

Abstract
Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF) is a promising electrocatalyst for the oxygen evolution reaction (OER) in alkaline solution. The OER activities of BSCF are gradually enhanced by prolonging the duration of electrochemical operation at OER potentials, but the underlying cause is not fully understood. In this study, we investigated the role of chemical operation, equivalent to immersion in alkaline solution, in the time-course of OER enhancement of BSCF. Interestingly, the time-course OER enhancement of BSCF was promoted not only by electrochemical operation, which corresponds to potential cycling in the OER region, but also by chemical operation. In situ Raman measurements clarified that chemical operation had a lower rate of surface amorphization than electrochemical operation. On the other hand, the leaching behavior of A-site cations was comparable between chemical and electrochemical operations. Since the OER activity of BSCF was stabilized by saturating the electrolyte with Ba2+, "chemical" A-site leaching was key to inducing the time-course OER enhancement on perovskite electrocatalysts. Based on these results, we provide a fundamental understanding of the role of chemical operation in the OER properties of perovskites.
Export citation and abstract BibTeX RIS
The oxygen evolution reaction (OER) is a very important process in energy-storage devices such as water electrolyzers 1 and rechargeable metal-air batteries, 2,3 but a serious problem is that the reaction rate is very sluggish. Therefore, many studies have been performed on the development of highly active and low-cost electrocatalysts. 4–7 Particularly, a family of perovskite oxides with the chemical formula ABO3 has attracted great interest as a promising electrocatalyst for the OER in alkaline solution because of its structural and compositional flexibility. 8–10
Ba0.5Sr0.5Co0.8Fe0.2O3−δ (BSCF), which exhibits quite high OER activity in alkaline solution, is considered to be a promising OER electrocatalyst. 11 Recent studies have clarified that the OER activity of BSCF gradually increases with an increase in the duration of operation at OER potential regions. Structural analyses using ex situ Raman spectroscopy, high-resolution transmission electron microscopy and X-ray absorption spectroscopy (XAS) revealed that the surface of electrochemically-cycled BSCF at the OER potential region becomes amorphized with conversion of the local structure of CoO6 units from corner-sharing to edge-sharing octahedra, which leads to the enhancement of pseudo-capacitance and OER activity. 12,13 In contrast, neither structural nor OER-activity changes were observed for conventional perovskite oxides such as LaCoO3. Furthermore, Fabbri et al. also showed by operando XAS that dynamic self-reconstruction at the surface of BSCF induced the formation of B-site oxyhydroxide at the OER potential region, which was responsible for its high OER activity. 14 These structural transformations are considered to be associated with A-site leaching and result in lattice OER, which is facilitated by the structural lability of BSCF with abundant oxygen defects.
Another unique property of BSCF is that its surface reacts chemically with electrolyte components such as OH− and H2O. In fact, Shen et al. have argued that A-site leaching at its surface induced by immersion in alkaline solution leads to exposure of the underlying Co/Fe spinel-like structure. 15 Moreover, oxygen vacancies in BSCF can uptake water (H2O + VO·· + OO × → 2OHO·). 16,17 These studies motivated us to examine the possible roles of "chemical" immersion in the "electrochemical" time-course OER enhancement of the BSCF electrocatalyst.
We investigated the dependence of time-course OER properties on the duration of immersion in alkaline solution and/or the choice of electrolytes. Interestingly, the OER activity of BSCF gradually increased not only with application of the OER potential (i.e., electrochemical operation) but also by immersing it in KOH solution (i.e., chemical operation). While a remarkable structural transformation was not observed after immersion in the alkaline solution, A-site leaching at the BSCF surface was observed, which could be associated with the time-course enhancement of OER activity by the chemical operation. Since the OER activity of BSCF became stabilized when A-site leaching was suppressed by saturating the KOH electrolyte with Ba2+, this study highlights the importance of A-site leaching for triggering the time-course OER enhancement of BSCF during electrochemical operation.
Experimental
Synthesis and characterization of electrocatalyst
BSCF and La0.5Sr0.5Co0.8Fe0.2O3−δ (LSCF) were synthesized by the sol-gel method as described previously. 11,18 For BSCF, stoichiometric amounts of metal nitrates (FUJIFILM Wako Pure Chemical, Japan) and glycine (Nacalai Tesque, Japan) were added to ultrapure water at 0.2 and 0.1 mol dm−3 (M), respectively. The solution was then heated until the evaporation of water, followed by calcination at 1100 °C in air for 24 h. For LSCF, stoichiometric amounts of metal nitrate (FUJIFILM Wako Pure Chemical, Japan) were dissolved in ultrapure water, and then citric acid (Nacalai Tesque, Japan) was added to the total metal cations at a molar ratio of 2:1. After a transparent solution was obtained, the solution pH was adjusted to between 6 and 8 by NH4OH (Nacalai Tesque, Japan). The solution was then evaporated, followed by calcination at 1000 °C in air for 2 h.
X-ray diffraction (XRD) patterns were collected by a RINT-2200 (Rigaku) with Cu Kα radiation. Specific surface area measurements by the Brunauer–Emmett–Teller (BET) analysis were conducted with a BELSORP-max (BEL Japan). In situ Raman spectroscopy was performed with a LabRAM HR-800vis KE (Horiba) at room temperature to observe structural changes at the BSCF surface during KOH immersion. X-ray photoelectron spectroscopy (XPS) was carried out with an ESCA-3400HSE (Kratos Analytical) equipped with an Al Kα X-ray source to observe the surface state of BSCF before and after KOH immersion. Quantification of dissolved BSCF and LSCF components in KOH electrolyte was conducted with an inductivity coupled plasma optical emission spectrometer (ICP-OES, ThermoFisher SCIENTIFIC iCAP7000).
Electrochemical measurement
OER activities of BSCF and LSCF were measured in 1.0 M KOH solution using a three-electrode cell with a rotating disk electrode (RDE) setup. Pt wire and a reversible hydrogen electrode (RHE) were used as the counter and reference electrodes, respectively. The working electrodes were prepared by drop-casting catalyst ink on a glassy carbon RDE at loading amounts of 250 μg cm−2 catalysts and 50 μg cm−2 Nafion (5 wt.%, Merck). Notably, carbon as a conductive material was not added to remove the influence of carbon on the OER activity of BSCF. 19,20 Cyclic voltammetry (CV) was performed in the potential range of 1.1 to 1.7 V vs RHE at a scan rate of 10 mV s−1 and at a rotation rate of 900 rpm in O2-saturated 1 M KOH solution. Ba2+-saturated KOH solution with a hydroxide concentration of 1.0 M was also used for comparison of CV. The OER current was normalized by the BET surface area of oxides.
A three-electrode beaker cell was also used to evaluate the leaching behaviors of BSCF and LSCF. The working electrode was fabricated by spray-coating perovskite catalysts and Nafion on carbon paper (TGP-H-030, Toray) at loading amounts of 2.5 and 0.25 mg cm−2, respectively. O2-saturated 1 M KOH solution, Hg/HgO electrode, and Pt mesh were used as an electrolyte, reference electrode, and counter electrode, respectively. For the chemical operation, the electrolyte sample for ICP-OES measurement was prepared by immersing the working electrode in KOH for 100 min. Electrochemical operation was performed by conducting CV measurements in the potential range of 1.1 to 1.7 V vs RHE at a scan rate of 10 mV s−1 for 50 cycles.
Results and Discussion
As shown in Fig. 1, the crystalline phases of synthesized BSCF and LSCF were confirmed to have a cubic perovskite structure by XRD, although a small amount of (La/Sr)2(Co/Fe)O4 with a Ruddlesden-Popper structure was present as an impurity in LSCF powder. The BET surface areas of BSCF and LSCF were approximately 0.16 and 2.0 m2 g−1, respectively.
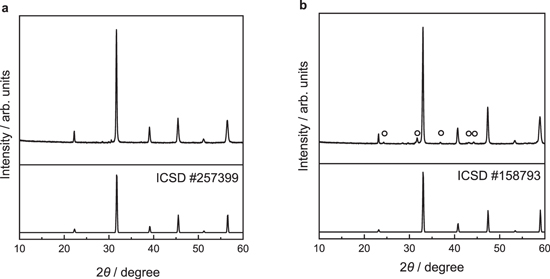
Figure 1. X-ray diffraction patterns of (a) BSCF and (b) LSCF. The circle symbols represent the diffraction pattern from (La/Sr)2(Co/Fe)O4 with a Ruddlesden-Popper structure.
Download figure:
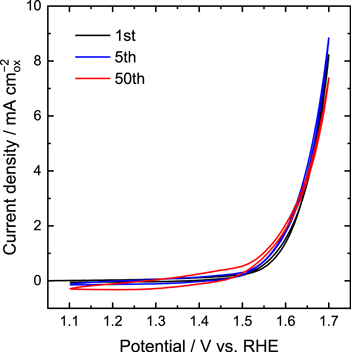
Standard image High-resolution imageCyclic voltammograms of BSCF and LSCF at the 1st, 5th, and 50th cycles are shown in Figs. 2a and 2b, respectively. The OER current of BSCF significantly increased during OER operation, which is consistent with a previous study. 12 The OER current at 1.7 V vs RHE at the 50th cycle was approximately 4.5 times larger than that at the 1st cycle. In contrast, no considerable increase in the OER current was observed for LSCF. Figure 2c shows Tafel plots of BSCF and LSCF at the 1st and 50th cycles obtained from average values of cathodic and anodic scans of the CVs. The value of the Tafel slope for BSCF at the 50th cycle was smaller than that at the 1st cycle, suggesting that OER kinetics became faster with an increase in the number of CV cycles. On the other hand, Tafel plots of LSCF did not change throughout CV.
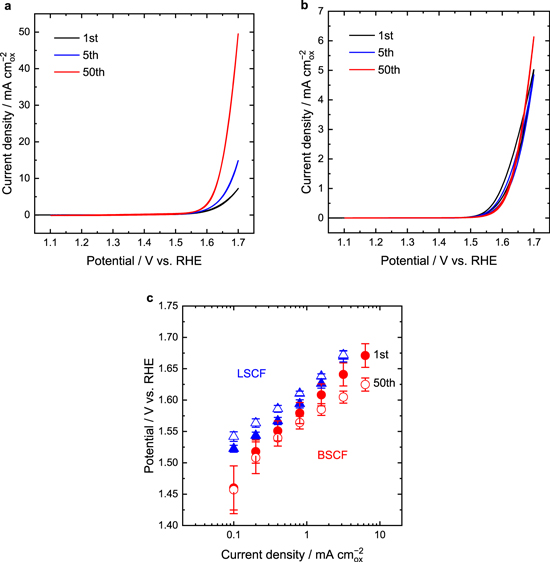
Figure 2. Cyclic voltammograms of (a) BSCF and (b) LSCF RDEs at selected cycles. (c) Tafel plots of BSCF and LSCF obtained from cyclic voltammetry. Error bars represent standard deviation from at least three independent measurements. Closed and open symbols represent the 1st and 50th cycles, respectively.
Download figure:
Standard image High-resolution imageTo identify the effects of chemical operation on the time-course enhancement of OER activity for BSCF, the influence of the duration of immersion in 1.0 M KOH solution was investigated. Figure 3a shows the results of intermittent CVs for BSCF. Here, we defined intermittent CV as a CV technique in which an open circuit potential (OCP) sequence is inserted between CV cycles for a specified period. Therefore, the duration of the OCP sequence was zero for ordinary CV (hereafter we call this "continuous CV"). The interval duration of the OCP sequence was set to 10 min after the 1st–3rd CV scans, and to 20 min after the 4th–7th CV scans. As expected, the OER activity of BSCF gradually increased with intermittent CV. Fig. 3b compares the increase ratios for OER currents at 1.7 V vs RHE between intermittent and continuous CVs. Interestingly, while the increase ratio of OER activity for the continuous CV at the 5th cycle was ca. 1.6, that for intermittent CV was ca. 3.0, indicating that the OER activity of BSCF could be further boosted by the addition of OCP sequences. The increase ratio at the 7th cycle with intermittent CV was as high as that at the 30th cycle with continuous CV. These results suggest that not only electrochemical operation in OER potential cycling but also chemical operation by simply immersing BSCF in KOH can produce a time-course enhancement of the OER activity for BSCF.
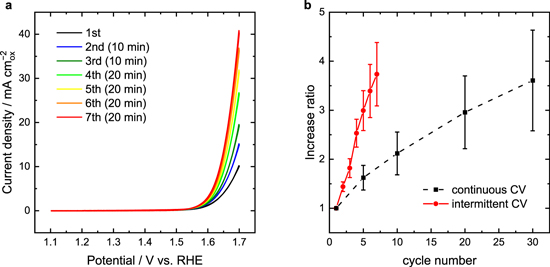
Figure 3. (a) Intermittent cyclic voltammograms of BSCF RDEs. Values in parentheses show interval duration between the CV cycles. (b) Increase ratio of OER currents at 1.7 V vs RHE as a function of cycle number obtained by continuous and intermittent CVs. Error bars represent standard deviation from at least three independent measurements.
Download figure:
Standard image High-resolution imageNext, the influence of the duration of immersion in KOH solution on OER activities was investigated to more clearly determine the effect of chemical operation. We established another CV protocol with the following sequence: (1) 1st CV cycling for one cycle; (2) OCP for a specified period (2, 10, 50, or 100 min); (3) 2nd CV cycling for one cycle. The resultant increase ratio of OER current at 1.7 V vs RHE at the 1st CV cycle to that at the 2nd cycle was calculated and is shown in Fig. 4. The increase ratio of the OER currents increased with an increase in the duration of the OCP period, suggesting that the chemical enhancement of OER activity for BSCF was positively correlated with the duration of alkali immersion. On the other hand, the increase ratio of the OER activity after immersion in alkaline solution for 100 min was still lower than that after the 50th cycle of continuous CV, which took 100 min. This suggests that, while chemical operation could contribute to the time-course enhancement of OER activity, the degree of OER enhancement during the chemical operation would be less than that during electrochemical operation.

Figure 4. Increase ratio of OER currents at different duration at OCP. The result from continuous CV for 50 cycles, which corresponded to 100 min, are also shown for comparison. Error bars represent standard deviation from at least three independent measurements.
Download figure:
Standard image High-resolution imageThe In situ behavior of the BSCF surface during immersion in 1.0 M KOH solution was analyzed by In situ Raman spectroscopy to discuss the correlation between the time-course OER enhancement and immersion in alkaline solution. Previously, it was reported that amorphization of BSCF after OER cycles led to enhanced OER activity. 12 As shown in Fig. 5, the Raman spectrum of pristine BSCF had a broad peak at approximately 675 cm−1, which was assigned to the internal motion of oxygen atoms in CoO6 and/or FeO6 octahedra. 21 In situ Raman spectra of BSCF just after immersion in KOH solution and after immersion for 120 min are also shown in Fig. 5. The peak at 675 cm−1 was still observed after immersion for 120 min, although it was somewhat broadened after 120 min. Transmission electron microscopy imaging and electron energy–loss spectroscopy techniques in previous work revealed that Co/Fe spinel–like structure was exposed accompanied with leaching Ba/Sr rich secondary phase by immersion into KOH solution. 15 However, the Raman spectra 22 attributed to Co/Fe spinel structure (approximately 420 and 620 cm–1) were not observed. This is probably because these peaks attributed to Co/Fe spinel structure are hidden by a broad peak of BSCF. Furthermore, in a previous work, 12 the Raman peak at 675 cm−1 almost disappeared after 50 cycles in 0.1 M KOH (i.e., 100 min), which was attributed to surface amorphization of BSCF. Accordingly, the In situ Raman result implied that the amorphization rate at the BSCF surface by the chemical operation would be slower than that by the electrochemical operation, which was consistent with the different increase ratios of OER activity between the chemical and electrochemical operations.
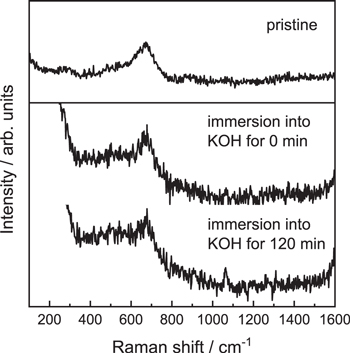
Figure 5. Raman spectra of BSCF before and during immersion in 1.0 mol dm−3 KOH solution.
Download figure:
Standard image High-resolution imageTo take a closer look at the effects of A-site leaching on OER properties, XPS and ICP-OES were carried out. Fig. 6a shows overlapped XPS Ba 3d and Co 2p spectra. Deconvolution of the XPS spectra clarified that Ba 3d spectra considerably decreased after immersion in alkaline solution for 100 min (from Ba/Co = ca. 0.86 to 0.12 by mol), suggesting that surface Ba2+ cation was leached by alkali immersion. To quantitatively evaluate the bulk-scale dissolution behavior of A-site cations, the amounts of dissolved La, Ba, Sr, Co, and Fe ions in 1.0 M KOH after immersion for 100 min or OER potential cycling for 50 cycles in 1.0 M KOH were verified by ICP-OES measurement (Fig. 6b). Here, dissolution ratio in this study was defined as the following equation:

While ca. 2−5% of the total A-site cations in BSCF were leached by alkali immersion, B-site cations were hardly leached, indicating that alkali immersion selectively promoted A-site leaching. Since the dissolution ratio of elemental Ba obtained from ICP-OES was much smaller than that obtained from XPS, it was suggested that A-site leaching during alkali immersion occurred mainly at the BSCF surface. On the other hand, while La cation in LSCF was hardly leached as expected, the dissolution rate of Sr was approximately half of that in BSCF. Higher dissolution ratios of Sr2+ in BSCF compared with LSCF might attribute to the soluble Ba/Sr rich phases on BSCF surface formed by reaction between BSCF and CO2 and/or H2O. 23,24 Furthermore, it was evident that the dissolution ratios of the components of BSCF and LSCF after immersion were surprisingly comparable to those after the OER for 50 cycles. This suggests that the A-site leaching after the OER for 50 cycles did not result from the electrochemical operation, but rather resulted from the chemical operation. Accordingly, it can be concluded that one main role of the chemical operation is the leaching of A-site cations rather than amorphization of the BSCF surface.
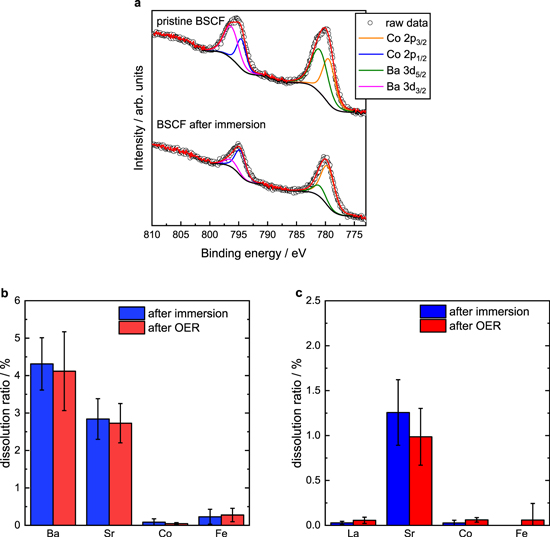
Figure 6. (a) Co 2p and Ba 3d X-ray photoelectron spectra of pristine BSCF and BSCF after immersion in KOH for 100 min. Dissolution ratios of (b) BSCF and (c) LSCF elements in electrolyte measured by ICP-OES after immersion and OER cycling.
Download figure:
Standard image High-resolution imageTo prove the importance of A-site leaching for the time-course OER enhancement, another CV measurement was performed in Ba2+-saturated KOH solution with a hydroxide concentration of 1 M (Fig. 7). The OER activity of BSCF did not increase during CV cycling in the Ba2+-saturated KOH solution. On the other hand, pseudo-capacity at potentials < 1.6 V vs RHE, which could be assigned to either or both Faradaic reaction of B-site oxyhydroxide or/and the electrochemical surface area, increased at 50th cycle. It is known that the structural change of BSCF can be associated with the lattice oxygen evolution reaction (LOER). The leached metal cations caused by LOER undergo either the re-deposition on the surface of oxides or the diffusion to the bulk electrolyte. 14,25 In case of 1 M KOH without Ba2+ addition, while A-site cations can leach and diffuse away from the electrode during immersion, B-site cations would be re-deposited on BSCF surface due to low solubility. On the other hand, in case of Ba2+-saturated KOH, both A-site and B-site cations would be re-deposited to form A-site carbonates and/or hydroxides and B-site oxyhydroxide. This result indicates that, the OER activity in Ba2+-saturated KOH could not become enhanced because the re-deposited A-site carbonate and/or hydroxide would block the surface of B-site oxyhydroxide. With respect to the structural change of perovskite catalysts, it has been proposed that transition-metal oxides with the O p-band center too close to the Fermi level are unstable at OER potential, and surface amorphization of perovskite catalysts is accompanied by A-site leaching during the OER. 12,14,26 In the present study, on the other hand, the time-course OER enhancement were suppressed by simply using Ba2+-saturated electrolyte even when we used the BSCF electrocatalyst, which had the O p-band center close to the Fermi level. Therefore, it is conceivable that "chemical" A-site leaching without re-deposition of A-site carbonates and/or hydroxides is essential for perovskite catalysts to trigger the time-course OER enhancement. In fact, since solubility products of Ba(OH)2 and La(OH)3 are approximately 10−2 and 10−20, respectively, 27,28 Ba2+ in BSCF was dissolved while La3+ in LSCF was not dissolved into 1.0 mol dm–3 KOH solution (Figs. 6b, 6c). The contrasting solubilities of A-site elements were in good agreement with the resultant time-course changes in their OER activities. We should note that although LSCF had much larger surface area, LSCF showed lower dissolution rate of Sr2+, implying that actual dissolution rate of Sr2+ per unit surface area would be much lower for LSCF. Therefore, the stable OER behavior of LSCF could be associated with the suppressed rate of inherent Sr2+ dissolution as well as the negligible rate of La3+ dissolution. Although further in-depth analyses will be required to clarify the correlation between the O p-band center relative to the Fermi level and the solubility of A-site cations, the present results suggest that the solubility of A-site cations should be a key descriptor to discuss the time-course enhancement of the OER activities of perovskite catalysts.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Cyclic voltammograms of BSCF RDEs at selected cycles in Ba2+-saturated KOH solution with the hydroxide concentration of 1 mol dm−3.
Download figure:
Standard image High-resolution image{kind=link}
Conclusions
In conclusion, we have shown that the time-course enhancement of the OER activity for BSCF occurs not only with electrochemical operation, which corresponds to potential cycling in the OER region, but also with chemical operation, which corresponds to immersion in alkaline solution. There was a positive correlation between the duration of immersion and the increase ratio of the OER activity of BSCF. Surface-scale and bulk-scale characterizations revealed that, while the amorphization rate of the BSCF surface during alkali immersion was lower than that during potential cycling in the OER region, the rates of A-site leaching at the BSCF surface were comparable. The suppressed time-course OER enhancement of BSCF in Ba2+-saturated KOH electrolyte demonstrated the importance of selective A-site leaching without re-deposition. This study highlights the crucial role of A-site leaching by chemical operation for triggering the time-course OER enhancement, which can lead to new design strategies for highly-active OER electrocatalysts.
Acknowledgments
This work was supported by JSPS KAKENHI Grant Number 19K15678.