Abstract
We present a procedure for growing thin films of an organic polyamid material based on a cyclic repetition of two consecutive, complementary, self-limiting surface reactions. The molecular compounds that react with the surface are dissolved in an organic solvent. This new method exemplifies how atomic layer deposition (ALD) and molecular layer deposition (MLD) can benefit from being transferred from the gas phase to the liquid phase, given that a broad variety of advantageous reagents are only available in dissolved form.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Atomic layer deposition (ALD) has established itself as a thin film deposition method with a broad range of applications, both in terms of functionalities and of materials.1,2 This technique is based on complementary, well-behaved gas-solid surface reactions with 'self-limiting' character. During ALD film growth, the chemical identity of the surface alternates between two distinct reactive states, so that a well-defined amount of material is deposited during each ALD cycle, independently of the amount of precursors delivered during each step. This feature renders ALD uniquely suited to coating structured substrates, including deep pores, with continuous films of homogeneous thickness.3 One very recent development of the ALD field has been the demonstration that the principles can be transferred from gaseous precursors to ones dissolved in liquid solvents,4 generalizing a number of previously existing but more narrowly focused techniques such as electrochemical ALD (or underpotential deposition), layer-by-layer (LBL) assembly of polyelectrolytes, and SILAR.5–12 Our previous paper demonstrates the principle of 'solution ALD' (sALD) and establishes reaction chemistry and procedures for the deposition of several inorganic oxides. The films obtained are comparable with corresponding 'gas-ALD' (gALD) layers not only in terms of growth rate and self-limiting behavior but also of the chemical identity and purity. The advantages of this novel deposition method are the mild (room-temperature) and experimentally facile (vacuum-free) reaction conditions, but also the wider range of potential precursors to be chosen from, since in sALD they do not have to be volatile.
In this paper, we demonstrate this particular advantage of sALD toward a class of materials which has proven challenging in gALD so far, namely, organic polymer films. Of course, a number of studies have shown the successful deposition of organic or hybrid films by gALD,13,14 but the so-called 'molecular layer deposition' has remained limited in scope due to the constraints that the small number of available volatile and reactive precursors represents. The difficulty becomes apparent if we consider a polyamide as an example (an important class since it includes biological as well as manufactured fibers such as silk, Nylon and Kevlar), Figure 1. Generating the amide bond between two monomers is possible upon reaction of an amine with an activated acyl unit (such as an acyl halide). However, a heterobifunctional precursor (such as an aminoacyl halide) reacts with itself in the gas phase before reacting with the solid surface, so that it causes undesired CVD-like deposition (CVD: chemical vapor deposition; case 2 in Figure 1). The alternative, which is in fact exploited in gALD and called 'molecular layer deposition' (gMLD), is the use of two homobifunctional reagents — for example, a diamine and a bis(acyl halide). The limitation of this approach is that each precursor molecule is likely to react with two neighboring reactive sites on the solid surface, which results in a non-reactive surface for the subsequent ALD/MLD step, situation 1.13,14
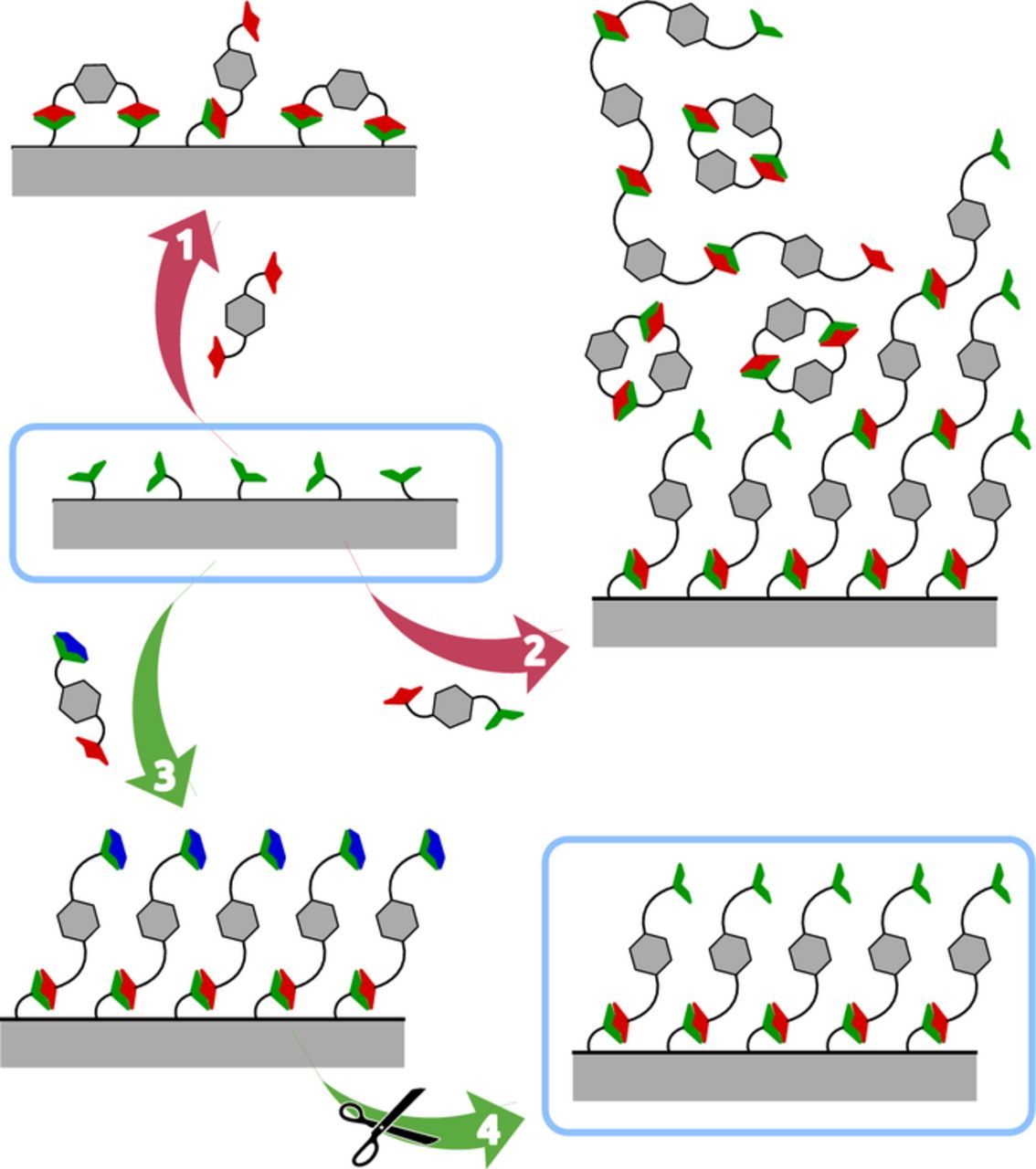
Figure 1. Schematic representation of various precursor types reacting with a solid surface: the complementary functional groups (e. g. amine, -NH2, and activated acyl, -COX) are displayed as green and red shapes, and protecting groups (e. g. the so-called Boc group) as blue caps. (1) Homobifunctional reagents passivate the surface irreversibly via double reaction. (2) Unprotected heterobifunctional precursors react in the homogeneous phase and causes uncontrolled deposition akin to chemical vapor deposition (CVD); (3) Only heterobifunctional reagents with one functional group "protected" (rendered temporarily unreactive) deliver the desired monolayer deposition. (4) Subsequent deprotection uncovers the initial, reactive surface type and yields ALD (or MLD) film growth.
Circumventing this limitation is possible by exploiting the wealth of activation and protection/deprotection strategies and reagents optimized in organic synthesis and solid-state synthesis of biopolymers, see 3 and 4.15 Since activating and protecting groups are typically large and bulky, and often charged, these strategies cannot be implemented in gALD/gMLD. We demonstrate here that they can be exploited in sALD, however: this establishes molecular layer deposition from solutions, sMLD.
Experimental
Instrumental methods
Scanning electron micrographs were taken on a JEOL JSM 6400 equipped with a LaB6 cathode and with an energy-dispersive X-ray spectroscopy (EDX) system based on an SDD detector. Spectroscopic ellipsometry data were collected from 370 to 1050 nm under 50°, 60° and 70° incidence angle with a SENpro spectroscopic ellipsometer from Sentech. Fits were performed using the model and software SpectraRay 4 provided with the instrument. Infrared spectroscopy was performed on an FTIR-8400S system from Shimadzu using a home-made sample holder. Optical transmission and reflectance spectroscopy were performed using an HR4000 spectrometer and a DH-2000-BAL deuterium and halogen light source from OceanOptics. The liquid quartz crystal microbalance (QCM) cell model "openQCM" was purchased from Novaetech. For mass spectrometry, the monolayer resulting from ALD deposition is naturally insufficient for direct (matrix-assisted) laser desorption ionization (LDI or MALDI) measurements. Thus, the sample was removed from the target plate under harsh conditions by hydrolysis with NaOH in MeOH. The resulting fragments of the polymer were analyzed by ESI. For a direct comparison, the tert-butyloxycarbonyl-protected (Boc-protected) monomer was used as a reference. It was dissolved and mixed with trifluoroacetic acid (TFA) for deprotection and diluted with MeOH. Electrospray ionization (ESI) experiments were conducted with a quadrupole time-of-flight (QqToF) mass spectrometer (micrOTOF-Q II, Bruker, Bremen, Germany).
Anodization
The nanoporous membrane substrates were prepared following a standard two-step anodization (electrochemical oxidation of aluminum in a protic solution) procedure carried out under 195 V in 1 wt% H3PO4 at 1°C.16 The underlying metallic aluminum was subsequently removed in CuCl2/HCl and the barrier oxide layer closing the pore extremities removed by warm H3PO4, whereby the pores were simultaneously widened.
sALD setup
Atomic layer deposition was carried out in a home-built microfluidic system (Figures 2 and 3). The sALD control electronics and software were designed and produced by ZUMOlab. The OEM peristaltic pumps (model 400A) and the flexible fluoroelastomer tubing were from Watson-Marlow. The Teflon tubes connecting the precursor solution flasks to the pumps and the pumps to the reactor were from VWR (228-0736, 0.5 mm ID) with Luer-Lock connections. The sALD precursor solutions were handled under inert atmosphere using standard Schlenk technique.17 The precursor solutions and pure solvent were kept in Schlenk flasks closed with rubber septa and connected to the tubing system using stainless steel cannulae (VWR). The reactor chamber used for coating planar samples was built from solid teflon, the one for nanoporous samples from stainless steel.4

Figure 2. Photograph of the sALD setup with in-line piezoelectric quartz crystal microbalance (QCM) for reaction monitoring.
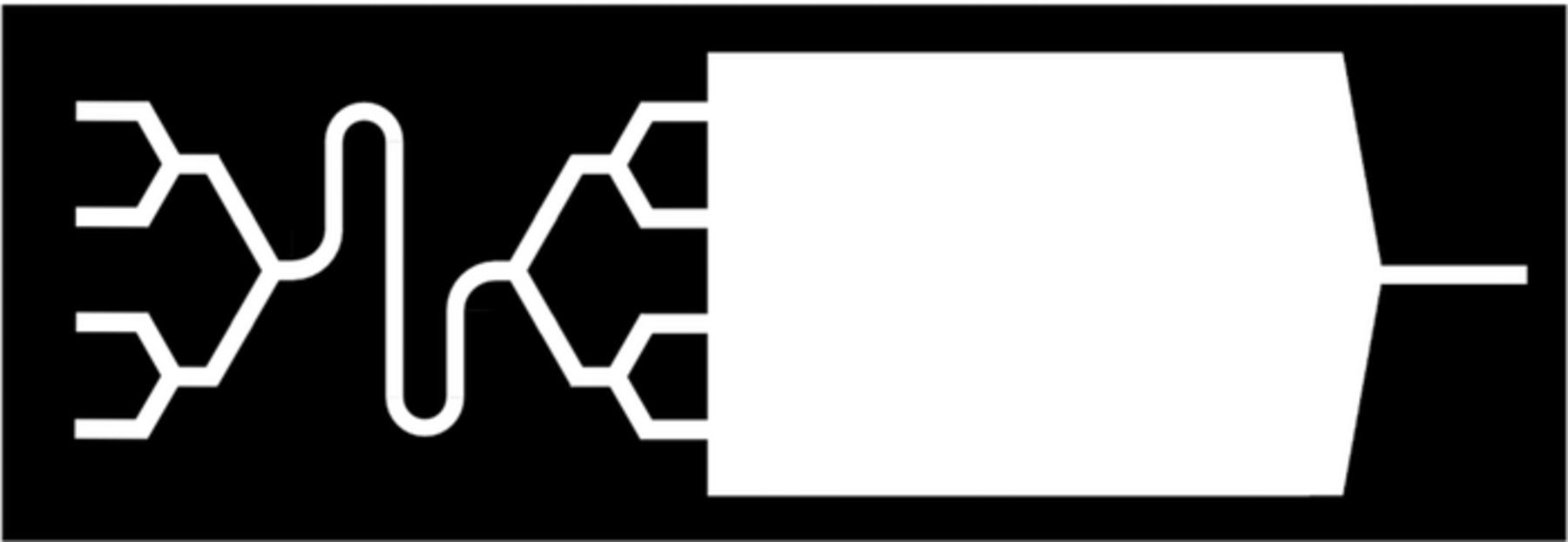
Figure 3. Sketch of the microfluidic reaction chamber with four inlets for two precursor solutions and the purging solvent.
Materials
Extra dry acetonitrile, 4-(tert-butyloxycarbonylamino)benzoic acid (Boc-pABA) and trifluoroacetic acid (TFA) were purchased from Acros organics. The acetonitrile used for purging the reaction chamber was dried over 3 Å molecular sieve. 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid (EDC) was purchased from Alfa Aesar. The aluminum used for the preparation of the porous aluminum membranes was purchased from SmartMembranes (99.99%). Phosphoric acid, copper(II) chloride dihydrate, chromium(VI) oxide, ethanol, hydrochloric acid, perchloric acid, and argon were purchased from commercial suppliers and used as received. Boron-doped [100] CZ silicon wafers with 200 nm thermal oxide were obtained from Silicon Materials. Water was purified immediately before use in a Millipore Direct-Q system.
Solutions
Two solutions were used as the precursors. Precursor A is a solution of Boc-pABA mixed with equimolar EDC in extra dry acetonitrile. Their reaction in solution generates the activated acyl functionality in situ. Precursor B is a solution of TFA in extra dry acetonitrile. The standard experimental parameters are listed in Table I. These parameters were varied in the course of the investigations as mentioned in the main text.
Table I. Standard parameters used for sMLD on planar samples. The pumps are run at their nominal power (100%) with 2.06-mm ID tubing.
| Chemical identity | Concentration (in MeCN, mM) | Duration (s) | |
|---|---|---|---|
| Precursor A | Boc-p-ABA | 2 | 10 |
| EDC | 2 | ||
| Precursor B | TFA | 4 | 10 |
| Purge | — | — | 20 |
Results and Discussion
Setup and reaction design
The principle of our setup, based on peristaltic pumps and a microfluidic reaction chamber, was published previously.4 We improved it in two important ways (Figure 2). Firstly, the connection of the four delivery lines (two for precursors and one for pure solvent) is now performed with a Luer system directly on the microfluidic setup, which eliminates the (leak-prone and poorly reproducible) Y-connectors in the tubing system. Instead, the nodes are integrated in the microfluidic device itself (Figure 3), which enables us to shorten the pulse and purge durations necessary. Secondly, we implemented an in-line monitoring with a piezoelectric quartz crystal microbalance. This possibility allows for an even more direct comparison of sALD chemistry with the gALD analogue methods (or sMLD with gMLD).
The MLD reaction will be based, along the principle delineated in Figure 1, on a heterobifunctional monomer as the skeleton for the first precursor, para-aminobenzoic acid (p-ABA). Here the amine and the acyl groups will provide the desired reactivity toward the amide linkage. To prevent direct CVD reaction, we initially mask the reactivity of the amino group with a tert-butyloxycarbonyl (Boc), a procedure which in organic synthesis is common and called 'protection'. Simultaneously, we enhance the reactivity of the acyl group using another common organic reagent, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimid (EDC). We note that this is not possible in gALD, because the large, charged molecule has no vapor pressure.
The Boc-p-ABA-EDC is now highly active for reaction with amine groups of the surface and builds one monolayer upon formation of amide bonds (Figure 4). Importantly, no double reaction is possible of one precursor molecule with two neighboring reactive surface sites. A Boc-terminated surface is formed, which is inert to further reaction until the so-called deprotecting agent trifluoroacetic acid (TFA) is introduced as the second precursor in the next step. Upon deprotection, the surface regains reactivity toward Boc-p-ABA-EDC in the next sMLD cycle (Figure 4).
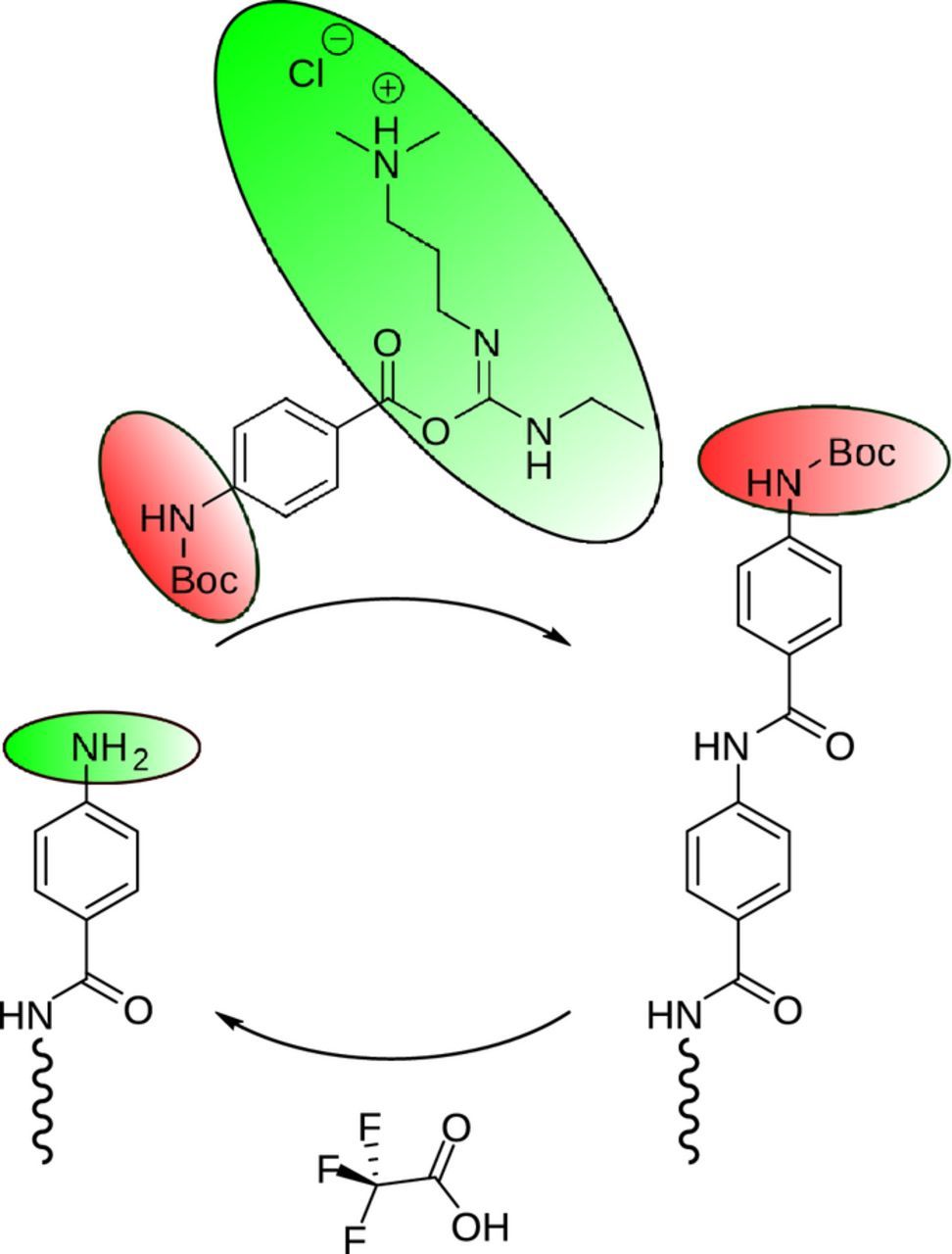
Figure 4. Scheme of the deposition reaction designed for sALD of a polyamide along the principles of Figure 1. The first precursor is heterobifunctional with one protected and one activated group. The second precursor only serves to deprotect and thereby render the surface reactive.
Demonstration of ALD/MLD character
In situ monitoring of thin film growth by piezoelectric (quartz) crystal microbalance (QCM) is a standard tool in many deposition methods, and in gALD it also serves as an investigative method into the nature of surface chemistry. We implemented a liquid-phase QCM, in which a frequency decrease is caused by mass uptake of the solid film (Figure 5). When acetonitrile solutions of the precursors Boc-p-ABA-EDC and TFA are flown over the QCM crystal in an alternating manner, an overall regular increase is observed corresponding to thin film growth at a constant rate (Figure 5a). A more detailed observation of each MLD cycle evidences the nature of each reaction step (Figure 5b). We explicitly avoid interpreting (and over-interpreting) the shape of the curve during pulses and purges (which are affected not only by chemisorption but also by possible effects of viscosity and streaming potential changes), and only focus on the frequency difference obtained at the end of each step. The first step, that is, the reaction of Boc-p-ABA-EDC with the surface, is accompanied by a large mass uptake (Δm > 0) as expected (red highlight). Subsequent treatment with TFA (blue highlight) then causes a mass loss (Δm < 0) due to deprotection of the amine from the Boc group.
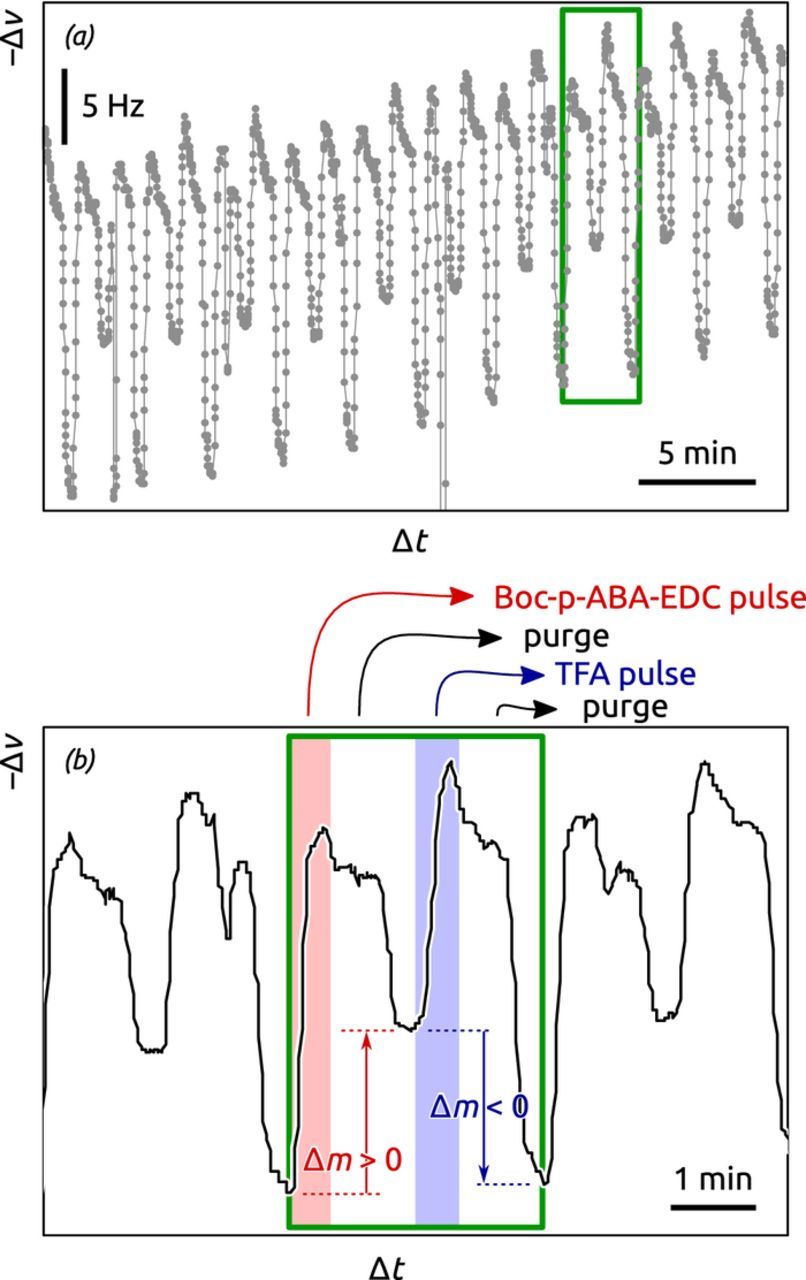
Figure 5. In situ quartz crystal microbalance data recorded during the deposition process depicted in Figure 4. (a) Overview of data (frequency decrease –Δν vs. time) of multiple deposition cycles. (b) Data analysis of the QCM data in one deposition cycle (green frame), highlighting the precursor pulses in red and blue, while the purging times are on the background.
The growth rate of this poly-pABA polymer is determined accurately by spectroscopic ellipsometry (Figure 6). Here, standard silicon wafers with 200 nm of thermal oxide are used as substrates. A systematic shift of the ellipsometry curves is observed if samples are compared after different numbers of sMLD cycles N, hinting at film growth (Figure 6a). Fitting the individual curves (under multiple incident angles) yields the thickness values presented in Figure 6b. A linear behavior is obtained (R2 > 0.9) with 1.0 (±0.1) Å per cycle growth rate and an intercept at the origin within experimental uncertainty (4 (±6) Å), indicating that nucleation on thermal silicon dioxide is facile.

Figure 6. Characteristics of poly-pABA sALD from Boc-pABA-EDC and TFA (at room temperature in acetonnitrile). (a) Individual spectroscopic ellipsometry traces (phase difference Δ(λ)) obtained on one Si/SiO2 wafer submitted to increasing numbers N of sALD cycles, evidencing the systematic film thickness increase. (b) Growth curve determined from the ellipsometry data, evidencing the linear growth with a rate of 1.0 Å per cycle.
Starting from standard deposition conditions summarized in Table I, a true demonstration of ALD (or MLD) characteristics is provided by the experimental independence of the growth rate with varying precursor dosage, which confirms the self-limiting behavior of the surface reactions. In our setup, this can be performed easily by varying the duration of the precursor pulses, while the purging duration is maintained constant (at 20 s, which is sufficient to remove excess precursor and physisorbed material). Figure 7a demonstrates that the growth rate remains constant at approximately 1.0 Å per cycle (within statistical error), and thereby proves the true ALD/MLD nature of this sMLD procedure. Furthermore, varying the purge step duration with constant precursor dosage (Figure 7b) yields 20 s as the minimal purging time which avoids direct contact of the precursors in solution, and thereby CVD behavior.
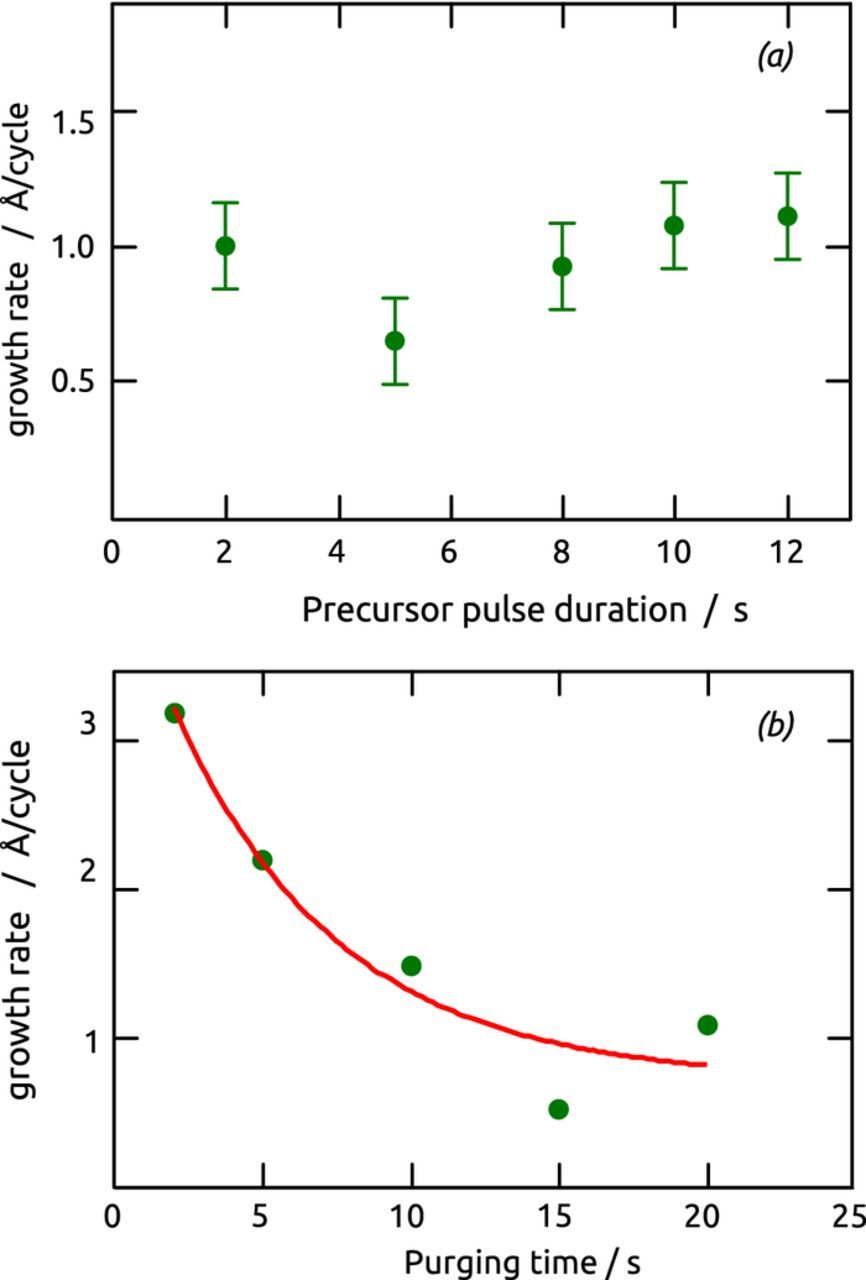
Figure 7. (a) Experimental dosage-growth behavior of the sMLD reaction between Boc-pABA-EDC and TFA. The growth rate is independent of the time during which each precursors are flown over the sample, which corresponds to varying the dosage. The datapoint at 5 s is an outlier. (b) Effect of the purging duration on growth: 20 s corresponds to a pure ALD behavior, shorter times may cause a CVD component.
Material characterization
The chemical identity of the film deposited is confirmed by electronic and vibrational absorption spectroscopies. The presence of aromatic units appears in UV-visible spectroscopy (Figure 8a) as a rather broad absorption edge near 440 nm, significantly red-shifted from that of the monomer Boc-p-ABA in MeCN solution. This red-shift, which is documented for 'aramid' fibers of similar chemical nature, is characteristic of the extended conjugation that occurs upon polymerization via the amide unit.18–20
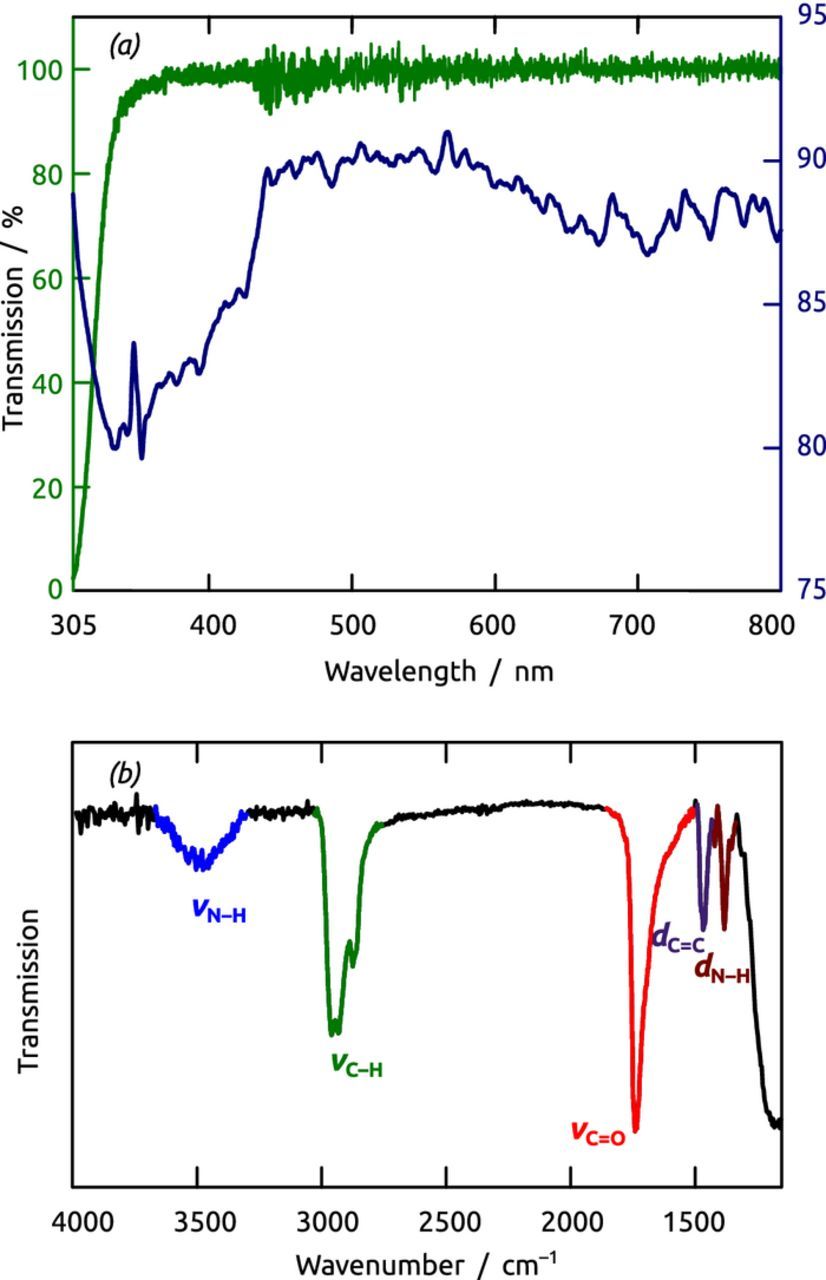
Figure 8. (a) Green: UV/visible transmission spectrum of Boc-p-ABA solution in acetonitrile (1 mM); blue: UV/visible transmission spectrum of a glass slide with 100 cycles of sMLD. (b) Infrared absorption difference spectrum of a poly-p-ABA coated nanoporous substrate. The spectrum displays the absorption peaks characteristic of the N–H (blue), C–H (green), C=O (red) and C=C (purple) stretching as well as the N–H bending modes (dark red).
The other functional units of the polymer chain are identified by infrared absorption spectroscopy (Figure 8b).21,22 The amide N–H stretch is found as a broad peak with a maximum at 3450 cm−1 (known as the 'amide A' peak in protein IR spectroscopy). The strong signals near 2900 cm−1 correspond to the C–H stretching modes. The C=O stretch ('amide I') is found at 1730 cm−1 as a very sharp and intensive peak (bright red), whereas the amide N–H bend (dark red) appears at 1380 cm−1. The aromatic C=C stretching mode is located at 1460 cm−1 (purple).
A final proof for the molecular identity of the sMLD film is provided by mass spectrometric investigation. The small absolute amount of material does not allow for matrix-assisted laser desorption ionization (MALDI), therefore one has to first bring the film into methanolic solution using base (10% NaOH) then use electrospray ionization (ESI) of the hydrolyzed units. Figure 9 exhibits a very similar pattern of peaks (green) separated by 158 mass units for the starting material Boc-p-ABA treated by a stoichiometric amount of TFA used as a reference (Figure 9a) and for the depolymerized film material (Figure 9b). The strong correlation of the signals shows that the sMLD film consists of the same monomeric units as the reference. A proof that the film is covalently polymerized (and does not merely consist of unreacted monomers) is provided by the absence of a signal at m/z 236 in Figure 9b, which corresponds to Boc-p-ABA and appears prominently in Figure 9a.
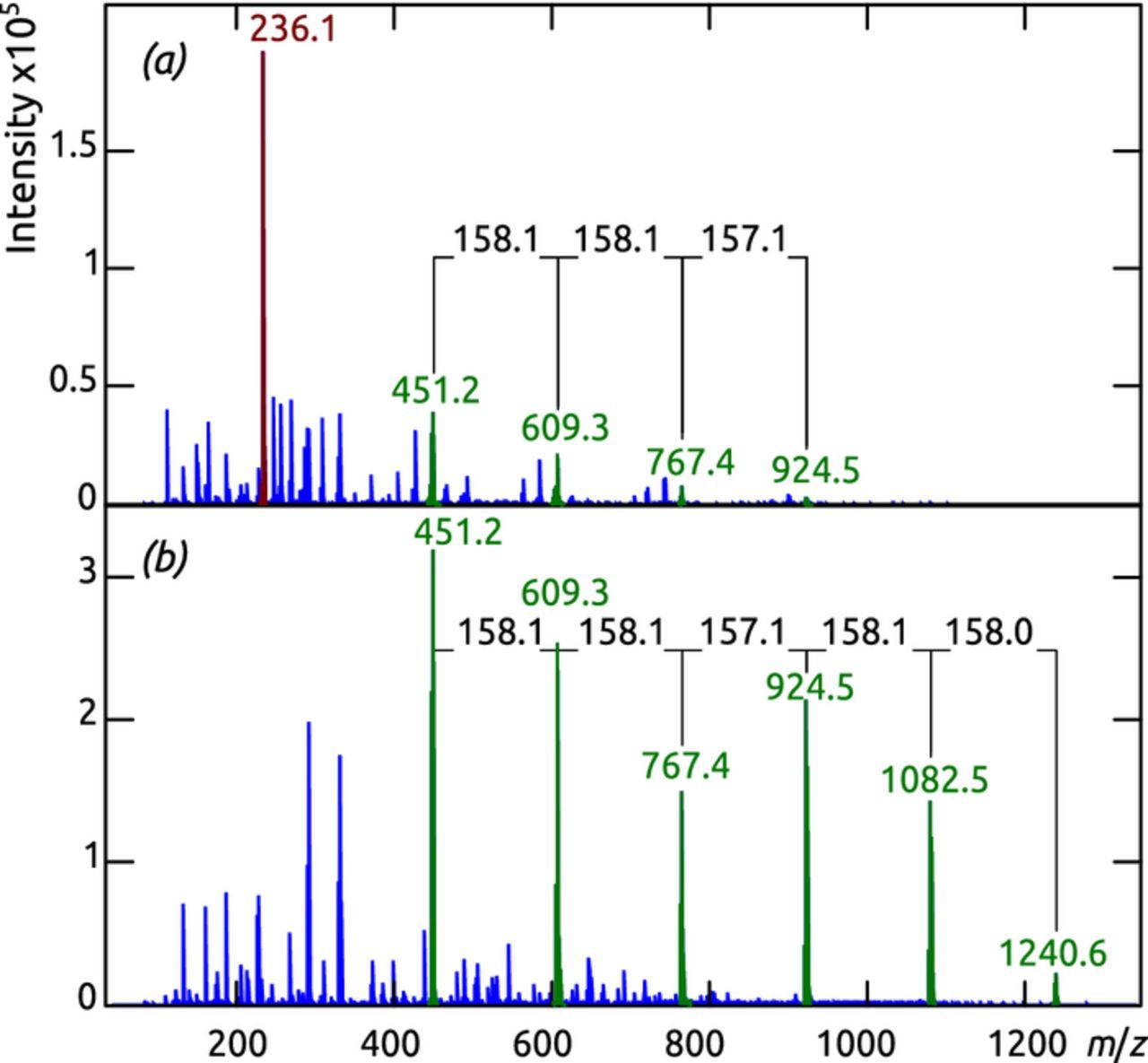
Figure 9. Negative-ion mode ESI QqTof mass spectra of the TFA-treated starting material Boc-p-ABA as the reference (a) and a methanolic solution obtained by hydrolyzing the sMLD film in NaOH (b).
Conclusions
Molecular layer deposition (MLD) has now been established from dissolved precursors. After the demonstration of solution ALD (sALD) as a novel thin film coating technique, this piece of work broadens the range of reaction chemistries accessible to ALD. Activation, protection and deprotection of organic functional groups are not possible in gas-phase thin film deposition (gALD/gMLD) because they rely on large and/or polar (ionic) species which cannot be brought into the gas phase in any significant amounts. sALD, however, circumvents this limitation and allows one to exploit the wide range of synthetic methodologies established in other areas of chemistry. In this particular example, an aromatic polyamide can be obtained as a thin film, with potential applications in the coating of fibers or other materials to be conferred with outstanding mechanical and fire-retardant properties.
However, the significance of the results goes beyond this, and we expect that sALD and sMLD will be recognized as alternatives to the established thin film coating techniques in the semiconductor, energy conversion, and textile fields, just to name a few. One can further envision that the very mild conditions of sALD and sMLD processing, combined with the versatility and specificity of available dissolved reagents, will likely enable advances in the emerging field of area-selective deposition and etching. For example, a protection/deprotection scheme similar to the one used in our work may be used in area-selective deposition, whereas alkylation could be exploited to render a surface inert in area-selective etching.
Acknowledgments
This work was supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant Agreement No. 647281, 'Solacylin') and by the Deutsche Forschungsgemeinschaft (DFG) in the SFB 953 'Synthetic Carbon Allotropes'.