper, and (ultra)faster.
Acknowledgments
The work described herein was demonstrated over a period >10 years. We gratefully acknowledge funding by the National Science Foundation (ERC ENG- 0310717; PHY-1125844; STC DMR-1548924), the Department of Energy (BES AMOS), the DARPA PULSE program (W31P4Q-13-1-0015), and through the Gordon and Betty Moore Foundation's EPiQS Initiative through Grant GBMF.
5.2. Back to the future—long wavelength lasers
Heide Ibrahim and François Légaré
Institut National de la Recherche Scientifique, Centre Énergie, Matériaux, et Télécommunications
Status
Until the end of the 1980s, the workhorse for strong-field laser science was the CO2 laser delivering high-energy pulses that could be as short as 600 fs (=20 optical cycles) [146]. Since the ponderomotive energy (Up) of charged particles in a laser field scales with the square of the laser wavelength, intense CO2 sources near 10 μm provide the capability to build compact, high-energy accelerators.
Despite the progress with CO2 lasers, the ultrafast scientific community has widely adopted the femtosecond titanium-sapphire (Ti-Sa) laser based on the chirped-pulse amplification (CPA) technique [147]. Nowadays, this commercial technology is widely available providing ultrashort pulse durations directly at the output of the amplifier (sub-15 fs), from PW peak power at a few Hz to repetition rates up to the 100 kHz level at lower energy per pulse. Furthermore, with the advances realized in the 1990s with frequency combs, hollow core fiber (HCF) compression techniques, and ultra-broadband chirped mirrors, carrier-envelope phase (CEP) stabilized NIR/VIS sub-cycle pulses are now available at kHz repetition rate and mJ level [148].
Ti-Sa lasers successfully drive secondary sources of photons and particles; using a variety of nonlinear optical processes, the spectral range from the THz [149] to hard x-rays becomes available. The latter is accessed via betatron radiation [150] while HHG can provide pulse durations down to tens of attoseconds in the XUV range [148]. This provides a unique rainbow of photons to pump, control, and image ultrafast dynamics in materials.
The future dream is to combine the benefits of large ponderomotive energies of IR lasers with the fs pulse duration obtained with Ti-Sa lasers. To partially bridge this gap, optical parametric amplifiers (OPAs) became extensively used over the last decade. Pumped by Ti-Sa lasers, OPAs provide mJ level pulses in the spectral range of 1.2 to 2.4 μm. Longer wavelengths from 3 to 30 μm can be reached using a second difference frequency generation step, however at much lower output pulse energy (∼100 μJ). The use of these OPAs has opened new research avenues in strong-field laser science, but has also provided a unique tool to scale the HHG cut-off, and thus to generate ultrashort soft x-ray pulses up to the 1 keV range of photon energies (see sections 5.1 and [129]). Pertot et al have recently used such a source to time resolve gas-phase molecular dynamics using transient x-ray absorption spectroscopy at the carbon K-edge and sulfur L-edges (see section 5.5 and [151]).
Current and future challenges
The bottleneck for all types of table-top laser driven x-ray sources has always been the achieved flux and photon energy. Increasing their performance has pushed the development of lasers delivering: a) higher peak and average power, with ps to fs pulse duration, in a challenging combination of b) CEP stabilized few-cycle pulses at long fundamental wavelength. These are the most promising tools to scale such HHG-based soft x-ray sources with attosecond pulse duration.
With this objective, few laboratories in the world, including the Advanced Laser Light Source ALLS (Varennes, Canada) and Riken (Tokyo, Japan) are working on upscaling conventional OPAs to the range of 10 mJ per pulse at up to 100 Hz repetition rate [152, 153]. In addition, few-cycle pulses near 2 μm, together with passive CEP stabilization have been generated with HCF compression [154]. Another approach to generate CEP stabilized few-cycle pulses in this spectral range is through optical parametric chirped-pulse amplification (OPCPA), pumped by Ti-Sa lasers [155]. With these OPA/OPCPA sources, half-cycle cut-offs in the water window spectral range have been observed, confirming the generation of isolated attosecond x-ray pulses in the water window [155, 156].
Amplifying these few-cycle pulses represents a major challenge in ultrafast optics as most amplifiers introduce gain narrowing leading to spectral losses. To maintain and efficiently amplify few-cycle pulses, our team has recently demonstrated the concept of frequency-domain optical parametric amplification (FOPA) [157]. Rather than amplifying the ultrashort pulses in the time domain, as performed in CPA and OPCPA, FOPA divides the task of amplification over multiple crystals installed in the Fourier plane of a 4f setup (see figure 19). In this plane, the pulses are of ps duration, suitable for amplification with Joule level ps pump pulses. After amplification, a subsequent transformation from frequency to time domain brings the pulses back to their input pulse duration. In a first attempt to demonstrate the concept of FOPA, two-cycle 1.8 μm pulses have been amplified from 0.1 to 1.5 mJ. Recently, FOPA has been modified and pumped by 250 mJ ps pulses, delivering ∼30 mJ two-cycle, 1.8 μm pulses [158], representing ∼2.5 TW of peak power. The current Fourier plane can be further upscaled in dimension to allow a Joule level of pumping to reach 10 TW few-cycle pulses. While this extension nicely demonstrates the capability of FOPA to reach high peak power few-cycle IR pulses, the current repetition rate is limited to 10 Hz as the pump laser is based on Ti-Sa technology.
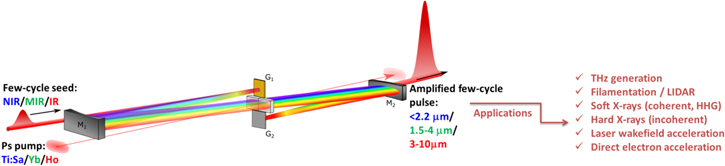
Figure 19. FOPA for different spectral regimes: NIR, pumped by Ti:Sa lasers, leading to amplified pulses up to 2.2 μm (blue); MIR, pumped by Yb lasers reaching 1.5–4 μm (green) to the IR up to 10 μm when pumped by Ho lasers (red). Possible applications are indicated on the right. The crystal between focusing mirrors M1, M2 and gratings G1, G2 indicates the Fourier domain of interaction.
Download figure:
Standard image High-resolution imageTo reach photon energies up to keV with HHG, a pump wavelength around 2 μm is still too short. Using a mid-infrared (MIR) laser based on OPCPA (10mJ, 80 fs, 3.9 μm, 20 Hz), phase-matched HHG up to 1.6 keV of photon energy has been observed [129]. For the other extreme of the electromagnetic spectra, the THz spectral range, scaling the Up allows the generation of high-field single-cycle pulses based on a two-color driven plasma source, where the energy per THz pulse scales nearly with  [159]. Combining both ends of the electromagnetic spectra into a single beamline would provide a unique tool to image and control ultrafast dynamics in materials. To enable this forefront science, the main challenge at the horizon of 2020 consists in scaling the average power of few-cycle MIR lasers.
[159]. Combining both ends of the electromagnetic spectra into a single beamline would provide a unique tool to image and control ultrafast dynamics in materials. To enable this forefront science, the main challenge at the horizon of 2020 consists in scaling the average power of few-cycle MIR lasers.
Advances in science and technology to meet challenges of today
With the progress realized over the last decade, it is clear that the MIR laser bears the potential to become the future workhorse for strong-field laser science. Ti-Sa lasers are simply not suitable to generate high-energy pulses above 2.5 μm, with high average power, so a technology change is required. For the future laser system we envision it is instructive to look into the technical details of the 3.9 μm 20 Hz OPCPA discussed in [129]. A Yb:KGW oscillator seeds two amplifiers: Amp1 (Yb:CaF2, fs) and Amp2 (Nd:YAG, ps). Following Amp1, IR seed pulses are generated through OPA and further amplified in an OPCPA with Amp2. While this MIR laser suffers from low average power, it offers the unique opportunity of scaling its repetition rate using a recently developed high average power Yb ps laser system. Cryogenically cooled Yb:YAG technology has recently proven 1 J per pulse at 500 Hz within 5 ps duration [160]. Considering the recent conversion efficiency obtained at 3.9 μm of ∼5%, a 50 mJ class MIR laser at 500 Hz is reachable based on existing Yb laser technologies.
While these high-energy MIR pulses are attractive to push the HHG cut-off to the keV spectral range, the generation of isolated attosecond pulses at high photon energies requires high-energy CEP stabilized few-cycle MIR pulses. For this, Fan et al have recently demonstrated the extension of the HCF compression approach to the MIR by compressing the output of an Yb:KGW pumped OPA, reaching CEP stabilized, few mJ level, 20 fs pulses at 3 μm [161].
Over the coming years, the challenge is to merge FOPA and Yb technologies: (i) generating the CEP stabilized MIR seed pulses, (ii) compressing these pulses to two-cycle duration in an HCF, to (iii) seed a FOPA pumped by high-energy Yb lasers. Our goal is to generate CEP stabilized 20 fs MIR pulses with >50 mJ at 500 Hz repetition rate, representing >35 W of average power. Through a cascade of nonlinear optical processes, and such a laser will provide bright CEP stabilized ultrashort pulses from the THz to the keV soft x-ray spectral range. Furthermore, high-energy MIR lasers are also ideal for direct electron acceleration to generate sub-cycle electron bunches [162] and incoherent sub-100 fs hard x-ray pulses [163] for ultrafast diffraction imaging.
Concluding remarks
With these modern table-top MIR lasers, it will be possible to use simultaneously different spectral ranges across a coherent rainbow of about 18 octaves, to manipulate and image ultrafast dynamics in materials (see figure 19). Two of the envisioned experiments are: (i) THz pulses provide a unique tool to control the fundamental properties of matter, such as magnetization, ferroelectricity, and optical properties. Combined with the soft x-ray pulses and electron bunches (or hard x-rays), THz induced transient dynamics (structural and electronic) will be imaged with high spatio-temporal resolution. (ii) In atomic and molecular physics, combining both ends of the electromagnetic spectra, THz and soft x-rays, will allow imaging ultrafast electronic dynamics, through THz streaking of the photoelectrons emitted by core-shell photoionization.
To come full circle and look ahead, the current development of high-energy ps Ho:YAG systems will enable us to further scale these MIR lasers up to 10 μm wavelength and beyond [164]. We are assisting the return of the long wavelength lasers, with ultrashort pulse duration, high peak and average power for a bright coherent rainbow generated table top.
Acknowledgments
The authors acknowledge valuable discussions with Drs Vincent Gruson and Bruno E Schmidt.
5.3. Applications of lab-scale, ultrashort XUV pulses
Marc Vrakking
Max-Born Institute, Berlin
Status
HHG was discovered in the late 1980s, in the course of experiments targeting a better understanding of the role of intermediate resonances in strong-field multiphoton ionization [165]. After a while, it was understood that many of the characteristic features of HHG, in particular the occurrence of a high-energy cut-off in the XUV spectrum, could be understood by a three-step picture, where (tunnel) ionization by an intense femtosecond laser sets an electron free, subsequent acceleration of the electron by the oscillatory laser electric field increases the kinetic energy of the electron and drives a re-collision process, and recombination leads to the production of XUV photons [120]. Although the potential benefits of HHG radiation for time-resolved spectroscopy were immediately realized, the application of HHG radiation in pump–probe experiments remained rare for more than a decade due to a number of unsolved technical problems that frustrated successful implementation.
Soon after the introduction of the three-step picture, it was realized that HHG produces the XUV radiation in a phase-locked manner that implies the production of attosecond laser pulses. The initial ionization step in the three-step picture depends highly nonlinearly on the laser electric field, and hence produces bunches of electrons with a duration much shorter than the duration of the driver laser optical cycle (2.7 fs for a Ti:Sa laser). As a result, the recombination that produces the XUV light also only occurs during a small fraction of the optical cycle. It follows that the production of attosecond pulses is inherent to the process of HHG.
The first demonstration of attosecond pulses in 2001 [166, 167] motivated experimental efforts by numerous research groups around the world. This led to substantial progress, not only regarding the generation, characterization and application of attosecond laser pulses, but, as a consequence, regarding the application of HHG radiation in general. Today, lab-scale HHG sources have developed into extremely versatile and powerful sources that can be used in a multitude of different ways as shown in figure 20, complementing research efforts that are undertaken in synchrotrons and free-electron lasers. I will review some of these applications, and describe where further developments in the next 5–10 years are likely to occur.
Figure 20. Evolution of CEP-stable few-cycle lasers, which are the most suitable driver lasers in attosecond experiments, towards higher pulse energy and higher repetition rates, as illustrated by a series of representative laser systems that were recently completed or are currently under development.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Attosecond XUV science
Although this may soon change, HHG has been, up to now, the only technique capable of producing attosecond laser pulses that can be used in pump–probe experiments. First attosecond pump–probe experiments targeting time-resolved electron dynamics focused on atomic systems (for a review see, e.g [168]). More recent experiments have addressed correlated two-electron dynamics [169], attosecond to few-femtosecond charge migration in molecules [170, 171] and electric field-induced charge separation in solids [172]. In almost all of these experiments, attosecond time resolution is accomplished by using the attosecond pulses in combination with NIR laser pulses (typically, a replica of the laser used to drive the HHG process) whose optical period is used as a clock with attosecond time resolution.

Figure 21. Time-resolved magnetic circular dichroism dynamics around the M2,3 edge of Co (60.8 eV). The nonlinear least-square fit reveals a mono-exponential decay with a demagnetization time constant of (92 ± 22) fs, convoluted with a 59 fs instrument response function. Reprinted figure with permission from [173], Copyright 2015 by the American Physical Society.
Download figure:
Standard image High-resolution imageIn the next few years attosecond science will likely develop in the following directions.
- –Attosecond XUV pump–attosecond XUV probe experiments: using multi-Terawatt (TW) Ti:Sapphire lasers or multi-TW few-cycle OPCPA, μJ-level attosecond pulses may in future be generated that can be focused to intensities high enough to ensure that the sample under investigation absorbs one or more photons from both the attosecond pump and the attosecond probe laser [174], greatly widening the scope of possibilities in attosecond experiments.
- –Attosecond experiments at high repetition rates: higher repetition rates, based on the implementation of fiber- or OPCPA-based laser architectures [175, 176], will permit the widespread application of coincidence methods in attosecond science. Given the large bandwidth of attosecond pulses, a multitude of processes are simultaneously excited and probed in an attosecond experiment, requiring a 'high-energy physics' approach to isolate the dynamics of interest.
- –Attosecond experiments at higher photon energies: attosecond pulses generated by HHG inherently operate in the XUV/x-ray wavelength domain, the domain where photoabsorption becomes increasingly core-level specific (and hence atom-specific). This allows experiments where XUV absorption of selected atoms is used as a site-specific 'detector' for particular electronic processes of interest. First results in this direction have been obtained in our laboratory at MBI (in preparation).
Femtosecond XUV science
Many of the benefits of HHG that were discussed above are beneficial beyond attosecond science, and are increasingly being exploited in femtosecond time-resolved experiments.
- –HHG as a tool for transient-absorption spectroscopy. Scaling of, in particular, the wavelength of the driver laser used in the HHG process has recently allowed increasing the high-energy cut-off of HHG spectra [129], and these days an increasing number of research groups are beginning to use extremely broadband XUV continua that extend into the water window, i.e. the wavelength region where light elements like C, N and O have their K-edges [151]. This permits exploiting atom-specific core-level absorption in experiments addressing diverse scientific interests such as chemical dynamics [177] and condensed phase physics [173, 178]. An example from our lab, revealing demagnetization dynamics in a thin magnetic film, measured by monitoring the absorption of circularly polarized harmonics around the Co M-edge, is shown in figure 21 [173].
- –HHG as a tool for time-resolved photoelectron spectroscopy. While the full XUV bandwidth generated in the HHG process supports attosecond pulses, the duration of single harmonics is in the femtosecond domain (typically about half of the duration of the HHG driver laser). In the last few years, time-compensating monochromators have been developed that allow the selection of a single harmonic without noticeable changes to the XUV pulse duration [179], providing a unique tool for time-resolved photoelectron spectroscopy, where XUV photon energies that exceed atomic and molecular ionization potentials provide universal detection of both ground and excited states [180]. Using this tool, we have, e.g., been able to investigate interference stabilization in multiple overlapping Fano resonances [181].
- –HHG as a tool for driving nonlinear processes and coherent diffractive imaging (CDI). While early HHG sources typically allowed the generation of focused XUV intensities <1011 W cm−2, significant progress in the last few years has allowed the production of intensities well beyond 1012 W cm−2. This has permitted the emergence of two significant and novel capabilities, namely (1) the ability to drive nonlinear ionization/excitation processes (i.e. laser–matter interactions where the sample absorbs more than one photon), and (2) the ability to use the HHG pulses for single-shot CDI measurements. As an example of (1), we have measured ionization and recombination dynamics in large rare-gas clusters [182, 183] and have observed XUV-induced Rabi oscillations in two-color double ionization of Ar [184], whereas as an example of (2), single-shot single-particle CDI measurements have been performed on sub-micron size helium droplets [100].
Advances in science and technology to meet challenges
Almost all the work in attosecond science up to now has been achieved using relatively standard, commercially available femtosecond laser amplifiers. However, substantially more will become possible in the near future, when—increasingly—pulse-compressed fiber lasers and inherently few-cycle OPCPA drivers will become available that are superior to commercially available laser amplifiers in many respects (average power, repetition rate, pulse duration, stability, etc). This will create breakthrough opportunities in the use of attosecond pulses for exploring electron dynamics in atoms, molecules, nanoparticles, liquids and solids, and will provide a further boost to the exploitation of HHG-based methods in numerous laboratories around the world.
5.4. Probing photoionization dynamics in atomic systems with attosecond pulses
Marcus Isinger, David Kroon, Mathieu Gisselbrecht and Anne L'Huillier
Physics Department, Lund University
Status
The electron dynamics following a photoionization event induced by a short XUV pulse is a topic that has attracted a lot of attention within the science community during the last few years. A series of recent experimental reports [185–193] showed that we now have the ability to characterize an escaping photoelectron wavepacket and its interaction with the ionic core. This also triggered theoretical studies of this interaction as well as of the effect of probing the system [187, 190, 194]. In this roadmap, we describe the status and challenges regarding both non-resonant [185–190] and resonant [191–193] photoionization dynamics in atoms. In the first case, we consider extremely fast dynamics, approximately tens of attoseconds; the corresponding bandwidth is large, a few eV or tens of eV; in the second case, the dynamics is typically several femtoseconds; we are discussing resonant features of the order of a fraction of eV.
Photoionization can be studied by pump/probe schemes in combination with photoelectron spectroscopy [185–193]. Pump/probe schemes often use either single attosecond pulses together with a rather intense infrared probe pulse [185, 186, 191] or an attosecond pulse train together with a weak infrared pulse [187–190, 192, 193]. We focus below on the latter technique, often referred to as RABITT [167] but we note that the results and challenges are quite general.
In ultrafast optics, an ultrashort pulse acquires a group delay when traveling through a dispersive medium, since the different frequency components of the pulse travel at different velocities. This group delay is the derivative of the spectral phase, dependent on the dispersion (and length) of the medium. Similarly, an electronic ionized wavepacket created by absorbing an attosecond pulse acquires a group delay when propagating through the atomic (or molecular) potential. This delay, early introduced by Wigner for scattering, is the derivative with respect to energy of the phase of the photoionization probability amplitude.
As with ultrashort optical pulses, measuring the group delay of an electronic wavepacket can be done by observation of interference between its different spectral components. This interference is achieved via optical cross-correlation using a two-photon process. In one arm of an optical interferometer, we generate high-order harmonics in a gas cell, while in the other arm, we finely adjust the delay of a small fraction of the IR laser field used for the generation. We combine the XUV and IR fields and focus them into an interaction chamber containing a gas target and a photoelectron spectrometer. Ionization of the gas by the odd-order harmonics leads to electron peaks with energy equal to nℏω−Ip, where n is an odd integer, Ip the ionization energy and ω the laser frequency. Additional absorption or emission of an IR photon leads to sideband peaks at energies nℏω−Ip, with n even. Because each sideband state can be reached by two quantum paths involving different harmonics, the probability amplitudes interfere and the sideband signal S oscillates as a function of delay τ between the XUV and IR fields as S = A + Bcos(2ωτ − Δϕ). The experiment consists in measuring the variation of the phase of these oscillations: Δϕ (radians) or Δϕ/2ω (seconds), which approximates the time delay τ2 = dϕ/dω.
Figure 22 shows the difference in the time delays corresponding to ionization in the 3s and 3p ionization of argon [187]. The calculations agree well with each other and with the experimental results for sideband 22 (34 eV) and 24 (37 eV) but deviate for sideband 26 (40 eV). This region is close to the Cooper minimum of the 3s shell, where correlation effects between the 3s and 3p subshells are important and affect the phase of the ionization wavepacket in a critical way. The difference between the calculations indicates the difficulty of describing accurately this spectral region.
Figure 22. Measured difference in photoionization time delays from the 3s and 3p shells in Ar (solid dots). The lines refer to different calculations. (Adapted figure with permission from [187], Copyright 2012 by the American Physical Society; additional theoretical result from [195]).
Download figure:
Standard image High-resolution imageIn figure 23, we examine the effect of resonance on the photoionization dynamics [191–193]. Here, we tune the laser wavelength so that 17ℏω spans the 3s−14p quasibound state and we show the phase variation of sideband 16 as a function of the 17th harmonic photon energy. The results shown in figure 23 [192] represent the phase distortion of the ionized electron wavepacket due to the presence of a discrete state, which decays by autoionization.

Figure 23. Phase variation of sideband 16 as a function of the energy of harmonic 17, close to the 3s−14p quasibound state. Theoretical results are indicated by green and red solid lines, whereas the experimental results are shown by black symbols. (Adapted by permission from Macmillan Publishers Ltd: Nature Communications [192], Copyright 2016).
Download figure:
Standard image High-resolution imageCurrent and future challenges
The delay τ mentioned above is actually the sum of three terms: τ2 = τXUV + τ1 + τcc. The first term represents the effect of the broadband XUV field. To access the ionization dynamics (τ1 + τcc), we need a precise characterization of attosecond pulses in an independent measurement. This is a major challenge, which we circumvent by performing two measurements simultaneously and taking the difference, as done in figure 22. As we use the same excitation pulses for ionization from the two subshells, τXUV is eliminated. Other experiments compare single and double ionization [188], or different gases [189–191]. The price to pay is that we cannot determine absolute photoionization delays, except if we assume that one of the delays can by sufficiently accurately calculated to serve as an absolute reference [186].
The next challenge is that our measurement utilizes a two-photon ionization process. In some conditions, the two-photon delay can be written as τ1 + τcc, where τcc is a correction to the photoionization time delay τ1 due to the influence of the probe field and the induced continuum–continuum transitions. In many cases, τcc can be accurately calculated (e.g. in the case shown in figure 22, its contribution amounts to −70 as). It becomes very large at low kinetic energies, thus preventing precise time delay measurements [188, 190]. In the small energy region considered in figure 23, both τXUV and τcc can be assumed to be constant and do not affect the measurement of the phase variation.
A third challenge appears when multiple angular channels contribute to the ionization probability amplitude. The measured angular-integrated signal then consists of an incoherent sum over the contributions. Unless one of the channels is dominant, the interpretation of the experimental results in terms of photoionization time delays (figure 22) [196] or phase retrieval (figure 23) becomes ambiguous. For example, the case of 3s → εp → εs or εd does not lead to any difficulty since there is only one channel in the intermediate state. By contrast, in the case of 3p → (εs, εd) → (εp, εf), two channels with different phase behaviors add incoherently, since both (εs, εd) contribute to the final state εp, while only εd leads to εf in the final state. Note that in the case presented above, the 3p → εd channel is mostly dominant.
To these challenges, which are coupled to the physics of the measurements, we should add more technical challenges related to the precision and stability of the optical interferometer, the laser repetition rate, which affects the length (and therefore reliability) of the measurements, the available spectral range and the resolution of the electron spectrometer.
Advances in science and technology to meet challenges
The scientific community is continuously, in various ways, striving to overcome these challenges. Using new laser technology based upon optical parametric amplification, compact and stable laser sources with repetition rate of a few 100 kHz, short pulse duration, a few hundreds of μJ energy and wavelength from the near to the mid-infrared range will soon be available for attosecond measurements in atomic and molecular physics such as those mentioned in this roadmap.
This increased repetition rate should make the use of two- or three-dimensional electron spectrometers more efficient. They will allow us to obtain angular information, solving in particular the issue of multiple channel contributions mentioned above. Coincidence techniques [188, 190, 191] will be used more routinely while keeping reasonable acquisition times, allowing, e.g., the determination of absolute delays by comparing with He [186], as well as the separation of multiple channels.
This roadmap would not be complete without mentioning the increasing theoretical effort to calculate accurate atomic and molecular temporal/phase data and to simulate the experimental measurements, investigating the validity of the approximations sometimes used to extract the physics information [194].
Concluding remarks
One of the most interesting applications of attosecond pulse technology is the possibility to measure phases (or phase derivatives, i.e. delays) of quantum mechanical probability amplitudes. Such measurements allow us to completely characterize electron wavepackets in the spectral or temporal domain, giving thus access to the temporal dynamics. The door is now open to the investigation of more complex problems such as the dynamics of electrons in inner shells or the migration of charge in molecules. With improved spectral range and temporal precision, we will get much more insight into the dynamics of correlated electrons in matter.
Acknowledgments
The authors acknowledge support from the European Research Council, the Swedish Research Council and the Knut and Alice Wallenberg Foundation.
5.5. Attosecond x-ray spectroscopy: from isolated molecules to the liquid phase
Hans Jakob Wörner
ETH Zürich
Status
The generation and characterization of attosecond pulses with durations reaching down to 43 as [197] has provided access to the dynamics of electrons, representing the most fundamental level of AMO physics and chemistry. Attosecond time-resolved spectroscopy is therefore expected to provide a deeper understanding of electronic motion, in particular of the effects of electron correlation, which defines many molecular properties. The two main branches of attosecond spectroscopy are (i) pump–probe measurements combining attosecond pulses with near-infrared femtosecond pulses and (ii) high-harmonic spectroscopy, which exploits the mechanism of HHG to record attosecond dynamics taking place in the generation medium. In both branches, important breakthroughs have been achieved, as summarized in recent reviews [198, 199]. Most recently, attosecond photoionization delays have been measured in molecules [200] and high-harmonic spectroscopy has been developed further to reconstruct charge migration in spatially oriented molecules on attosecond timescales [171]. As shown in figure 24, an electron hole created on an iodine atom by strong-field ionization was found to migrate across the iodoacetylene cation in less than 1 fs.
Figure 24. Reconstruction of charge migration in the iodoacetylene cation from high-harmonic spectroscopy. Figure adapted from [171] with permission from AAAS.
Download figure:
Standard image High-resolution imageCurrent and future challenges
All of the fundamental advances in attosecond science have so far been realized in the XUV. The traditionally used titanium:sapphire laser systems indeed limit the practically applicable radiation to photon energies below ∼100 eV. In this spectral region spectroscopy is complicated by the high density of molecular valence states and its interpretation is challenging due to the delocalized nature of the associated electronic wave functions. A detailed understanding of molecular spectroscopy is however a prerequisite for both the planning and the interpretation of attosecond time-resolved measurements. This challenge has been addressed by extensive computational work in the past (see e.g. [170, 171]), but will clearly impose a limit on the complexity of the samples that can be investigated in the future.
This challenge is one reason why frequency-domain spectroscopy of complex systems is often realized in the x-ray domain [201]. Short-wavelength radiation gives access to inner shells that have element-specific binding energies. X-ray measurements at atom-specific absorption edges therefore provide information about both the electronic and the structural environment of the probed atom. An extension of attosecond spectroscopy to the x-ray domain therefore appears to be the most promising future direction in this area of research.
Two families of light sources are actively being developed to achieve this goal. High-harmonic sources, which are being extended to the soft x-ray domain [129, 202, 203], are in principle clearly capable of achieving attosecond durations and provide pulses that are perfectly synchronized with the driving laser pulse. These sources, however, remain relatively weak. FELs are much more intense, but struggle to achieve very short pulse durations, as well as a good temporal synchronization with laser pulses.
Understanding the attosecond dynamics of electrons in isolated molecules is an important first step, which is still far from being fully realized. Nevertheless, an even more important goal is to understand the dynamics of molecules in solution, where most chemical and biophysical processes take place. Moving attosecond spectroscopy from the gas phase to the liquid phase represents a huge challenge. It is, however, necessary to obtain the information required to understand the dynamics of molecules in their natural environment, which will in general significantly differ from that of isolated molecules in the gas phase.
Advances in science and technology to meet challenges
The weakness of high-harmonic sources in the soft x-ray domain has so far prevented their application to time-resolved measurements. Very recently, the first soft x-ray transient-absorption measurements have been realized on the femtosecond time scale [151] (see also section 5.6). Figure 25 shows the time-dependent x-ray absorption spectra at the carbon K-edge that trace in time the dissociation dynamics of  created by strong-field ionization, to
created by strong-field ionization, to  and F. The lowering of molecular symmetry from Td to D3h manifests itself as a splitting of the dominant absorption band. This progress has been enabled by a relatively modest increase in the average power of the driving mid-infrared laser source to ∼2.5 W by using a 20 W Ti:Sa laser system to pump an optical parametric amplifier. Much higher average powers will be achievable using OPCPAs pumped by high-average-power lasers. The approach followed by our group is to combine a recently developed 500 W Yb pump laser [204] with the principle of FOPA (see section 5.2) [157] to directly amplify two-cycle mid-infrared pulses to an average power of 50 W. Such a laser source will enable the development of many attosecond time-resolved spectroscopies in the water window (284–543 eV) and beyond.
and F. The lowering of molecular symmetry from Td to D3h manifests itself as a splitting of the dominant absorption band. This progress has been enabled by a relatively modest increase in the average power of the driving mid-infrared laser source to ∼2.5 W by using a 20 W Ti:Sa laser system to pump an optical parametric amplifier. Much higher average powers will be achievable using OPCPAs pumped by high-average-power lasers. The approach followed by our group is to combine a recently developed 500 W Yb pump laser [204] with the principle of FOPA (see section 5.2) [157] to directly amplify two-cycle mid-infrared pulses to an average power of 50 W. Such a laser source will enable the development of many attosecond time-resolved spectroscopies in the water window (284–543 eV) and beyond.

Figure 25. X-ray absorption spectroscopy with a high-harmonic source. The dissociative strong-field ionization of CF4 was studied at the K-edge of carbon. Figure adapted from [151] with permission from AAAS.
Download figure:
Standard image High-resolution imageThe second challenge, the extension of attosecond science to the liquid phase, has also been successfully addressed in the XUV spectral region [205]. The development of high-vacuum-compatible liquid microjets with diameters of tens of micrometers had previously enabled photoelectron spectroscopy to be performed on liquids and solutions [206]. The combination of a liquid microjet with an attosecond light source has enabled our group to demonstrate attosecond time-resolved photoelectron spectroscopy. This new technique was recently applied to measure photoemission delays between liquid and gaseous water.
Attosecond time-resolved spectroscopy in the liquid phase will greatly benefit from an extension to the x-ray domain because water is highly transmissive in the water window where the absorption edges of carbon (284 eV) and nitrogen (410 eV) lie. This will enable attosecond transient-absorption spectroscopy to be performed on liquid samples. A crucial development towards this goal is the recent invention of a flat microjet that enables the generation of thin 'liquid sheets' with thicknesses below 1 μm [207].
New opportunities will also arise from the combination of HHG- and FEL-based light sources. A particularly attractive route will consist in combining the specific advantages of both types of light sources. We envision the construction of an experimental AMO end station at the Swiss-FEL that combines narrowband, intense FEL pulses to ionize the sample from specific core shells with ultra-broadband attosecond supercontinua covering the spectral range from the ultraviolet to the water window. These probe pulses, combined with energy-dispersed transient-absorption spectroscopy, will enable the detection of electronic and structural dynamics at many absorption edges simultaneously.
Although this discussion has mainly focused on transient-absorption spectroscopy, other forms of attosecond x-ray spectroscopy will emerge (see also sections 3.1 and 3.2). The broad bandwidth of HHG sources is optimally exploited in energy-dispersed transient absorption because the observed linewidth is only limited by the lifetime of the final state of the x-ray transition. The same principle however extends to Auger spectroscopy, such that one could envision attosecond time-resolved x-ray measurements based on Auger electrons. X-ray emission and RIXS will also offer intriguing new directions, especially when operated close to the limits imposed by the uncertainty principle.
Concluding remarks
Recent progress in HHG- and FEL-based light sources has tremendously advanced ultrafast science. These developments now bring attosecond x-ray spectroscopy within reach. This extension will considerably broaden the scope of attosecond science by extending its applicability to complex molecules, liquid-phase systems, and even heterogeneous systems, e.g. those employed in photovoltaic devices. Such measurements will provide the most detailed, element specific and thus spatially resolved, insight into electronic dynamics in complex systems. A combination of HHG- and FEL-based light sources exploiting the specific advantages of each will open up additional possibilities to address the most fundamental dynamics of matter.
Acknowledgments
The author acknowledges the invaluable contributions of all his colleagues and co-workers who have contributed to the work summarized here over the past six years, in particular Peter Kraus, Yoann Pertot, Inga Jordan, Martin Huppert, Denitsa Baykusheva, Aaron von Conta, Bruno Schmidt, François Légaré, Cédric Schmidt and Jean-Pierre Wolf. Funding from an ERC starting grant (contract #307270-ATTOSCOPE), the Swiss National Science Foundation and ETH Zürich is gratefully acknowledged.
5.6. Attosecond science: from transient absorption to multidimensional spectroscopies
Stephen R Leone
University of California, Berkeley
Status
Benefitting from the technical advances of high harmonic generation and free-electron lasers, attosecond pulses as short as 53 attoseconds in the XUV or x-ray spectral regions, are reported [208]. This enables the rich field of attosecond science, investigations that achieve the shortest possible measurements of time dynamics in atoms, molecules, and solids [168]. The field already provides novel measurements of Auger decay [209], autoionization [210], electron re-collision [171, 211], electronic superpositions [212], and charge migration in molecules [170]. Remarkable results for photoemission delays from orbitals, electronic timescales of dielectric properties [213] and band gap excitation [214] provide compelling evidence that attosecond methods will continue to address new frontiers in dynamics.
Transient absorption in the XUV or x-ray region is uniquely suited to the investigation of core-level transitions and higher lying states near and above ionization continua. Core-level transient-absorption spectroscopy [215], a method that is element specific, and sensitive to oxidation states and spin states, is applicable to a wide range of chemical elements and species. At photon energies up to 300 eV, the state-of-the-art in x-ray transient-absorption dynamics (based on femtosecond high harmonics) has been used to measure excited-state ring opening of cyclohexadiene [216] (figure 26) and strong-field ionization of CF4 [151]. The strong-field ionization of an atom to two different spin-orbit states reveals that there can be several hundred attosecond time differences in the appearance of one spin-orbit state versus the other [217], despite the difficulties of using few-femtosecond phase-locked optical pulses together with 100 attosecond pulses. In solids, excitations across band gaps in basic semiconductors provide a clear separation of timescales for electronic processes versus phonon-mediated events [218], allowing mechanisms and timescales to be established for insulator-to-metal transitions [219].

Figure 26. X-ray fs identification of the key transition state labeled 2 A, blue points, for ring opening of cyclohexadiene. From [216] with permission from AAAS.
Download figure:
Standard image High-resolution imageBy selecting a nonintuitive order of the pump and probe pulses, i.e. introducing the XUV pulse first, superpositions of electronic and vibrational states can be created, which are probed as they evolve in time by using time-delayed optical pulses that modify the polarization of the medium via coupling to neighboring states [212]. Time decays can be obtained for autoionization processes, and even high-resolution spectroscopic information is acquired from the coherent superpositions of electronic and vibrational states. Controlled modification of lineshapes from Lorentz to Fano [220] is found to be a natural result of the phase imparted by the optical pulse. One- and three-photon interferences are beautiful examples of quantum mechanical multiple slit interferences that can be probed. Each measurement is subject to considerable rigorous theoretical interpretations, to ferret out the discrepancies and artifacts that can arise when attempting to make time measurements at the shortest possible timescales.
Current and future challenges
Attosecond methods are not without their challenges [221]. While short pulses are achievable in the XUV and x-ray portions of the spectrum, the fluxes are usually extremely weak. To perform any type of traditional pump–probe measurement requires marriage of attosecond pulses with few-cycle carrier-envelope-phase stabilized optical pulses. As a result, unless the measurements involve a field-dependent process, such as strong-field excitation or ionization, or photoelectron streaking detection, the methods can be limited by the time duration of the optical pulses. The extreme bandwidth of the XUV pulses also requires creative solutions to resolve spectral features. This is achieved by spectrally resolving the XUV pulse after the interaction of the pump and probe pulses in the sample, as is traditionally done in white-light transient-absorption spectroscopy. Transient-absorption measurements at synchrotrons and in laboratory table-top systems in the XUV and x-ray were developed first, and the method was only applied to attosecond transient absorption in 2010 [222]. Since that time, many researchers have adopted the transient-absorption technique as a favored method for probing dynamics.
Despite the successes with transient-absorption and photoelectron streaking methods, there are many desirable scientific goals that have not yet been attained. While tantalizing results are emerging, charge migration [170] in molecules, materials, and across junctions represents a grand challenge of the attosecond field. Similarly, the exploration of passage through conical intersections, which can in principle be probed by few- to sub-femtosecond time dynamics, would provide a major breakthrough. The investigation of important problems such as 2D materials, topological phase, and superconducting films offer important potential advances for the attosecond field. Pump–probe transient spectroscopies are simply the most basic version of measurements made in the optical region. Multidimensional spectroscopies [223] remain an important goal of the field to peer beneath broadened and obscured spectral features with coherent methods. Core-hole lifetimes are at the heart of many short time dynamical processes, and those timescales could stymie the desired goals of obtaining ultrashort electronic timescales in molecules and solids. Progress needs to be made in many of these areas to address new scientific frontiers.
Advances in science and technology to meet challenges
There are several advances that signify great promise for the kinds of breakthrough investigations that will solidify a permanent place for attosecond spectroscopy in the field of time dynamics. The introduction of shorter-pulse free-electron lasers and more powerful high-harmonic laser systems will permit true attosecond–attosecond measurements in the near future. By splitting a pulse or synchronizing two attosecond pulses, it will be possible to excite a molecule or a material with one attosecond pulse and to probe the core levels of the excited state with another pulse. Instruments at several locations are seeking the requisite pulse energies of about 1 μJ to 1 mJ. The availability of higher photon energies, such as in the 300–500 eV range, will greatly enhance the chemical and physical systems accessible to investigation, such as carbon, nitrogen, and oxygen, greatly extending the range of molecular or materials properties that can be studied.
The recent advance of wave-vector matched, noncollinear attosecond four-wave mixing of XUV pulses with optical pulses is a propitious step towards multidimensional coherent spectroscopies [224] (figure 27). Already the noncollinear FWM method provides a significant advantage for unraveling what states are coupled by the optical pulse to generate the phase-matched signals. The background free nature of the signals gives exquisite detail on electronic and vibrational coherences in atoms and molecules. If these methods, which already resolve the frequencies of the XUV pulses, can be extended to simultaneously spectrally resolve the frequencies of the optical pulses, true multidimensional coherent spectroscopies in the XUV will be realized. Moreover, when these techniques are applied to solids or liquids, core-hole lifetimes, which are notoriously short in condensed phase media, will be extracted for the first time.
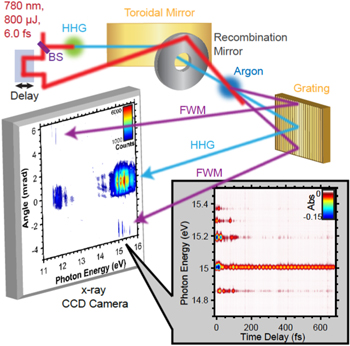
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 27. Background free noncollinear FWM in argon, XUV + two 800 nm photons. From [214] with permission from AAAS.
Download figure:
Standard image High-resolution image{kind=link}
Attosecond transient reflectivity in the XUV and x-ray regions of the spectrum offers promise to provide the real and imaginary parts of the complex dielectric function in excited-state materials [225]. By introducing circularly polarized light fields for pump and probe, it will be possible to investigate the topological phase in excited states or symmetry-protected spin systems. The real part of the dielectric function can derive information about the renormalization of the Coulomb potential after photoexcitation (polarization), while the imaginary part shows the increase or decrease of loss channels, i.e. changes in absorption. In the same way that transient reflectivity revolutionized the study of semiconductor band gap physics, materials will soon be viewed through the element selective, oxidation state, and spin-state specific 'eyes' of XUV and x-ray transitions.
Concluding remarks
Measuring important electron dynamical timescales, attosecond science is the next breakthrough technology beyond femtosecond spectroscopy, which was often limited to the slower motions of nuclei. Borrowing from important principles of core-level spectroscopy at synchrotrons, the field can address oxidation states, spin, and element specificity, with shorter timescales than previously possible. Attosecond science is on a threshold of becoming a mainstream topic that will address many important problems in charge-state dynamics.
Acknowledgments
The author gratefully acknowledges support from the National Science Foundation, the Department of Energy, the Army Research Office, the Air Force Office of Scientific Research, and the Defense Advanced Research Projects Agency, as well as important input from group members Peter Kraus, Andrew Attar, Erika Warrick, and Michael Zuerch.