

Abstract
A KI-catalyzed indirect electrochemical oxidative method for the synthesis of sulfenylated 4-hydroxycoumarins via cross-coupling of 4-hydroxycoumarins and aryl thiols at a low potential has been reported. The electrocatalytic activity of KI for sulfenylation of 4-hydroxycoumarin was investigated by cyclic voltammetry. In situ FTIR data reflected the structural change of functional groups during the reaction process. The mechanism of electrochemical sulfenylation involved the generation of intermediate 1,2-bis(4-chlorophenyl)disulfane has been revealed by control experiments. Various sulfenylated 4-hydroxycoumarins were obtained under the optimum reaction conditions in moderate to excellent yields with good functional group tolerance.
Export citation and abstract BibTeX RIS
Direct C–S bond formation is vital to synthesize many sulfur-containing compounds which are widely existed in bioactive compounds, natural products, pharmaceuticals and functional materials.
1–4
Many compounds containing C–S bond in their core structures display a wide variety of biological activities, such as tubulin inhibitory properties, antimalarial, anti-inflammatory and anti-HIV activities.
5–8
Sulfenylated coumarins are one of the important sulfur-containing compounds, and several synthetic approaches for sulfenylation of 4-hydroxycoumarins have been developed.
9–13
For example, a direct sulfenylation of 4-hydroxycoumarins with arylsulfonyl hydrazides catalyzed by CuBr·Me2S under refluxing condition (100 °C) has been reported and the sulfenylated products were obtained in good yields (Scheme
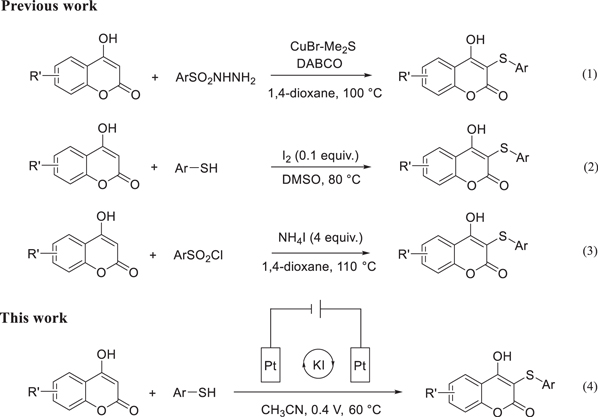
Scheme 1. Construction of 3-sulfanyl-4-hydroxycoumarins.
Download figure:
Standard image High-resolution imageIodine, as a catalytic mediator for organic chemical reactions, has increasingly aroused general interest due to its low cost, easy availability, and non-toxicity.
15–21
An efficient method for synthesis of sulfenylated 4-hydroxycoumarins from aryl thiols and 4-hydroxycoumarins in the presence of I2/DMSO has been reported (Scheme
Electrosynthesis has been regarded as an efficient and environmentally friendly synthetic method, which could form C–S bond through the electrochemical oxidative cross-coupling without transition-metal catalyst and external oxidant. 23–26 Notably, the anodic oxidation of iodide ions in an electrochemical reaction was a green and sustainable method to replace equivalent oxidants in an traditional organic reaction. 27,28 In 2018, Sun et al. established an indirect electrochemical method to synthesize selenyl heteroarenes through iodide-catalyzed electrolytic selenation. 29 Pan et al. synthesized 3,5-disubstituted-1,2,4-thiadiazoles from dimerization of thioamides with NH4I as the redox catalyst and supporting electrolyte. 30 Our group established an indirect electrochemical system for sulfenylation of indoles with disulfides to produce 3-sulfenylindoles through C–S formation catalyzed by KI at 0.4 V. 31 Indirect electrooxidation method for synthesis of aryl sulfides from aryl thiols and electron-rich arenes catalyzed by KI at low potential has also been realized. 32 Under the optimal reaction conditions, polymerization of indoles and the excessive oxidation of thiols could be effectively avoided.
Compared with arylsulfonyl hydrazides,
33
sodium arylsulfinates
34
and arylsulfonyl chlorides,
35
aryl thiols were promising sulfenylating reagents with a wide range of substrates and good substrate stability.
36
Herein, aryl thiols were chosen as the sulfenylating reagents to react with 4-hydroxycoumarins through an indirect electrochemical oxidation reaction to synthesize sulfenylated 4-hydroxycoumarins (Scheme
Experimental
General information
Unless otherwise noted, all commercially available solvents and chemicals were purchased from commercial suppliers and were used without further purification. 4-Hydroxy-6-methyl-2H-chromen-2-one (1b) and 4-hydroxy-6-methoxy-2H-chromen-2-one (1c) were synthesized according to the procedure of literature. 37 1H NMR and 13C NMR spectra were recorded with DMSO-d6 as the solvent and tetramethylsilane (TMS) as the internal standard at a Bruker Avance III HD spectrometer at 500 MHz for 1H NMR and 125 MHz for 13C NMR.
Electrochemical characterization
Cyclic voltammetry was performed in CH3CN solution (15 ml) using NaBF4 (0.1 mol l−1) as the supporting electrolyte in 25 ml undivided cell to determine the redox properties of each compound. A three-electrode system was adopted in the experiments, containing L-type Pt electrode (3 mm in diameter) as the working electrode, platinum sheet (2.25 cm2) as the counter electrode, and Ag/Ag+ electrode (0.1 mol l−1 AgNO3 in CH3CN) as the reference electrode. Potential was applied using CHI600e electrochemical workstation (CH Instrument Inc., Austin, TX, USA).
In situ FTIR spectroscopy experiments were carried out on Nicolet 670 FTIR spectrometer equipped with a MCT-A detector cooled by liquid nitrogen. The working electrode was a Pt disk (6 mm in diameter). A three-electrode spectro-electrochemical cell with CaF2 window at the bottom was used. IR spectra was calculated as:
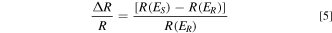
In the above equation, R (E S ) and R (E R ) were the single beam spectra separately received at the sample potential (E S ) and the reference potential (ER). 38–40 The reference potential (E R ) was selected at 0 mV. Each spectrum was collected at 8 cm−1 resolution at different potentials. ΔR/R represented the potential difference of the single-beam spectra collected at different potentials.
Synthesis of 4-hydroxycoumarins
4-Hydroxycoumarins had been prepared according to Scheme
Scheme 2. Synthesis of 4-hydroxycoumarins.
Download figure:
Standard image High-resolution imageElectrolysis experiments
Electrolysis experiments were performed on Vertex Potentiostat/Galvanostat. Both the working electrode and the counter electrode were platinum electrodes (1.5 cm × 1.5 cm). The reference electrode was Ag/Ag+ electrode (0.1 mol l−1 AgNO3 in CH3CN). 4-Hydroxycoumarin (1a, 0.5 mmol, 81.1 mg), 4-chlorothiophenol (2a, 0.75 mmol, 108.5 mg), KI (0.05 mmol, 8.3 mg) and 0.1 mol l−1 NaBF4/CH3CN solution (15 ml) were added into 25 ml undivided cell. The electrolysis reactions were operated at 0.4 V under 60 °C for 6 h. The resulting mixture was concentrated under reduced pressure and purified by column chromatography on silica gel using petroleum ether/ethyl acetate (5:1) as eluent to afford 3-((4-chlorophenyl)thio)-4-hydroxy-2H-chromen-2-one (3aa) as a white solid in 94% yield.
Analytical data
4-Hydroxy-6-methyl-2H-chromen-2-one ( 1 b). —Yield: 71%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 12.43 (s, 1H), 7.58 (d, J = 1.15 Hz, 1H), 7.42–7.40 (m, 1H), 7.22 (d, J = 8.45 Hz, 1H), 5.56 (s, 1H), 2.35 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 165.6, 162.0, 151.7, 133.4, 133.1, 122.8, 116.1, 115.5, 90.9, 20.3.
4-Hydroxy-6-methoxy-2H-chromen-2-one ( 1 c). — Yield: 64%; Light red solid; 1H NMR (500 MHz, DMSO-d6) δ 12.49 (s, 1H), 7.29 (d, J = 9.3 Hz, 1H), 7.22–7.19 (m, 2H), 5.59 (s, 1H), 3.80 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 165.3, 162.0, 155.3, 147.9, 120.3, 117.6, 116.2, 105.0, 91.3, 55.6.
3-((4-Chlorophenyl)thio)-4-hydroxy-2H-chromen-2-one ( 3 aa). — Yield: 94%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.98–7.96 (m, 1H), 7.74–7.70 (m, 1H), 7.44–7.39 (m, 2H), 7.34–7.32 (m, 2H), 7.24–7.21 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 168.6, 160.8, 153.1, 135.2, 133.8, 130.2, 129.0, 128.0, 124.4, 124.3, 116.5, 115.8, 94.1.
3-((4-Fluorophenyl)thio)-4-hydroxy-2H-chromen-2-one ( 3 ab). — Yield: 90%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.97–7.96 (m, 1H), 7.72–7.69 (m, 1H), 7.42–7.39 (m, 2H), 7.30–7.28 (m, 2H), 7.15–7.12 (m, 2H); 13C NMR (125 MHz, DMSO-d6) δ 168.7, 161.4, 160.7 (d, J = 201.3 Hz), 153.5, 134.1, 131.8 (d, J = 2.5 Hz), 129.5 (d, J = 6.3 Hz), 124.9, 124.7, 116.9, 116.5 (d, J = 18.8 Hz), 116.2, 95.5.
3-((4-Bromophenyl)thio)-4-hydroxy-2H-chromen-2-one ( 3 ac). — Yield: 73%; Light yellow solid; 1H NMR (500 MHz, DMSO-d6) δ 7.98–7.96 (m, 1H), 7.73–7.70 (m, 1H), 7.46–7.39 (m, 4H), 7.16 (d, J = 7.2 Hz, 2H); 13C NMR (125 MHz, DMSO-d6) δ 169.2, 161.3, 153.6, 136.3, 134.2, 132.3, 128.8, 124.9, 124.7, 118.8, 117.0, 116.3, 94.3.
4-Hydroxy-3-(p-tolylthio)-2H-chromen-2-one ( 3 ad). — Yield: 71%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.96–7.94 (m, 1H), 7.71–7.67 (m, 1H), 7.42–7.37 (m, 2H), 7.13–7.08 (m, 4H), 2.23 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 167.9, 160.8, 152.9, 135.2, 133.5, 132.2, 129.7, 127.0, 124.3, 124.2, 116.4, 115.6, 95.3, 20.4.
4-Hydroxy-3-((4-methoxyphenyl)thio)-2H-chromen-2-one ( 3 ae). — Yield: 71%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.95–7.94 (m, 1H), 7.69–7.67 (m, 1H), 7.40–7.37 (m, 2H), 7.29–7.26 (m, 2H), 6.90–6.87 (m, 2H), 3.71 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 167.8, 161.3, 158.7, 153.3, 134.0, 130.5, 126.3, 124.8, 124.7, 116.9, 116.1, 115.3, 97.2, 55.7.
3-((2-Chlorophenyl)thio)-4-hydroxy-2H-chromen-2-one ( 3 af). — Yield: 75%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.99–7.98 (m, 1H), 7.75–7.72 (m, 1H), 7.47–7.40 (m, 3H), 7.22–7.15 (m, 2H), 6.97–6.96 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 169.5, 160.8, 153.2, 135.2, 133.8, 129.5, 127.7, 126.3, 125.8, 124.4, 124.2, 116.6, 116.0, 92.1.
3-((3-Chlorophenyl)thio)-4-hydroxy-2H-chromen-2-one ( 3 ag). — Yield: 94%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.97–7.95 (m, 1H), 7.73–7.69 (m, 1H), 7.43–7.38 (m, 2H), 7.32–7.27 (m, 1H), 7.23–7.15 (m, 3H); 13C NMR (125 MHz, DMSO-d6) δ 168.9, 160.9, 153.1, 138.8, 133.8, 133.7, 130.1, 125.5, 125.4, 124.8, 124.5, 124.2, 116.5, 115.8, 93.5.
4-Hydroxy-3-(naphthalen-2-ylthio)-2H-chromen-2-one ( 3 ah). — Yield: 95%; Light yellow solid; 1H NMR (500 MHz, DMSO-d6) δ 8.01–7.99 (m, 1H), 7.85–7.80 (m, 3H), 7.74–7.70 (m, 2H), 7.47–7.40 (m, 4H), 7.38–7.36 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 168.5, 160.9, 153.1, 133.6, 133.5, 133.4, 131.1, 128.6, 127.6, 126.9, 126.6, 125.5, 124.9, 124.4, 124.2, 123.9, 116.5, 115.8, 94.4.
4-Hydroxy-3-(thiophen-2-ylthio)-2H-chromen-2-one ( 3 ai). — Yield: 39%; Light yellow solid; 1H NMR (500 MHz, DMSO-d6) δ 7.94–7.92 (m, 1H), 7.68–7.64 (m, 1H), 7.52–7.51 (m, 1H), 7.38–7.35 (m, 2H), 7.29–7.28 (m, 1H), 6.99–6.98 (m, 1H); 13C NMR (125 MHz, DMSO-d6) δ 166.8, 160.7, 152.8, 133.6, 133.2, 132.2, 129.4, 127.5, 124.4, 124.3, 116.4, 115.6, 98.2.
4-Hydroxy-6-methyl-3-((4-chlorophenyl)thio)-2H-chromen-2-one ( 3 ba). — Yield: 77%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.75 (d, J = 1.0 Hz, 1H), 7.52–7.51 (m, 1H), 7.34–7.30 (m, 3H), 7.22–7.20 (m, 2H), 2.39 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 169.0, 161.4, 151.7, 135.7, 135.1, 134.1, 130.6, 129.4, 128.4, 124.4, 116.8, 115.8, 94.4, 20.8.
4-Hydroxy-6-methoxy-3-((4-chlorophenyl)thio)-2H-chromen-2-one ( 3 ca). — Yield: 74%; White solid; 1H NMR (500 MHz, DMSO-d6) δ 7.38–7.36 (m, 2H), 7.34–7.33 (m, 2H), 7.31–7.28 (m, 1H), 7.22 (d, J = 1.7 Hz, 1H), 7.21 (d, J = 1.6 Hz, 1H), 3.83 (s, 3H); 13C NMR (125 MHz, DMSO-d6) δ 168.8, 161.4, 156.0, 147.9, 135.6, 130.6, 129.4, 128.5, 122.1, 118.3, 116.5, 106.4, 94.8, 56.2.
Results and Discussion
Electrochemical analysis
Electrochemical sulfenylation of 4-hydroxycoumarin (1a, 0.5 mmol, 81.1 mg) with 4-chlorothiophenol (2a, 0.75 mmol, 108.5 mg) catalyzed by KI (0.05 mmol, 8.3 mg) in NaBF4/CH3CN solution (0.1 mol l−1, 15 ml) was studied by cyclic voltammetry. Cyclic voltammgrams of 1a, 2a and KI in the solution of NaBF4/CH3CN were collected respectively with potentials ranged from −0.6 V to 0.8 V vs Ag/Ag+ electrode at the scan rate of 50 mV s−1, and results were shown in Fig. 1. Two typical oxidation peaks of iodide ions were appeared at −0.05 V and 0.32 V in curve b, which corresponded to the oxidation of I− to I3 − and I3 − to I2, respectively. 41 As shown in curve a, no obvious redox peak was observed, indicating that 1a was difficult to be oxidized in the potential range of −0.6 V–0.8 V. While oxidation current of 2a started to increase gradually at about 0.38 V (curve c).
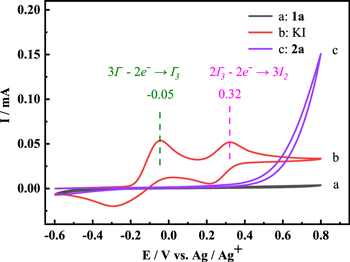
Figure 1. Cyclic voltammograms recorded at the scan rate of 50 mV s−1 in 0.1 mol l−1 NaBF4/CH3CN with (a) 1a (0.5 mmol); (b) KI (0.05 mmol); (c) 2a (0.75 mmol).
Download figure:
Standard image High-resolution imageFig. 2 (A) explored the reaction of 2a in the presence of KI in NaBF4/CH3CN solution. The oxidation current for the mixture of 2a and KI (curve e) increased sharply compared with curves b and c, and the potential of oxidation peak moved from 0.32 V to 0.54 V. It indicated I2 had high catalytic activity and 2a could be rapidly oxidized in the presence of I2.
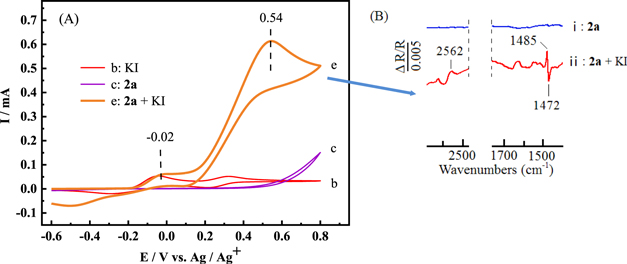
Figure 2. (A) Cyclic voltammograms recorded at the scan rate of 50 mV s−1 in 0.1 mol l−1 NaBF4/CH3CN with (b) KI (0.05 mmol); (c) 2a (0.75 mmol); (e) 2a (0.75 mmol) and KI (0.05 mmol). (B) In situ FTIR spectra collected at the potential of 300 mV in 0.1 mol l−1 NaBF4/CH3CN solution with (ⅰ) 2a (1.5 mmol); (ⅱ) 2a (1.5 mmol) and KI (0.1 mmol).
Download figure:
Standard image High-resolution imageIn situ FTIR spectroscopy was an effective technique to study the surface reaction characteristics of the electrode surface. To further explore the consumption of reactants and the generation of intermediates or products in the reaction process, in situ FTIR spectroscopy was introduced. In situ FTIR spectra collected during the reaction of 2a and KI in 0.1 mol l−1 NaBF4/CH3CN solution at the potential of 300 mV were shown in Fig. 2 (B). The positive-going bands represented the consumption of reactants, and the negative-going bands represented the formation of intermediates or products. 42–46 The mixture of 2a and KI in NaBF4/CH3CN solution (curve ⅱ) showed obvious infrared signals compared with 2a in NaBF4/CH3CN solution (curve ⅰ). The positive-going bands at 2562 cm−1 and 1485 cm−1 were respectively attributed to the S-H stretching vibration and the aromatic ring skeleton vibration of 2a, which indicated that 2a was consumed. The negative-going band 1472 cm−1 could be attributed to the characteristic peak of the aromatic ring skeleton vibration of 1,2-bis(4-chlorophenyl)disulfane produced by 2a. It showed that the corresponding 1,2-bis(4-chlorophenyl)disulfane was generated during the reaction. 47
When KI was added into the NaBF4/CH3CN solution with 1a, the corresponding cyclic voltammetric curve was shown in Fig. 3 (A). As compared with curve b, the oxidation current of the mixture of 1a and KI (curve d) had a slight increase, which indicated that 1a had a small voltammetric response at the electrode in the presence of I2. To further verify the result of cyclic voltammetry, we collected in situ FTIR spectra of 1a and KI in 0.1 mol l−1 NaBF4/CH3CN solution with the time. In order to clearly observe the change of spectra signals, the in situ FTIR spectra was collected at 500 mV. As shown in Fig. 3 (B), with the increase of scanning time, the negative-going band of 977 cm−1 could be clearly observed which was the signal of C-I bond generated from the reaction of 1a and I2. The reaction of 4-hydroxycoumarin and I2 to form 4-hydroxy-3-iodo-2H-chromen-2-one was shown as the Eq. 6 48 :
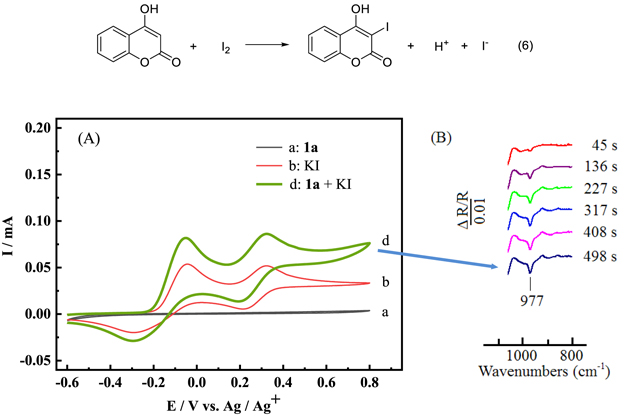
Figure 3. (A) Cyclic voltammograms recorded at the scan rate of 50 mV s−1 in 0.1 mol l−1 NaBF4/CH3CN with (a) 1a (0.5 mmol); (b) KI (0.05 mmol); (d) 1a (0.5 mmol) and KI (0.05 mmol). (B) In situ FTIR spectra collected during the reaction of 1a (1 mmol) and KI (0.5 mmol) in 0.1 mol l−1 NaBF4/CH3CN solution at the potential of 500 mV.
Download figure:
Standard image High-resolution imageWhen 1a, 2a and KI were added into the NaBF4/CH3CN solution, oxidation peak current at about 0.5 V (curve f in Fig. 4 (A)) increased compared with curve e, but the oxidation peak current at about −0.1 V (curve f) did not increase. It suggested that the electrochemical oxidation process of I2 and 4-hydroxycoumarin was restrained when 2a was added to the solution of 1a and KI, and the product was mainly formed through the intermediate disulfane instead of 4-hydroxy-3-iodo-2H-chromen-2-one.
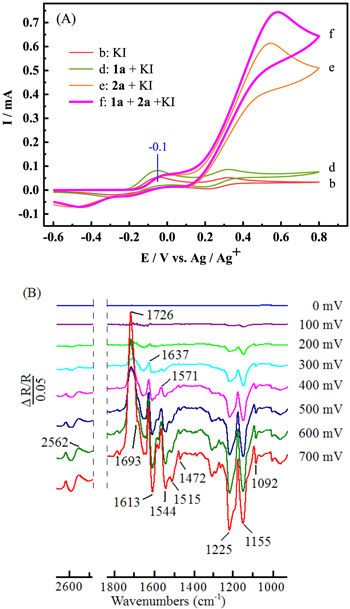
Figure 4. (A) Cyclic voltammograms recorded at the scan rate of 50 mV s−1 in 0.1 mol l−1 NaBF4/CH3CN with (b) KI (0.05 mmol); (d) 1a (0.5 mmol) and KI (0.05 mmol); (e) 2a (0.75 mmol) and KI (0.05 mmol); (f) 1a (0.5 mmol), 2a (0.75 mmol) and KI (0.05 mmol). (B) In situ FTIR spectra collected during the reaction of 1a (1 mmol) with 2a (3 mmol) in the presence of KI (0.1 mmol) in 0.1 mol l−1 NaBF4/CH3CN solution at potentials varied from 0 mV to 700 mV.
Download figure:
Standard image High-resolution imageFigure 4 (B) showed the changes of functional groups during the reaction of 1a, 2a and KI in 0.1 mol l−1 NaBF4/CH3CN solution at potentials varied from 0 mV to 700 mV by in situ FTIR spectroscopy. With the increase of potential, starting from 300 mV, the infrared signals of positive-going bands (2562, 1726, 1637, 1571 cm−1) and negative-going bands (1693, 1613, 1544, 1515, 1472, 1225, 1155, 1092 cm−1) could be clearly observed. The positive-going band at 2562 cm−1 was attributed to the S-H stretching vibration of 2a, which indicated that 2a was consumed. The positive-going bands at 1726 cm−1 and 1637 cm−1 could be attributed to the C=O stretching vibration and C=C stretching vibration of the six-membered lactone ring of 1a, respectively. In addition, the positive-going band located at 1571 cm−1 could be attributed to a characteristic peak of the aromatic ring skeleton vibration of 1a. 49,50
When ArS- was attached to 1a, due to the conjugation effect of benzene ring and lone pair electrons of sulfur, the infrared bands of the corresponding C=O and C=C stretching vibration would red-shift. 51–54 Therefore, the negative-going band at 1693 cm−1 was assigned to the C=O stretching vibrations of 3aa. However, it was not obvious because of the coverage by the positive-going band at 1725 cm−1. The 1613 cm−1 of the negative-going band could be attributed to the C=C stretching vibrations of 3aa. The signals of 1544 cm−1, 1515 cm−1 and 1472 cm−1 could be attributed to the characteristic peaks of the aromatic ring skeleton vibration of 3aa. 49,50 The signal located at 1092 cm−1 might be caused by the synergistic vibration of C–S bond and C–Cl bond. Two strong negative-going bands around 1225 cm−1 and 1155 cm−1 could be assigned to the symmetric and asymmetric stretching of aryl ether group of 3aa. 49,50 Therefore, 3aa was synthesized while 1a and 2a were consumed during the electrochemical process.
Determination of optimal conditions for electrolysis experiments
We commenced electrolysis experiments by employing 1a and 2a as the model substrates to optimize the reaction conditions (Table I). The initial studies began with a reaction of 1a (0.5 mmol, 81 mg) and 2a (0.75 mmol, 108 mg, 1.5 equiv.) in 0.1 mol l−1 NaBF4/CH3CN solution containing 10 mol% KI at a constant voltage of 0.6 V at 60 °C in a 25 ml undivided cell. The reaction process was monitored by HPLC. It was found that 1a was almost completely consumed in 6 h. The desired C–S coupling product 3-((4-chlorophenyl)thio)-4-hydroxy-2H-chromen-2-one (3aa) was obtained in 89% yield (entry 1). The reaction temperature played an important role in this transformation. Decreasing the temperature from 60 °C to 45 °C and 25 °C, the desired product 3aa was given in 41% and 25% yields, respectively (entries 2 and 3). Therefore, to get a satisfactory yield, 60 °C was necessary. The effect of reaction potential was also investigated and the yield of desired product 3aa was increased from 89% to 94% when the reaction potential was reduced from 0.6 V to 0.4 V (entry 4). High potential would promote the side reaction of 1a and I2 to form 4-hydroxy-3-iodo-2H-chromen-2-one. 48 While the reaction potential was dropped to 0.2 V, the yield of 3aa was only 28% (entry 5). So, 0.4 V was suitable in the following experiments.
Table I. Optimization of the reaction conditions. a)
| Entry | 2a (equiv.) | KI (mol%) | Temperature (°C) | Potential (V) | Yield b) (%) |
|---|---|---|---|---|---|
| 1 | 1.5 | 10 | 60 | 0.6 | 89 |
| 2 | 1.5 | 10 | 45 | 0.6 | 41 |
| 3 | 1.5 | 10 | 25 | 0.6 | 25 |
| 4 | 1.5 | 10 | 60 | 0.4 | 94 |
| 5 | 1.5 | 10 | 60 | 0.2 | 28 |
| 6 | 1.5 | 5 | 60 | 0.4 | 62 |
| 7 | 1.5 | 0 | 60 | 0.4 | 0 |
| 8 | 1.2 | 10 | 60 | 0.4 | 80 |
| 9 | 1 | 10 | 60 | 0.4 | 74 |
a)Reaction conditions: 0.5 mmol of 1a, 0.1 mol l−1 NaBF4/CH3CN solution (15 ml), a 25 ml undivided cell, reaction time 6 h. b)Yields of isolated products based on 1a.
The effect of the amount of catalyst on the reaction was also studied. It was ideal to achieve high yield of 3aa with low amount of KI. However, when the amount of KI was decreased from 10 mol% to 5 mol%, the yield of 3aa was decreased from 94% to 62% (entry 6). Furthermore, 3aa could not be formed in the absence of KI (entry 7). Thus, 10 mol% KI was necessary for the next experiments. In addition, the loading of 2a had a significant impact on the reaction yield. When the loading of 2a was decreased from 1.5 equiv. to 1.2 equiv. or 1 equiv., the desired product was obtained in 80% and 74% yields, respectively (entries 8 and 9). It could be seen that excessive 2a was beneficial to the sulfenylation reaction. 55 Hence, combining the above experimental results, 1a with 1.5 equiv. of 2a in the presence of 10 mol% KI in 0.1 mol l−1 NaBF4/CH3CN solution at constant potential 0.4 V at 60 °C for 6 h were chosen as the optimized reaction conditions for the synthesis of 3aa.
Scope of sulfenylation of 4-hydroxycoumarins with aryl thiols
With the optimized reaction conditions in hand, the substrate scope and universality of this electrochemical sulfenylation with different 4-hydroxycoumarins 1 and aryl thiols 2 were explored (Scheme
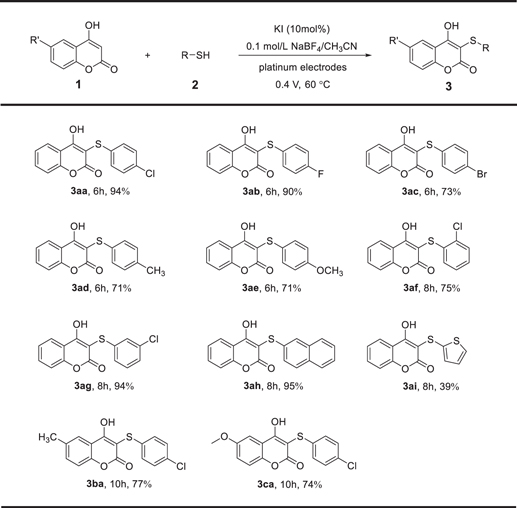
Scheme 3. Reactions of 4-hydroxycoumarins (1) and aryl thiols (2). Reaction conditions: Pt anode, Pt cathode, 0.1 mol l−1 NaBF4/CH3CN (15 ml), 1 (0.5 mmol), 2 (0.75 mmol), KI (0.05 mmol), 0.4 V, 60 °C. Isolated yields.
Download figure:
Standard image High-resolution imageSubsequently, 4-hydroxycoumarins 1b and 1c were used as the substrates to react with 2a, and the corresponding sulfenylated 4-hydroxycoumarins 3ba and 3ca could be obtained in moderate yields in 10 h. It was worth noting that 1b and 1c could not be completely converted to their desired products in 10 h. By prolonging the reaction time, the yields of 3ba and 3ca could be further improved.
Mechanism investigation
To ascertain the mechanism of this sulfenylation reaction, we conducted several control experiments (Scheme
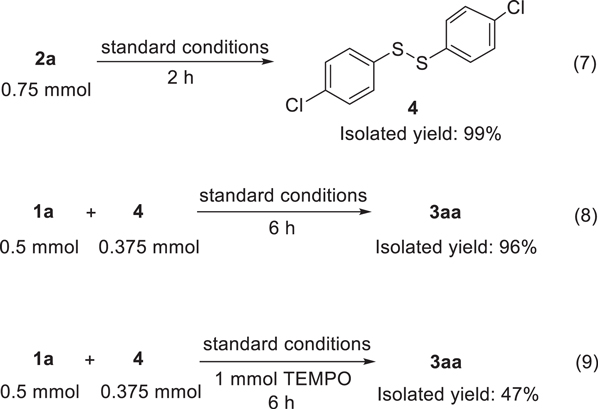
Scheme 4. Control experiments. Standard conditions: Pt anode, Pt cathode, 0.1 mol l−1 NaBF4/CH3CN (15 ml), KI (0.05 mmol), 0.4 V, 60 °C. Isolated yields.
Download figure:
Standard image High-resolution imageBased on the above control experiment results and previous reports,
32,57,58
two possible mechanisms for this electrochemical sulfenylation of 1a were proposed in Scheme

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 5. Plausible mechanism.
Download figure:
Standard image High-resolution image{kind=link}
Pathway B reflected the non-radical process of this reaction. Initially, the oxidation of 2a under the electrophilic molecular iodine afforded the disulfide 4. The reaction of 4 with I2 formed an electrophilic species 4-Cl-PhSI (6). 32,57,58 The intermediate 6 underwent the electrophilic attack of 1a to generate the intermediate 8. Finally, product 3aa was obtained by releasing HI from intermediate 8.
Conclusions
In summary, we have developed an efficient KI-catalyzed electrochemical system for the synthesis of sulfenylated 4-hydroxycoumarins at a low potential from 4-hydroxycoumarins and aryl thiols. The electrocatalytic activity of iodine for sulfenylation of 4-hydroxycoumarin was explored by cyclic voltammetry. Furthermore, the possible mechanism was proposed by combining cyclic voltammetry, in situ FTIR and control experiments results. The present methodology had a broad substrate scope and generated the corresponding products in moderate to excellent yields, which provided a convenient route for the synthesis of sulfur-containing compounds.
Acknowledgments
We are grateful for the generous financial support of the National Natural Science Foundations of China (21773211 and 21776260).