Abstract
Glutamate is the principal excitatory amino acid in the vertebrate nervous system and is responsible for learning and memory. Understanding of these complex biological processes can be gained through experimentally accessible systems of glutamate detection. In this work, the microclinical analyzer (μCA) was used with a sensitive and stable glutamate sensor and model neuronal cells for quantitative glutamate detection under physiological relevant shear. Glutamate was detected by immobilized glutamate oxidase on a screen-printed electrode array. The sensor's linear range spanned glutamate's physiological to pathophysiological concentration range, and the biologically relevant sub-second to month temporal range. After 11 hours of use, the sensor retained 91 ± 1% of its signal, and it was able to be stored for a month without a significant decrease. When model neuronal cells were integrated into the μCA bioreactor and exposed to glutamate, they initially took up 210 ± 100 μmoles of glutamate, which increased to 390 ± 50 μmoles during their second exposure. These data suggest that the neurotransmitter uptake systems were functional and may be upregulated. The dynamic and durable μCA platform offers an experimentally accessible system of glutamate detection that can be used to monitor glutamate metabolism and signaling.

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Glutamate is one of the 20 canonical amino acids that together provide the structural and enzymatic foundation of proteins. Alone, glutamate plays a very different role and is itself an excitatory signaling molecule widely distributed throughout the central nervous system. In fact, glutamate is the most prevalent neurotransmitter and its function is essential for proper neurocognition, learning, and memory. Overactivation of glutamate receptors causes excitotoxicity, a pathological process whereby neurons are damaged or killed, which may result in neurodegeneration.1,2 Because of its central role in metabolic and cognitive processes, a strong effort has been put toward developing methods to detect glutamate.2–5
Accurate detection of glutamate can be accomplished using many techniques, including spectrometry, spectroscopy, and electrochemistry. The benefits of spectrometry and spectroscopy include sensitivity and selectivity. However, mass spectrometry requires chromatographic separation or vacuum preparation, which decreases the temporal resolution of the system.4 Although spectroscopic techniques, such as the iGluSNFR,2 offer impressive temporal resolution, they require optical transparency thereby limiting the scope of samples that can be analyzed. In contrast, electrochemical sensors require almost no sample manipulation and can be placed directly in the area of interest, allowing, for example, detection of glutamate and dopamine signaling in the brain.6–8 Electrochemical sensors are also versatile: They can be made on the nanoscale,1 have been 3D-printed,9 and can be inexpensive.10,11 The highly translatable nature of electrochemical techniques has already been demonstrated with the advent of Smart Bands12 and iSTAT meters (Abbott Point of Care Inc.).13,14 Detecting glutamate has many advantages: It advances our knowledge of the effects of pesticide exposure, explains some of the erratic behaviors seen after traumatic brain injuries, informs our dietary considerations (MSG), and, perhaps most importantly, functions as a window to understanding human memory and consciousness.
The majority of electrochemical glutamate sensors are enzymatic sensors, beneficial because of their selectivity and sensitivity.15,16 The enzymes on which these sensors are based are selective by their very nature. In the heterogeneous milieu of cells, they successfully identify their corresponding metabolite by relying on macromolecular substrate recognition. Similarly, enzymes are adapted to be sensitive at physiological concentrations. The signal they generate is enhanced by their ability to interact with their metabolite in a way that is non-destructive to the enzyme.17 Just within the subcategory of oxidase enzymes, glucose-, lactate-, choline-, cholesterol-, and glutamate-oxidase have all been used for electrochemical detection.17–19
Other benefits to enzymatic sensors include their biocompatibility, portability, compatibility, and relative low cost. Screen printing is an inexpensive and reliable way to fabricate electrodes that, when enzymatically modified, are also sensitive and selective.20 Furthermore, the biocompatibility of both enzyme and some screen-printed materials, such as platinum, makes them suitable for implantable devices. Their scalability qualifies them for portable, point-of-care analysis. Still, electrochemical analysis of biological systems presents some challenges—including enzyme activity loss and biofouling of the electrode—that compromise quantitative analysis.3,21 Despite the benefits of enzymatic sensors, these challenges require sensor calibration for accurate quantification of the analyte.
The microclinical analyzer (μCA) provides automated sensor calibration and increases ease of use. By integrating a microfluidic chamber with an automated pump and valve system, the μCA can automate sensor calibration, increasing ease of use by decreasing user intervention and increasing sample throughput.17,22 In addition, microfluidics allow for low sample volumes (26 μL sample chamber), enhancing the ability to detect small metabolic changes.5,23 This versatile platform can easily integrate cells or other enzyme sensors to monitor cellular bioenergetics in real-time.17
In addition to increasing the analytical power, microfluidic systems also help recapitulate physiological conditions.24 Microfluidic systems can add flow, and, as a result, mechanical forces. This feature alone has spawned an entire area of research into organotypic cultures. An organotypic culture is a 3D-culture of heterotypic cells and mechanical features25 that aims to mimic an organ or organ system.26 These platforms are more representative of an in vivo system than traditional cell cultures and are used for drug development, predictive toxicology, and basic research. A number of noteworthy examples exist including a liver bioreactor that models detoxification,26,27 a fetal membrane model used to study preterm birth,28 and a neurovascular unit that recreates transit across the blood-brain barrier.29 The ideal platform for studying glutamate would be sensitive and selective, recapitulate physiological conditions, and allow for quantification.
In this work, a sensitive, stable, and selective sensor was developed and incorporated into the μCA (Figure 1) to detect glutamate metabolism of model neuronal cells. This system automated sensor calibration and integrated cell culture through the use of microfluidics, allowing for quantitative, real-time measurements of cellular bioenergetics.
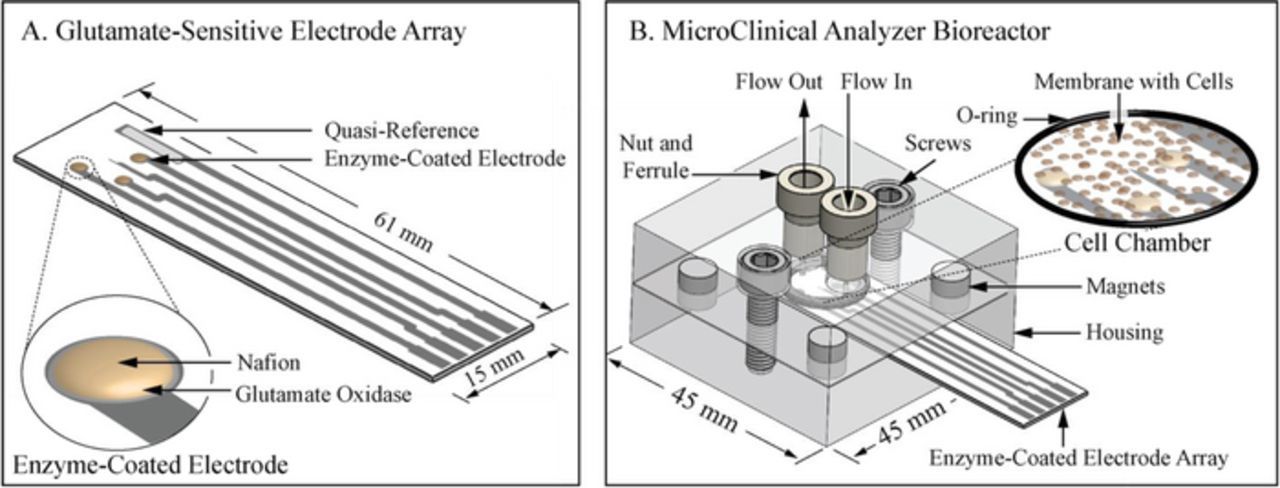
Figure 1. Drawings and schematic of A) the glutamate-sensitive electrode array and B) the μCA bioreactor. A) The electrode array was screen-printed and modified as shown with glutamate oxidase and Nafion on three working electrodes and Ag/AgCl on the quasi-reference before being inserted into B) the μCA bioreactor. The bioreactor housing plates separate so that the enzyme-coated electrode array and membrane-immobilized cells can be inserted or removed. The magnets align the housing, electrode array, and membrane, then the chamber is compressed by tightening the screws. Media was perfused through tubing (not pictured) that connects the housing's cell chamber through nuts and ferrules to the media reservoirs, pumps and valves (not pictured).
Materials and Methods
Material procurement
All cells used in these experiments were PC12 cells purchased from American Type Culture Collection (Manassas, VA). For cell culture and electrode modification, penicillin, streptomycin, T25 flasks, collagen type I, trypsin-EDTA, glutamate oxidase, bovine serum albumin, and glutaraldehyde were purchased from Sigma Aldrich (St. Louis, MO). Dulbecco's modified eagle's medium (DMEM) and Roswell Park Memorial Institute medium (RPMI) 1640 was purchased for cell culture from ThermoFisher (Waltham, MA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Flowery Branch, GA). Nafion was purchased from Alfa Aesar (Ward Hill, MA). Transwells were purchased from Corning (Corning, NY). The μCA housing was designed by VIIBRE (The Vanderbilt Institute for Integrative Biosystems Research and Education, Nashville, TN) fabricated from polmethylmethacrylate by the Vanderbilt Microfabrication Core (VMFC) that VIIBRE operates. Buna-N rubber O-rings, and screws purchased from McMaster-Carr (Elmhurst, IL). Nut-and-ferrule fluid connections were purchased from IDEX (Oak Harbor, WA). Tygon tubing was purchased from Cole Parmer (Vernon Hills, IL). Magnets were purchased from K&J Magnetics (Pipersville, PA). Screen-printed electrodes were designed in house and purchased from Pine Research (Durham, NC). The rotary planar peristaltic micropump (RPPM, US patents 9,874,285 and 9,725,687 and applications claiming priority from US patent application 13/877,925), five-port valves (VIIBRE, US patent 9,618,129), microcontrollers, and computer software were designed by and purchased from VIIBRE/VMFC. The CH 1440 potentiostat was purchased from CH Instruments (Austin, TX).
Electrode modification
Glutamate sensitive electrodes were made by drop-casting glutamate oxidase enzyme on a screen-printed electrode array.17 The electrode array features five electrodes; three platinum disk electrodes and two band electrodes. The three platinum disk electrodes (A = 1.8 mm2) were used for enzymatic detection of glutamate. The larger of the two band electrodes (A = 19 mm2) was used as an Ag/AgCl quasi-reference, while the other, designed for use as an oxygen detector (A = 0.08 mm2), was not used. Glutamate oxidase from Streptomyces was dissolved in 800 mg/mL bovine serum albumin (BSA) in minimal buffer (2 mM PBS, pH 7) to 10 mg/mL and stored for up to one month at −18°C until use. When required, these glutamate oxidase solutions were thawed, glutaraldehyde was added to 0.25% wt/v, and the mixture was vortexed for approximately five seconds. After mixing, the glutamate oxidase solution was quickly drop-cast by pipetting 1.0 μL onto the platinum disk electrode surface. The solution was allowed to dry for 1 hour before a 1.0 μL coat of Nafion (5% v/v) was deposited on top of it. After Nafion deposition, the sensors were allowed to dry for an additional 45 minutes before calibration. If not used immediately, electrodes were stored in the dark and in buffer (2 mM PBS/120mM KCl, pH 7) at 4°C. All electrode preparation was done at ambient conditions.
Limit of detection and limit of linearity
The limit of detection and limits of linearity were determined by performing calibrations with the μCA. For all calibrations, glutamate oxidase modified electrodes were sealed against the polymethylmethacrylate closed-cell housing, (external dimensions w = 43 mm, l = 43 mm, h = 23 mm, internal dimensions r = 6 mm, h = 0.23 mm), with an O-ring, aligned with magnets, and compressed with 10–32 × 5/8-inch screws. Calibrations were performed by monitoring the current generated by 21 calibrants ranging from 1 μM to 5 mM glutamate in buffer (2 mM PBS, 120 mM KCl, pH 7). Calibrants were sampled through a five-port valve pulled by the RPPM at a flow rate of 100 μL/min and monitored by a CHI 1440 potentiostat held at 0.6 V vs. Ag/AgCl until a steady state was reached (∼3 min). Each calibrant was followed by buffer to provide a baseline. The limits of linearity, limit of detection, and sensitivity of the electrode were determined using a linear regression of the calibration data. The maximum limit of linearity was set when the addition of the next calibration point altered the previous slope by more than 5%. The limit of detection was calculated by adding the signal of the blank to three times the error of the blank (y values) and dividing by the slope of the calibration curve. The sensitivity of the electrode was determined by dividing the slope of the curve by the area of the disk electrode. A non-linear regression was performed using data from the 10 μM to 10 mM to determine the maximum rate of reaction for the enzyme, Vmax, and the concentration at which the enzyme is 50% saturated, Km.
Sensor stability during use and storage
To determine the stability of the sensor over experimentally relevant time periods, the current generated by 300 μM glutamic acid in buffer (2 mM PBS, 120 mM KCl, pH 7) was monitored (4 min) every hour for eleven hours and compared to the original signal. In between glutamate measurements, the baseline was set by measuring the current with the electrode in buffer (56 min). Storage capacity was determined by comparing the signal of the electrode before and after storage. For these experiments, three different working electrodes were monitored for four minutes in glutamic acid solution and four minutes in buffer for ten cycles before and after being stored in buffer at 4°C for a week. Electrodes were tested once a week for a month.
Effect of nafion on interference
To determine the utility of Nafion to exclude interference, the effect of ascorbic acid on glutamate detection was investigated. For these experiments, electrodes were prepared as above by drop-casting the glutamate oxidase solution (1.0 μL) onto the working electrodes. However, before applying the Nafion film, the current generated by 300 μM glutamic acid in buffer (2 mM PBS, 120 mM KCl, pH 7) was monitored (4 min) on three different working electrodes with increasing amounts of ascorbic acid (0, 1, 10, 100 1000 μM). A Nafion film was then applied (1.0 μL, 5% v/v) to the same electrodes and, after drying (45 min), the effect of ascorbic acid on the signal was investigated at the same conditions.
Microclinical analyzer fabrication
The μCA bioreactor was designed in-house and fabricated by Vanderbilt Institute for Integrative Biosystems Research and Education (VIIBRE) at Vanderbilt.17 The screws, magnets, O-rings, membranes, nuts, and ferrules can be purchased commercially. The screen-printed electrode was designed in-house and fabricated by Pine Research. The μCA bioreactor opens to insert or remove the electrode and cells from the internal chamber (A = 113 mm2, V = 26 μL) and interfaces the pump, valve (with calibrants, treatments and media), analytics (electrode), and biological system of interest (PC12 cells immobilized on a membrane) with Tygon tubing (internal r = 0.25 mm). The VIIBRE five-port rotary valve automated treatments with up to five different solutions and the VIIBRE RPPM accommodated flow speeds from low μL/min to mL/min.
Cell culture
PC12 cells cryopreserved in liquid nitrogen were thawed, added to warmed DMEM culture media, and spun down (180 x g, 7 min). The supernatant was discarded and the pellet was triturated with 1 mL of DMEM with 5% FBS, 0.1 mg/mL penicillin, and 100 units/mL streptomycin. The resulting suspension was brought to ∼1 million cells/mL and transferred to two collagen-coated T25 flasks. All cell culture flasks and transwells were coated in collagen type I overnight at room temperature before adding cells. The cells were grown to confluency (∼10 days) at 37°C, 5% CO2, trypsinized, (0.25% wt/v trypsin-EDTA) and plated onto two T25 flasks and a six-well PET-track etched 3.0 μm transwell. These cells were grown to confluency (10–14 days) before use. All cellular experiments and cell culture were conducted at 37°C and 5% CO2 within an incubator.
Potassium chloride and glutamate treatment
Confluent PC12 cells in a six-well transwell were maintained in glucose-free RPMI media for twelve hours before being transferred into the μCA bioreactor. Before addition of the cells, the electrodes were calibrated as above with modifications. Calibrants (RPMI, 20–1000 μM glutamate, 100 mM KCl, pH 7) were passed over the electrodes (20 μL/min, 37°C) and were electrochemically monitored until a steady state was reached (∼10 min). After calibration, a transwell membrane with immobilized PC12 cells was removed from its ridged plastic support with a paring knife and transferred to the μCA bioreactor. To transfer the membrane, the housing was opened and a membrane was placed on top of the electrode with the cells facing up. A second 0.3 μm membrane was placed on top to secure the cells in place. The two membranes were aligned with the electrode and the O-ring by the magnets on either side of the housing and compressed with the screws to seal the cell chamber. During treatment, the bioreactor chamber was amperometrically monitored by three different glutamate-sensitive electrodes along the cell-containing membrane at 0.6 V vs. Ag/AgCl. First the cells were allowed to equilibrate in RPMI for 30 min. RPMI with 100 mM KCl was then passed over the cells for 30 min. Finally, RPMI with 1.0 mM glutamate and 100 mM KCl was passed over the cells for 30 min before the treatment cycle repeated. After these treatments, the cells were allowed to recover for 30 min under flow in RPMI. Flow was stopped for two seconds in between treatments to prevent pressure backup during the valve change, otherwise a flow of 20 μL/min was maintained. After both treatment cycles, the membranes were removed and the electrodes were calibrated again. All treatments were done at 37°C and 5% CO2 in an incubator.
Discussion
Glutamate, the principal excitatory amino acid, plays a key role in neurocognition, yet it is excitotoxic in high quantities. Developing methods to detect glutamate is essential for understanding its biochemistry. In this work, a sensitive and stable glutamate sensor was developed and incorporated into the microclinical analyzer (μCA). This easy-to-use and versatile format (Figure 1) was then used for glutamate microphysiometry by adding model neuronal cells to the analytical chamber and measuring their response to the amino acid.
As evidence of the μCA's biological relevance, the glutamate sensor spanned the physiological to pathophysiological temporal range. The biologically relevant temporal range of glutamate parallels its scope of function. In traumatic brain injuries, extracellular excitotoxic glutamate builds up over the course of hours and can take days or weeks to return to normal levels. In synaptic signaling, glutamate is released from and taken up by neurons in less than a second. Therefore, a sensor that can respond on the sub-second time scale but is also stable for weeks is required to monitor this wide temporal range. Here, the sensor had a sub-second response time, making it suitable for monitoring rapid synaptic signaling. After 11 hours of use, the sensor retained 91 ± 1% of its original signal, demonstrating optimal stability over experimentally relevant time periods. During this time, the enzymatic sensor had a consistent (R2 = 0.96) and therefore predictable signal loss of 0.8 ± 0.1% per hour, which may be due to decreasing enzyme activity.3,21 Overall, the sensor showed the fast response time and long-term stability necessary for monitoring both glutamate signaling and trauma-induced excitotoxicity.
As further evidence of the sensor's biological relevance, the sensitivity of the electrode spanned the physiological to pathophysiological concentration range. Within the brain, glutamate concentration is thought to range from low micromolar under normal conditions up to hundreds of micromolar in conditions of stress, such as stroke.30,31 The sensitivity of the disk electrode (19 ± 1 μA mM−1 cm−2) enabled detection down to 8 ± 1 μM and quantitation down to 11 ± 1 μM (Figure 2). The upper limit of quantitation, beyond which the electrode began to saturate and was no longer able to linearly quantitate glutamate, was 1 mM. Between these limits (11–1000 μM), the sensor response was linearly quantifiable, making our sensor suitable for measuring physiological to pathophysiological concentrations.
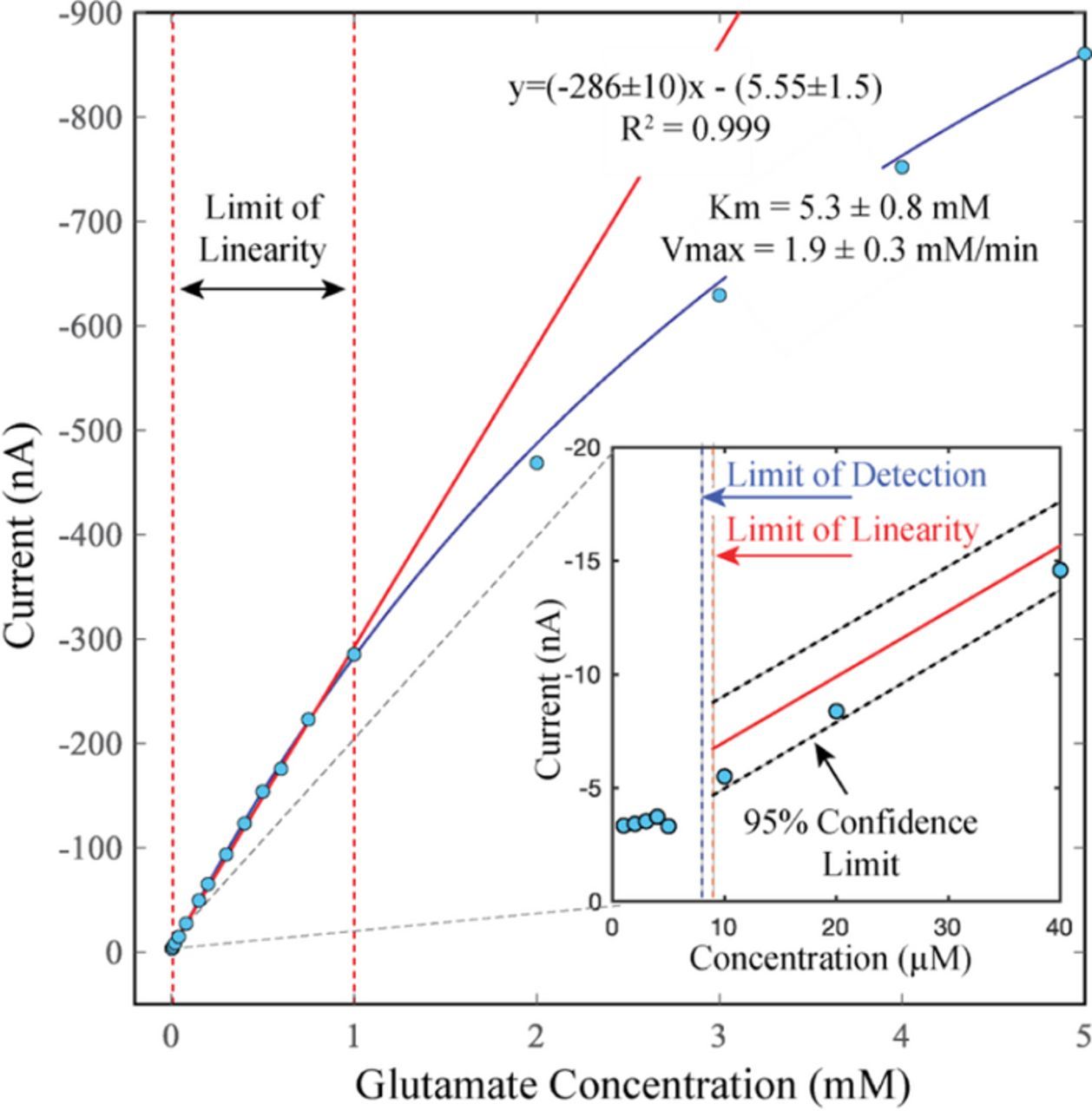
Figure 2. Representative calibration curve showing limits of linearity, limit of detection, Vmax and Km. The linear range, from 11 to 1000 μM glutamate, is demarcated by the dashed red lines and the slope is represented by the solid red line [y = (−286 ± 10)x–(5.55 ± 1.5), R2 = 0.999]. Divergence from linearity can be seen at 2 mM, where the data deviates from the linear red line. The Vmax and Km were calculated by non-linear regression (blue curve) to be 1.9 ± 0.3 μA/min and 5.3 ± 0.8 mM, respectively. Inset: zoom-in of the lower glutamate concentrations delineating the limit of detection vs. the lower limit of linearity. The limit of detection, indicated by the dashed blue line, was 8 ± 1 μM glutamate, the lower limit linearity (limit of quantitation) was 11 ± 1 μM glutamate. A confidence interval of 95% is represented by the dashed black lines. Experiments were performed in buffer (2 mM PBS, 120 mM KCl, pH 7) at ambient conditions, n = 3.
Even at high concentrations, small changes in glutamate could be observed. At the upper limits of linearity (1 mM), this system could detect a 1.5% (15 μM) difference in glutamate concentration with 95% confidence. Together with the low limit of quantitation (11 μM), the high-resolution of this system makes it a powerful analytical tool for measuring glutamate dynamics.
The sensor was designed to decrease interference by addition of Nafion. To examine the ability of Nafion to exclude interferents, the effect of ascorbic acid on glutamate detection was investigated both with and without a Nafion film. Ascorbic acid, a common interferent linked to glutamate physiology, introduced a large change in current (1.04 ± 0.05 nA/μM, n = 3) without the use of Nafion. With Nafion, this slope decreased (0.29 ± 0.04 nA/μM of ascorbic acid, n = 3), thereby mitigating ascorbic acid's interference. At 300 μM glutamate, the interference of 1 μM ascorbic acid decreased significantly with the use of Nafion (9.45 ± 0.08% without Nafion vs. 7.39 ± 1.14% with Nafion, n = 3, P < 0.1). This ability to exclude interference increased sensor accuracy and robustness for electrochemical microphysiometry.
This sensor also demonstrated optimal stability during storage. After one month in storage, the sensor retained 123 ± 11% of the signal. This may be due to a combination of the storage method (wet storage), the use of Nafion (antifouling), and the enzyme entrapment technique (crosslinking), which all stabilize enzyme sensors.6,17 The dynamic and durable nature of this sensor make it well suited for integration into the μCA to measure cellular microphysiometry.
The μCA bioreactor and microfluidic system were designed for enhanced analytical power (Figure 1). Although previous studies have also used microfluidics for glutamate detection, the cells were spatially removed from the electrode in these cases.23 Here, the cells and sensor were brought in close proximity within the bioreactor. The low limit of quantitation of the glutamate sensor combined with the small volume of the μCA bioreactor (V = 26 μL) allowed for monitoring small changes in glutamate such as those seen in synaptic signaling. This configuration improved analytical power by increasing signal-to-noise ratio and time resolution.32
In addition to increasing analytical efficiency, the microfluidic system also imposed shear stress on the cells, thereby mimicking physiological conditions. At a flow rate of 20 μL/min, the cells experience a calculated shear force of 32 mN/m2 (assuming 25°C, η = 0.89 cP, laminar flow), similar to that in the brain. The μCA can accommodate flowrates from low μL/min to mL/min to monitor cellular response under a range of physiological conditions.
Here, the μCA was used with PC12s, a model neuronal cell line, to track glutamate microphysiometry upon exposure.33 PC12s, derived from rat pheochromocytomas, have been used extensively to study glutamate metabolism, toxicity, and cell signaling.34 Within the μCA bioreactor, glucose-starved PC12 cells took up 210 ± 100 μmoles of the provided glutamate (35 ± 16% or 7 ± 3 μmoles/min), which increased to 390 ± 50 μmoles (65 ± 9% or 13 ± 2 μmoles/min) during the second exposure (Figure 3). This uptake was not seen when cells were cultured in glucose-containing media. Extracellular exposure to glutamate triggers uptake via EAAC, GLT-1 and GLAST proteins. Once inside the cell, glutamate may act as a fuel substrate for the Krebs cycle and restore bioenergetic homeostasis following prolonged PC12 depolarization and glucose starvation. That the amount of glutamate taken up by PC12 cells increased during a second treatment suggests that these neurotransmitter uptake systems were functional and may increase to mitigate excitotoxicity. Other cell types and enzyme sensors can be used with this system to monitor cellular microphysiometry in response to a range of drugs and environmental toxins.
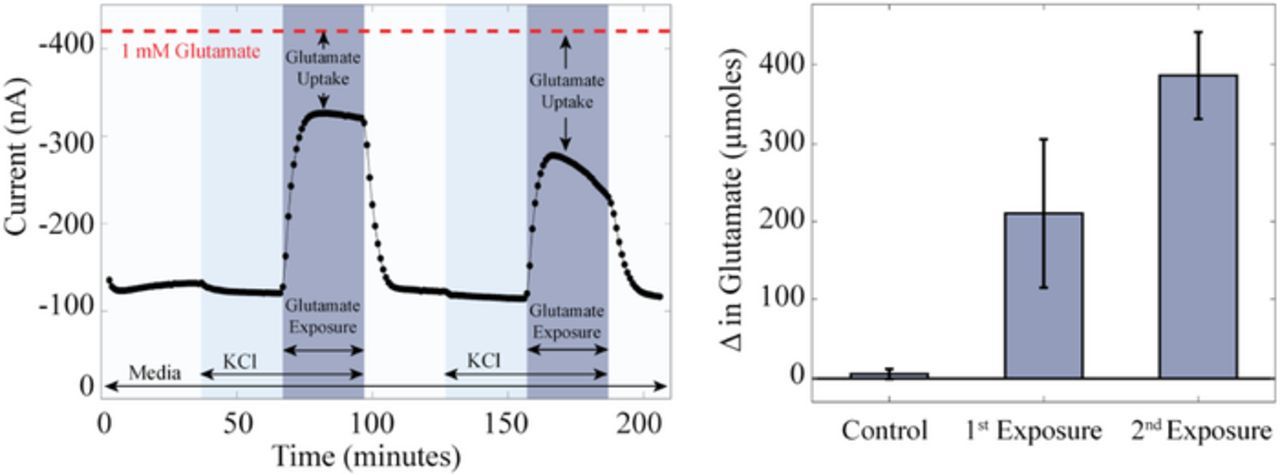
Figure 3. Representative amperometric i-t curve during a typical four-hour experiment (Left), and average uptake of glutamate by cells during glutamate exposure (Right). Left) During a typical four-hour experiment PC12 cells were perfused with two cycles of media (glucose-free RPMI, white pillars), 100 mM KCl (light blue pillars), and 100 mM KCl with 1.0 mM glutamate (dark blue pillars) followed by media for 30 min each. The dashed red line indicates the current generated by the addition of 1.0 mM glutamate with no cells present. Right) On average, cells took up 210 ± 100 μmoles (35 ± 16%) of the glutamate, which increased to 390 ± 50 μmoles (65 ± 9%) during the second exposure. All experiments were performed at 37°C, 5% CO2, and 20 μL/min in media (glucose-free RPMI) and are represented as the mean (n = 8) and standard error of the measurements, control vs. 1st exposure: p ⩽ 0.062: control vs. 2nd exposure: p ⩽ 0.0002: 1st exposure vs. 2nd exposure: p ⩽ 0.068.
Glutamate detection can aid in understanding excitotoxicity following traumatic brain injury, synaptic signaling, and perhaps even memory and consciousness. Glutamate can be detected electrochemically with almost no sample manipulation, increasing time resolution and sample throughput. Electrodes can be inexpensive (e.g. screen printed) and, when enzymatically modified, are sensitive and selective.20 The μCA is an easy-to-use and versatile sensor format that can monitor cellular response under a range of physiological conditions and exhibits desirable signal-to-noise ratio and ample time resolution. Patients and clinicians have already benefited from the highly translational nature of electrochemistry and expanding the repertoire of analytes and analytical power of these systems may facilitate the automation and personalization of medicine.
Conclusions
In this work, a glutamate sensor showed the fast response time (<1s), large linear range (11–1000 μM), and long-term stability (weeks) necessary for monitoring both glutamate signaling and trauma-induced excitotoxicity. PC12 cells were incorporated into the microclinical analyzer for real-time monitoring of cellular glutamate microphysiometry. On average, glucose-starved PC12 cells took up 210 ± 100 μmoles of the provided glutamate, which increased to 390 ± 50 μmoles during the second exposure. Other sensors or cell types could be incorporated into this system to track cellular response to a variety of drugs and environmental toxins.
Acknowledgments
This work was supported in part by IARPA grant number 2017-17081500003, EPA grant number 83573601, NIH training grant number ES007028, and using the resources of the Vanderbilt Microfabrication Core operated by the Vanderbilt Institute for Integrative Biosystems Research and Education. We thank Kazi Tasneem for her modeling of the shear stress, professors John P. Wikswo and BethAnn McLaughlin for their critical review of the manuscript, and Jennifer McKenzie, Ph.D. and David K. Schaffer, M.S. for their contributions to the microclinical analyzer design and fabrication.
ORCID
Ethan S. McClain 0000-0001-7677-0114
David E. Cliffel 0000-0001-8756-106X