

Abstract
High harmonic generation (HHG) delivering attosecond pulse duration with photon energy in the extreme ultraviolet spectral range has been demonstrated as a robust table-top coherent light source, allowing for the observation and manipulation of ultrafast process within the shortest time window ever made by humans. The past decade has witnessed the rapid progress of HHG from a variety of solid targets and its application for photoemission spectroscopy in condensed matter. In this article, we review the HHG in solids and the understanding of the underlying physics of HHG, which allows all-optical band structure reconstruction. We also introduce combinations of HHG source and photoemission spectroscopy, such as angular-resolved photoemission spectroscopy and photoemission electron microscopy. With the capacity of exploring a wide momentum space and high temporal resolution, the extension of attosecond science to the field of condensed matter physics will lead to new insights into the fundamental ultrafast dynamics in novel quantum materials.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Future perspectives
Probing the electronic structure is of fundamental interest in physics and material science, because of its essential role in determining the physical property of the material. The emergence of high harmonic generation (HHG) and subsequent birth of attosecond science in the dawn of the 21st century provides a unique opportunity to study electron dynamics in their intrinsic time scale. HHG has been demonstrated as a robust extreme ultraviolet (XUV) light source for photoemission spectroscopy, with the power to study nonequilibrium quantum dynamics in a wide momentum space in time-resolved measurements, though compromise has to be made between temporal and spectral resolution. Great efforts have been made, especially in the past few years, to build high flux, high repetition rate HHG sources worldwide that can overcome the low efficiency and space-charge effect in preliminary HHG based time-resolved experiments in materials. All this progress has paved the way for the collaboration of attosecond and condensed matter communities in the future. However, there is still a way to go to explore new methods towards understanding the basic dynamics in charge, spin and phonon interaction in ultrashort time scales. It can be noted that most results reviewed in this manuscript is in the XUV spectral range. The undesirable scaling of the yield of high harmonics with driving laser wavelength has limited the extension of attosecond science to the x-ray range over the past years. The recent advances in mid-inferred driving laser and attosecond pulse characterization in the water window range may lead to a breakthrough in the study of core-level electron dynamics. Another breakthrough may follow the recent demonstration of the high peak-power attosecond pulse up to the gigawatt-level, a firm step towards attosecond-pump-attosecond-probe experiments and nonlinear attosecond optics.
1. Introduction
The nonperturbative light–matter interaction, when the ultrashort pulsed laser intensity exceeds 1014W cm−2, leads to the emission of a comb of harmonics of the driving laser. Such a nonlinear process called HHG was first observed in the late 1980s [1, 2]. Unlike perturbative harmonics, HHG usually has a broad plateau region with comparable efficiency and a sharp cut-off. The special phenomenon was well explained by the semi-classical model of the single atom response by Krause et al [3] and Corkum [4]. It is often referred to as the Three-step model in which the electron (a) is tunnelling ionized, (b) accelerated in the strong field and (c) recombines with the core and emits a high energy photon. A quantum-mechanical theory based on strong field approximation is then formed by Lewenstein et al [5], which enables an accurate description of the HHG process. HHG provided a new approach to create table top coherent XUV light sources. Moreover, it was soon predicted that the spectral structure of HHG corresponds to a train of attosecond pulses and potentially supports the generation of an isolated attosecond pulse (IAP) [6, 7]. In 2001, the attosecond pulse train (APT) and IAP were experimentally observed [8, 9]. It opened the door to the investigation of ultrafast dynamics in their natural time scale. Since then, attosecond pulses have been extensively applied in the study of ultrafast dynamics in various areas, such as the photoionization time delay in atoms [10–12], molecules [13, 14] and solids [15, 16], the electron correlation of Fano resonances [17, 18] and Auger decay [19], charge migration [20] and photodissociation [21] in molecules and the instantaneous control of dielectric properties with a light field [22].
HHG in gases has been the most accessible source for attosecond pulses and has laid the foundation of attosecond physics. Nevertheless, the past decade has seen rapid progress in the generation of harmonics in solid materials. Since the first experimental demonstration in 2011 [23], HHG in solids has been investigated in different material systems, for example in semiconductors [24, 25], dielectrics [26, 27], nanostructures [28], topological materials [29–31] and monolayer materials [32, 33]. With the potential of probing electronic dynamics and band structure in solid materials, as well as being a promising solution for compact XUV sources with a high repetition rate [34], HHG in solids opened a new era of attosecond science. Moreover, HHG has been demonstrated to be a novel source for photoemission spectroscopy such as in condensed matter, with the power of exploring the full Brillouin zone with high temporal resolution [35, 36]. The rest of this paper will be organized as follows: in section 2, we will first highlight some of the major progress in solids state HHG, including the experimental demonstration and interpretation of HHG in various novel materials and the application in the reconstruction of band structure and imaging valence electron. In section 3, we will briefly introduce the principle of angular-resolved photoemission spectroscopy (ARPES) and the extension of APRES towards time-resolved measurements. Time-resolved ARPES (tr-ARPES) setup, employing high repetition rate, high average power narrowband HHG as the probe and its application is reviewed. In section 4, we will introduce an alternative tr-ARPES technique, which is termed as atto-ARPES [37]. The combination of attosecond metrology [38, 39] and ARPES allows the study of the photoemission process in condensed matter with attosecond resolution. In section 5, we will overview the photoemission electron microscopy (PEEM) technique and the effort towards imaging surface dynamics with attosecond resolution. In the last section, we will give a summary of and prospects for ultrafast dynamics in condensed matters.
2. High harmonics spectroscopy in solids
Not long after the theoretical foundation of gas-phase HHG is established, Faisal et al, predicted that semiconductors can be used to generate harmonics at the peak field intensity of 1012 W cm−2 [40], which was prominently lower than that required by HHG in gases (∼1014 W cm−2). Early HHG experiments using solid targets were limited in the perturbative regime [41, 42]. In 2011, Ghimire et al, reported the first observation of non-perturbative HHG in bulk crystals [23], as shown in figure 1. A 3.25 μm, 100 fs laser is focused onto a 500 μm thick ZnO crystal at a peak intensity of 5 TW cm−2. The high harmonics is measured up to 25th order (9.5 eV), far beyond the ZnO bandgap. In addition, the cut-off energy was found to scale linearly with the peak intensity that deviates from the quadratic scale in the semiclassical three step model. On the other hand, the scaling of the individual harmonic yield with laser peak intensity was also highly non-perturbative. The results suggested that HHG in solids cannot be solely interpreted by the recollision model as in atoms and molecules.
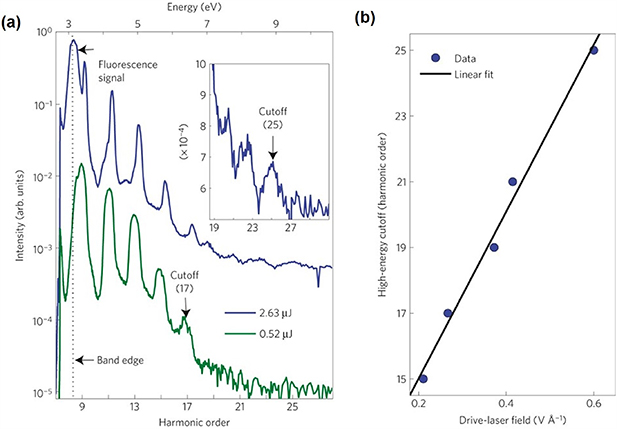
Figure 1. (a) HHG spectra from a 500 μm thick ZnO crystal for driving pulse energies of 0.52 μJ (green) and 2.63 μJ (blue). The magnified view of the cutoff is shown in the inset for the 2.63 μJ driving pulse. The approximate band edge of the ZnO is indicated by the black dotted line. (b) The scaling of HHG with drive-laser field, the black line is the linear fit of the experimental data (blue dots). Reproduced from [23], with permission from Springer Nature.
Download figure:
Standard image High-resolution imageA simple two step model was first used to describe the HHG signal in ZnO [23, 43], comprising ionization from the occupied valence band to an unoccupied conduction band, and subsequent acceleration by the strong laser field that produces a nonlinear current as the source of the intraband emission of harmonics. Besides the intraband emission, numerical results suggested that interband emission via recombination between electron–hole pairs may also contribute to the HHG yield [44, 45], especially when the photon energy is above the band gap. The interband source provided a link between gas-phase and solid-phase HHG, but it was often neglected in early works. Vampa et al, introduced a weak second harmonic of the driving laser to perturb the HHG process in ZnO [24]. The modulation of the even harmonic signal, due to the break of symmetry, allowed the distinction between the inter- and intraband contributions. The results provided evidence that the interband HHG dominates in the ZnO HHG process. Furthermore, the link between gas- and solid-phase harmonics implied the possibility of applying the method of gas-phase HHG spectroscopy [46, 47] to solid materials, to probe the band structure and ultrafast dynamics in strong field process. Including the interband emission, HHG in solids can be qualitatively described in a three-step recollision picture, both in real and reciprocal space, as illustrated in figure 2. It involves tunnel ionization that creates electron–hole pairs; propagation that leads to intraband emission; and the interband recombination [48].
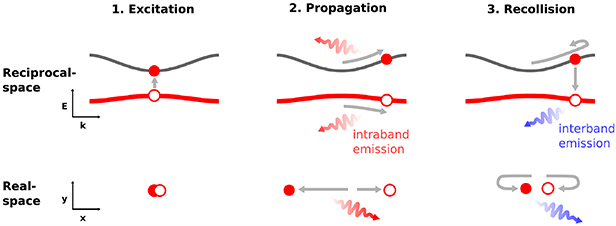
Figure 2. Sketch of the three-step recollision model for HHG in solids. It comprises (1) excitation, (2) propagation, and (3) recollision in reciprocal space (upper panels) and in real space (lower panels). Reprinted with permission from [48] © The Optical Society.
Download figure:
Standard image High-resolution imageIn subsequent research, HHG in solids was investigated in a variety of materials. As the conventional source of HHG, the comparison of high harmonics in solid and gas phase rare gas elements can lead to new insight into the fundamentally different mechanisms of the two types of HHG. Ndabashimiye et al found that HHG in weakly bounded van der Waals solid Ar and Kr [49] exhibit multiple plateau, exceeding the cut-off limit in a single-atom model in gas-phase HHG. The behaviour was also proposed by Wu et al in theoretical research [50]. The first plateau was due to transitions between the valence band and the lowest conduction band, while the second plateau was from the contribution of the higher-lying bands.
In polycrystalline SiO2 files, Luu et al demonstrated broadband coherent XUV radiation up to 40 eV [26]. The spectrum was well reproduced by the intraband current, based on numerical simulation solving semiconductor Bloch equations [44, 51]. The conversion efficiency of HHG in SiO2 was found to fall within the same range (10−7–10−6) as that in gases, whereas the total yield was 8 times higher than in gases under identical conditions. The fine waveform control enabled by the light field synthesizer [52–54] allowed them to explore the ultrafast control of multi-PHz-scale currents via manipulation of the HHG spectra with different waveforms. It implied the great potential in photonics and electronics towards a new frequency regime.
The remarkable properties of strong nonlinear electromagnetic response and isotropic band structure in graphene have been proposed as an ideal source for HHG [55, 56], especially in efficient THz harmonic generation [57]. Yoshikawa et al reported the HHG up to 9th order in graphene using a mid-infrared laser with a peak power of 1.7 TW cm−2 (30 MV cm−1) in 2017 [32]. The essential elliptic dependence of HHG in graphene can be explained in a semimetal regime [58], as illustrated in figure 3 in comparison to that in an AC Zener tunnelling regime, due to zero bandgap. Instead of tunnelling ionization, the electron–hole pairs were created when the bandgap become closed in an external light field. In 2018, Hafez et al show that the extremely efficient terahertz harmonics can be generated by introducing free background Dirac electrons via doping in a weak THz field of tens of KV cm−2 [33]. The weak driving field is about one order of magnitude smaller than the usual field in ultra-high speed transistors.
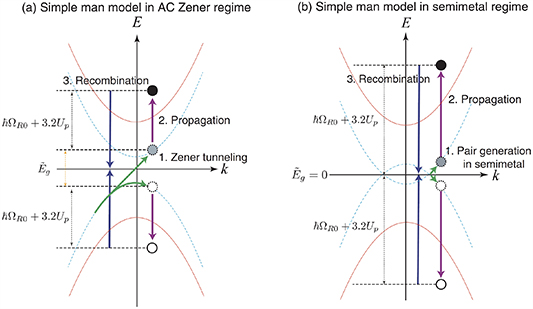
Figure 3. Schematic diagram of the three step model in (a) AC Zener regime and (b) semimetal regime. In both regimes, the cut off energy of the HHG is  , where
, where  is the photon energy and
is the photon energy and  is the ponderomotive potential. The XUV photons are produced via (1) the generation of carriers, (2) propagation, and (3) recombination band dispersion with (blue dotted lines) and without (red solid lines) external electric field are shown. The main difference is the excitation of the carriers. The carriers are excited by Zener tunnelling in the AC Zener regime while they are excited when the band gap becomes closed in the field in the semimetal regime. Reproduced from [58]. CC BY 3.0.
is the ponderomotive potential. The XUV photons are produced via (1) the generation of carriers, (2) propagation, and (3) recombination band dispersion with (blue dotted lines) and without (red solid lines) external electric field are shown. The main difference is the excitation of the carriers. The carriers are excited by Zener tunnelling in the AC Zener regime while they are excited when the band gap becomes closed in the field in the semimetal regime. Reproduced from [58]. CC BY 3.0.
Download figure:
Standard image High-resolution imageAs predicted in a few theoretical works, three-dimensional topological insulators exhibit unique properties that support strong-field-driven dynamics including HHG [59, 60]. Therefore, high harmonic spectroscopy can be used to characterize the topological properties and non-equilibrium electron dynamics in the bulks that were rarely explored in the time-resolved regime. Despite the theoretical attempts, HHG in topological insulators was demonstrated experimentally very recently in 2020 [29]. Bai et al produced HHG in the topological insulator BiSbTeSe2 in a reflective geometry. Both odd- and even-orders of harmonics up to the 9th order were observed. The odd-order harmonics originated from the intraband charge current, whereas the even-order harmonics were from the spin current, which is a topological signature of the field-driven Dirac fermions on the surface of the material. Shortly after that, Schmid et al independently reported HHG in the topological insulator Bi2Te3 [31]. Terahertz radiation from 25 THz to 42 THz with a peak electric field of up to 29 MV cm−1 was used as the driving pulse. By tuning the central frequency below and above the bandgap Eg, they found that in the condition of hνTHz < Eg and moderate THz field, the light driven Dirac currents in the surface governed the HHG without the need for strong field interband transition as in the bulk. The carrier-envelope phase (CEP) dependent HHG signal revealed the presence of the Dirac point as well as the influence of Berry curvature [61, 62].
The signature of the band structure encoded in the HHG spectrum in solids allowed the investigation of electrical properties in a particular material. It was conventionally studied by photoelectron spectroscopy, especially ARPES [63]. The interband recollision process that mapped the band dispersion can be used to reconstruct the band structure. As an extension of [24], Vampa et al reconstructed the band structure from the modulation of the interband even-order harmonic signal in ZnO with a resolution of better than 0.2 eV, which is comparable to tr-ARPES [35] but in an all-optical approach. In addition, Luu et al [26] and Lanin et al [64] used the intraband currents as the probe of the band structure. The below-bandgap nature of intraband HHG removed the limitation of material absorption of the above-band gap interband HHG. As an alternative method to ARPES, high harmonic spectroscopy offers the opportunity to study a material in extreme conditions, such as high pressure and high magnetic field, where the photoelectrons are difficult or impossible to detect, while maintaining high temporal and spatial resolution.
In 2020, Lakhotia et al demonstrated the direct imaging of valence electrons in solids with a spatial resolution of 26 pm [65]. The idea was adapted from atomic-scale diffraction microscopies in the framework of scattering, available when the crystal potential has weak perturbation compared to the laser field. The intensity of the harmonics in the scattering picture can be written as:
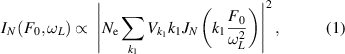
where  is the field strength,
is the field strength,  is the driving laser frequency,
is the driving laser frequency,  is the electron density,
is the electron density,  and
and  are the projections of the reciprocal space vectors and the Fourier components of the potential on laser polarization. By measuring the harmonic signal as a function of laser field strength, the potential (figures 4(a)–(c)) and thus the electron density (figures 4(d)–(f)) in real-space (and reciprocal-space by inverse Fourier transform) can be retrieved. Combining with time-resolved spectroscopy, the imaging of valence electrons with picometer resolution may give direct and instantaneous access to the chemical, electrical and topological properties of materials.
are the projections of the reciprocal space vectors and the Fourier components of the potential on laser polarization. By measuring the harmonic signal as a function of laser field strength, the potential (figures 4(a)–(c)) and thus the electron density (figures 4(d)–(f)) in real-space (and reciprocal-space by inverse Fourier transform) can be retrieved. Combining with time-resolved spectroscopy, the imaging of valence electrons with picometer resolution may give direct and instantaneous access to the chemical, electrical and topological properties of materials.

Figure 4. (a), (b) The 1D valence potential (blue curves) when the polarization direction of the laser is aligned with the [110] (a) and [100] (b) axes. The coloured spheres represent F− (grey) and Mg+2 (orange) ions. (c) The 2D valence potential of MgF2. The Mg+2 ion is in the centre, surrounded by F− ions. (d) Valence electron density derived from (c). (e) Electron density of MgF2 simulated from density function theory. (f) The green curve shows the electron density derived from (a) along the F–Mg–F axis of the MgF2 crystal. Black dashed lines indicate the valence radius rh and principal maximum rmax of the ionic radii. Reproduced from [65], with permission from Springer Nature.
Download figure:
Standard image High-resolution image3. HHG based tr-ARPES
Despite the pioneering exploration by high harmonic spectroscopy mentioned above, the most applicable method to probe band structure and electron dynamics in materials is ARPES. Based on the photoelectric effect, ARPES provides the simultaneous measurement of electron energy and momentum, namely the dispersion  , that are encoded in the angular dependence of the photoelectron kinetic energy. According to quantum theory, the kinetic energy
, that are encoded in the angular dependence of the photoelectron kinetic energy. According to quantum theory, the kinetic energy  and the projection of momentum on the surface of the material
and the projection of momentum on the surface of the material  are given by [66]:
are given by [66]:
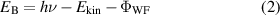
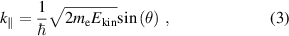
where  and
and  are the electron binding energy and mass,
are the electron binding energy and mass,  is the photon energy,
is the photon energy,  is the work function of the material and
is the work function of the material and  is the angle with respect to the surface normal. The conservation of the parallel component of k enables the direct band structure measurement on the material surface, while the perpendicular component does not conserve due to the broken translation symmetry. Nevertheless, it is still possible to determine the value of k⊥ for a 3D material in the case where the photon energy dependence of photoemission that accounts for the periodic dispersion along this direction is measured.
is the angle with respect to the surface normal. The conservation of the parallel component of k enables the direct band structure measurement on the material surface, while the perpendicular component does not conserve due to the broken translation symmetry. Nevertheless, it is still possible to determine the value of k⊥ for a 3D material in the case where the photon energy dependence of photoemission that accounts for the periodic dispersion along this direction is measured.
The first attempts to map energy bands by ARPES were experimentally demonstrated in the 1970s [67, 68]. Since then, ARPES has played a leading role in the probing of electrical propensities, especially in high temperature cuprate superconductors since the late 1980s [69–71]. In spite of the success in condensed matter physics, ARPES in equilibrium has limitations, that the electronic states to be measured should be initially occupied and that the nonequilibrium quantum dynamics that attract fundamental interest cannot be captured. Therefore, the fast development towards the simultaneous energy and momentum measurement with temporal resolution, e.g. tr-APRES, became a frontier of condensed matter physics in recent years. A schematic of the tr-APRES is illustrated in figure 5. In a pump-probe measurement, ultrashort femtosecond pulses are usually used as the pump to trigger the nonequilibrium states in the material, and another photon source with controllable delay is used as the probe to ionize the photoelectrons. The electrons are then collected by an electron energy analyser, either hemispherical [72] or angle-resolved time-of-flight [73].
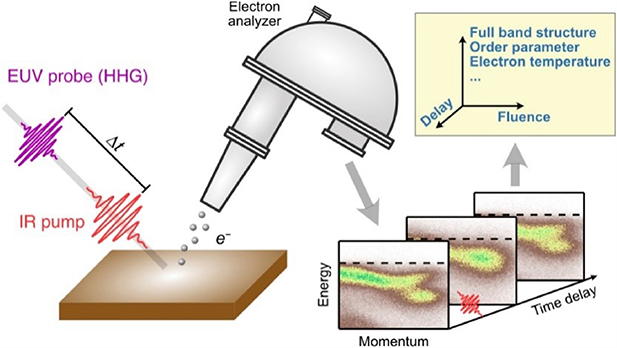
Figure 5. Schematic of tr-ARPES. A femtosecond IR pulse is used as the pump to excite the material and an attosecond XUV pulse is used to probe the change at the controllable time delay Δt and laser fluence (input energy). The measured full band structure, order parameter and electron temperature can be used to investigate the dynamics in materials. Reproduced from [37]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageThe choice of probe light is critical in an APRES experiment. The photon energy should be large enough, generally more than 5 eV, to overcome the work function of the material. In addition, a narrowband light is preferable to achieve high energy resolution. Therefore, a continuous-wave (CW) or quasi CW laser with a photon energy of more than 5 eV (249 nm in wavelength) became a popular light source for high-resolution ARPES. It was realized by a cascade of up-frequency conversion such as second harmonic generation or sum frequency generation of the infrared laser. Such a deep ultraviolet (UV) or vacuum ultraviolet (VUV) laser usually has a photon energy of around 6 eV and a high resolution of <1 meV could be achieved [74, 75]. The photon energy determines the accessible momentum window. According to equation (3), for a typical UV probe at 6 eV and work function from 4 to 5 eV, the maximum wave vector is about 0.6 Å−1. It can often reach the important region of high temperature cuprate superconductors and topological insulators that are located in the centre of the Brillouin zone. However, this is not always the case. For instance, the Dirac cone of graphene is located at 1.7 Å−1 [66], which is far out of reach using a VUV laser source. Moreover, in tr-APRES, the pulse duration of the light source is also important. It means a trade-off between temporal and spectral resolution.
To cover a large momentum space (e.g. high photon energy) with a short pulse duration and relatively narrow bandwidth, HHG turned out to be a promising light source for tr-APRES. The first tr-APRES based on HHG was demonstrated in 2007 [76]. A 1 kHz, 35 fs, 1.4 mJ Ti:sapphire laser was used for producing high harmonics, as well as acting as the pump laser in tr-APRES. The single harmonic at 41.85 eV, corresponding to the 27th order, was selected by a double mirror monochromator. The mean free path of photoelectrons produced by such a photon energy is typically <10 Å, in contrast to the tens of ångstroms mean free path using a VUV light source [36], making it rather suitable for a material surface. The bandwidth of the transmitted discrete harmonic was about 800 meV that supports a pulse duration as short as 3 fs. However, their tr-APRES instrument was limited by the low repetition rate and photon flux of the HHG source. In an APRES experiment, a high repetition rate light source is desirable to reduce the data acquisition time and the effect of space charge, while the typical few-cycle Ti:sapphire laser used for HHG driving pulse usually has moderate repetition rate of 1–10 kHz. The low conversion efficiency of the HHG process (regularly in the range of 10−7 and 10−6) is also not ideal for ensuring a high signal-to-noise ratio and the quality of the data statistics. On the other hand, the excitation fluence per pulse would be limited at too high a repetition rate due to the low pulse energy. Moreover, the tight-focusing required in high repetition rate HHG may further decrease the conversion efficiency due to phase mismatch and a small interaction volume [77, 78]. The trade-off between these factors made tr-APRES a demanding technique to fulfil.
The advances in high average power laser techniques [79–81] and phase-matching in a tight-focusing regime [82, 83] in the past decades, as well as combining the up-frequency conversion, since HHG yield scales with wavelength as ∼λ−(5−7) [84, 85], enables the high repetition rate, high photon flux HHG source [86, 87]. In a recent work, Klas et al reported 1 MHz, 3 × 1015 photons s−1 output at 26.5 eV using the double frequency of a 1030 nm Yb fibre laser as the driving pulse [88]. Such a HHG source has promoted the HHG-based tr-APRES towards a high repetition rate, tunable frequency and high photon flux. To name a few, Buss et al presented a tr-APRES setup with a 22.3 eV photon at 50 kHz repetition rate. The energy and temporal resolution were 80–100 meV and 65 fs, respectively, obtained at 1–2 × 1010 photons s−1 on the sample, while the brightest harmonic was 3 × 1013 photons s−1 [89]. The loss of energy was induced by the transmission filters and XUV optics. The measurements of quasiparticle dynamics in MoSe2 and photo-induced phase transitions in 1 T-TiSe2 were performed in this setup. Liu et al proposed to take advantage of a long pulse duration and short wavelength laser to generate directly the narrowband individual harmonic, to avoid the use of a monochromator. The increased photon efficiency and tuneable repetition rate of up to 150 kHz enabled an energy resolution of 21.5 meV [90]. Another advantage of the lack of a monochromator is the flexibility to conduct attosecond photoionization time delay measurement in a material similar to that in atoms and molecules. This will be highlighted in the next section. An alternative approach to achieve a high repetition rate is the cavity-enhanced HHG [91, 92], as illustrated in figure 6. The pulse, directly from a multi-MHz femtosecond oscillator, recycles in a passive resonator to achieve peak power comparable to that of a kHz laser system that can drive in-cavity HHG. Mills et al demonstrated a tr-ARPES based on cavity-enhanced HHG at 60 MHz, which is almost two orders of magnitude higher than in a single-pass HHG source [93], with photon flux of 1011 photons s−1 on the sample over a tuneable energy range from 8 to 40 eV.

Figure 6. Schematic of an enhanced cavity XUV source based on a Ti:Sapphire laser. A diffraction grating mirror (GM) XUV is used as the output coupler to spatially separate the HHG from a Xe gas target. A piezo transducer (PZT) is used to control the cavity length. The input coupler (IC) and two photodetectors (PD1 and PD2) are used to collect the feedback signal in the stabilization. Reproduced from [91]. © IOP Publishing Ltd. All rights reserved.
Download figure:
Standard image High-resolution imageThe implementation of an HHG based tr-ARPES allows access to a wide momentum space with a ∼100 fs temporal resolution. In the transient electronic structure of superconductors, for instance BaFe2As2 and FeSe [94, 95], significant modulations of the Fermi surfaces were observed. It might be associated with photon-induced superconductivity. As a competition state of superconductivity, the studies of a charge density wave (CDW) might lead to a better understanding of the mechanism of high Tc superconductors. The ultrafast dynamics after photoexcitation associated with CDW was investigated. The photon-induced CDW-to-metal transition exhibited significant non-equilibrium behaviour that was attributed to reduced lattice fluctuations [96]. Supported by a theoretical analysis [97], the impact of the electron band structure on the self-energy in quasiparticle excitation was revealed. In cooperation with time-resolved magneto-optical Kerr effect measurements, tr-APRES was also utilized in the measuring of ultrafast magnetic phase transition in Ni [98]. The rapid response of the spin system within 20 fs was observed whereas the demagnetization occurred in 176 fs with a long time lag. The results indicated the existence of a non-equilibrium transient magnetic state during demagnetization.
4. Photoemission time delay in condensed matters: atto-APRES
One of the most fundamental questions regarding the dynamics of the photoelectric effect is how long does the electron take to leave the atom after interaction. The implementation of attosecond pulses, especially the characterization technique of attosecond streaking [38] and RABBIT [39] (Reconstruction of Attosecond Beating by Interference of Two-photon Transitions), gives access to this question. Rather than a classical particle, the time of photoionization is defined associated with the electron wave packet. After ionization, the electron wave packet experienced a group delay in the potential, as shown in figure 7. The group delay, often referred to as Wigner time delay, can be defined as the derivative of the scattering phase η with respect to energy  . It was first introduced by Wigner in the scattering theory [99]. Since photoionization can be also interpreted as a 'half scattering' process, the common concept of the Wigner delay will help to describe the dynamics of photoionization. Initially, the attosecond metrology aimed at the accurate characterization of the attosecond pulses, since the photoelectron wave packet is to a large extent a replica of the attosecond pulse. The spectral phase that accounts for the dispersion of the attosecond pulse (atto-chirp) is retrieved via XUV + IR cross-correlation. In an analogy to the dispersion of the pulse in a dispersive medium, the group delay induced by the potential is also embedded in attosecond metrology that was often ignored when characterizing the attosecond pulse profile.
. It was first introduced by Wigner in the scattering theory [99]. Since photoionization can be also interpreted as a 'half scattering' process, the common concept of the Wigner delay will help to describe the dynamics of photoionization. Initially, the attosecond metrology aimed at the accurate characterization of the attosecond pulses, since the photoelectron wave packet is to a large extent a replica of the attosecond pulse. The spectral phase that accounts for the dispersion of the attosecond pulse (atto-chirp) is retrieved via XUV + IR cross-correlation. In an analogy to the dispersion of the pulse in a dispersive medium, the group delay induced by the potential is also embedded in attosecond metrology that was often ignored when characterizing the attosecond pulse profile.

Figure 7. Sketch of the Wigner time delay. The electron wave packet (green) acquires a phase shift due to scattering of the potential, and thus a group delay compared to a reference free electron (orange). Reprinted figure with permission from [100], copyright (2017) by the American Physical Society.
Download figure:
Standard image High-resolution imageThe absolute time delay associated with XUV + IR two-photon transition was not trivial to determine, due to the difficulty in the accurate retrieval of the delay between XUV and IR, atto-chirp and in establishing a free electron reference. Most measurements aimed at determining the relative delay between different ionization channels. Using the attosecond streaking or RABBIT technique, time delay measurements were performed in various targets like atoms, molecules and solids [10, 12, 13, 15], particularly in complex processes involving electron correlation, such as in Cooper minima [11, 101], Fano resonances [18, 102] and spin–orbit coupling [103].
As the first proof-of-principle experiment, Cavalieri et al extended the attosecond metrology from gas-phase to condensed matter for the first time [15]. The photoemission time delay between the electrons ordinate from the 4 f core state and the conduction band state of a Tungsten crystal was measured to be 110 ± 70 as, as shown in figure 8. The mean free path and propagation in the material played an important role in determining the emission time in solids, unlike that in gases, which can be well attributed to the Wigner delay and IR-induced continuum-continuum transition time delay [104]. Similar measurements in well-defined magnesium films indicated that the process comprises the free-particle-like propagation to the surface and the interaction with the screened streaking light field on the material-vacuum interface [105, 106]. In a recent work, Siek found that in addition to the propagation, an intra-atomic interaction, before the electron moved to the neighbouring atoms and started to feel the crystal structure, has to be considered [107]. The intra-atomic delay was angular momentum dependent, similar to that in isolated atoms in gases.
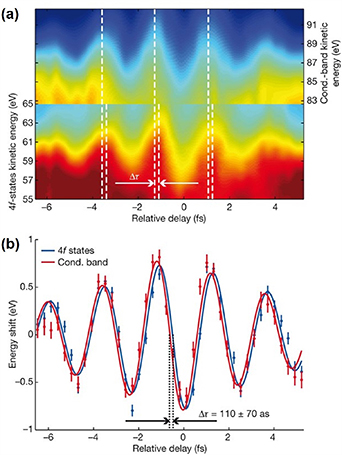
Figure 8. (a) The 4 f and conduction-band streaking spectrograms. Only the spectral region of the 4 f and conduction band photoelectrons are shown for visual comparison. A delay time shift is indicated by the white dashed lines at each fringe maximum and minimum. (b) Centre of mass analysis of the spectrograms. Vertical error bars are calculated from the noise in the measured spectrograms. The 4 f spectral region is scaled by a factor of 2.5 for visual comparison. The data points are fitted with a sinusoid curve in blue (4 f) and red (conduction band) lines. Reproduced from [15], with permission from Springer Nature.
Download figure:
Standard image High-resolution imageThis early research employed time-of-flight (TOF) spectrometers and attosecond streaking metrology. The principle of TOF spectrometers ensures high energy resolution of <100 meV only in the low energy region (typically <10 eV). In streaking metrology, the spectrum of IAP usually spans over more than 10 eV, further limiting the energy resolution in probing the dynamics in specific bands. However, it was already demonstrated that a high spectral resolution can be achieved with high temporal resolution using RABBIT metrology [12] that can be applied in photoelectron spectroscopy. Another solution is attosecond transient absorption spectroscopy (ATAS) [108] in which the temporal resolution is independent of the spectral resolution [109]. Recently, attosecond transient reflection spectroscopy was also proven to be an equivalent approach [110, 111]. It offers better applicability than ATAS in solids because the thickness of the sample in ATAS is limited to being in the order of tens of nanometers. It has also been demonstrated that the photoemission time delay revealed complex angular distribution [112], especially in the presence of resonance [18], due to the coupling of multiple transition channels. The lack of angular resolution manifests to a loss of coherence in the case where the radial and angular amplitude of different transition channels are entangled. Therefore, tr-ARPES combining RABBIT metrology is ideal to perform complete the characterization of the electron wave packet in condensed matters. A schematic of the atto-ARPES is illustrated in figure 9(a). The technique involves a comb of harmonics, instead of a broadband continuum in attosecond streaking and the narrowband individual harmonic selected by a monochromator in conventional HHG based tr-ARPES. RABBIT metrology is based on the idea of quantum interferometry. The two-photons' transitions paths (a) absorption of high harmonic photon  and simultaneous absorption of infrared (IR) photon
and simultaneous absorption of infrared (IR) photon  and (b) absorption of
and (b) absorption of  and simultaneous emission of IR photon ωL
results in the same sideband
and simultaneous emission of IR photon ωL
results in the same sideband  . The interference pattern as a function of XUV and IR delay
. The interference pattern as a function of XUV and IR delay  can be written as:
can be written as:
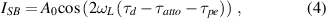
where  is intensity,
is intensity,  is the group delay induced by atto-chirp and
is the group delay induced by atto-chirp and  is the delay in photoemission. The first two terms can be cancelled when calculating the relative phase difference (e.g. the group delay) of different bands using the same harmonics. The photoemission time delay is thus retrieved from the sideband oscillation.
is the delay in photoemission. The first two terms can be cancelled when calculating the relative phase difference (e.g. the group delay) of different bands using the same harmonics. The photoemission time delay is thus retrieved from the sideband oscillation.
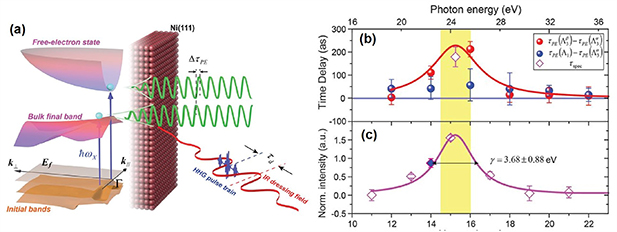
Figure 9. (a) Illustration of the atto-ARPES. Different photoelectron states can be accessed by an attosecond pulse train. The photoelectron spectrum is modulated by an IR dressing field via XUUV + IR two-photon transition. (b) Photoemission time delays  and
and  as a function of photon energy. The red solid line is a Lorentzian curve with the same linewidth as in (c). (c) Spectral intensity of the
as a function of photon energy. The red solid line is a Lorentzian curve with the same linewidth as in (c). (c) Spectral intensity of the  initial band as a function of photon energy. A linewidth of 3.68 eV is measured for the resonance. From [113]. Reprinted with permission from AAAS.
initial band as a function of photon energy. A linewidth of 3.68 eV is measured for the resonance. From [113]. Reprinted with permission from AAAS.
Download figure:
Standard image High-resolution imageTao et al presented the comparison of resonant  and nonresonant
and nonresonant  photoemission time in a Ni(111) crystal [113] in their atto-ARPES setup. The nonresonant transition from initial state
photoemission time in a Ni(111) crystal [113] in their atto-ARPES setup. The nonresonant transition from initial state  was used as the zero-time resonance. The
was used as the zero-time resonance. The  exhibited a strong variation around the resonant photon energy up to 212 ± 30 as, whereas the nonresonant
exhibited a strong variation around the resonant photon energy up to 212 ± 30 as, whereas the nonresonant  was smooth over the whole spectrum (figure 9(b)). The peak of the resonance coincided with the peak of the time delay variation (figures 9(b) and (c)) and the resonant time delay was similar to the photoelectron life extracted from the linewidth of the resonance, implying that the time delay can be interpreted as the photoelectron lifetime from a Bloch wave in the material to a free-electron wave, in close analogy to the shape resonance lifetime measurement performed in gases [13]. The angular dependence that became accessible by ARPES indicated the strong effect of band-structure on photoemission time delay, in contrast to some previous results [105, 106]. In a following work, the photoelectron emission time of Cu(111) was compared with that from the same bands as Ni(111) [114]. The ∼100 as time life difference in the resonant transition can be explained only if the spin dependent electron-electron scattering in the half-unoccupied d bands of ferromagnetic material Ni was taken into account. Therefore, the cooperation of time-, energy-, polarization-, and angle-resolving power of atto-ARPES has been demonstrated to give a unique insight into the electron interaction in quantum materials.
was smooth over the whole spectrum (figure 9(b)). The peak of the resonance coincided with the peak of the time delay variation (figures 9(b) and (c)) and the resonant time delay was similar to the photoelectron life extracted from the linewidth of the resonance, implying that the time delay can be interpreted as the photoelectron lifetime from a Bloch wave in the material to a free-electron wave, in close analogy to the shape resonance lifetime measurement performed in gases [13]. The angular dependence that became accessible by ARPES indicated the strong effect of band-structure on photoemission time delay, in contrast to some previous results [105, 106]. In a following work, the photoelectron emission time of Cu(111) was compared with that from the same bands as Ni(111) [114]. The ∼100 as time life difference in the resonant transition can be explained only if the spin dependent electron-electron scattering in the half-unoccupied d bands of ferromagnetic material Ni was taken into account. Therefore, the cooperation of time-, energy-, polarization-, and angle-resolving power of atto-ARPES has been demonstrated to give a unique insight into the electron interaction in quantum materials.
5. Atto-PEEM: towards attosecond-resolved surface imaging
As another important method in photoemission spectroscopy, PEEM provides high resolution imaging using low energy electrons that are emitted from the surface of the material excited by a light source whose photon energy exceeds the work function of the sample. The emitted electrons are then accelerated by an extractor field and imaged by the objective of the microscope, as illustrated in figure 10(a). A spatial resolution down to the few-nanometer scale can be achieved [115, 116]. PEEM does not rely on the generation of a high energy electron beam, in contrast to other electron microscopies such as scanning electron microscopy or transmission electron microscopy. It is thus more appropriate to image the surface dynamics like the relaxation, transport, and recombination of charge carriers with high temporal resolution by the optical pump-probe approach.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. (a) Schematic of the atto-PEEM setup. The label BS denotes the beam splitter, FM the focusing mirror, RM the recombination mirror, and TM the toroidal mirror. (b) Energy and (c) temporal structure of the attosecond pulse train used in atto-PEEM. (d) The full PEEM image of the electron emission spot and (e) the profile along the blue dashed line in (d), excited by the XUV laser pulse at high intensity. Reprinted from [128], with the permission of AIP Publishing.
Download figure:
Standard image High-resolution image{kind=link}
Instead of the conventional excitation light source such as UV-discharge lamps and synchrotron radiation, the femtosecond laser pulse was introduced into the time-resolved PEEM (tr-PEEM) in the early 2000s. Schmidt et al demonstrated the first tr-PEEM in 2002 employing a 82 MHz pulsed Ti:sapphire laser delivering a 9 nJ pulse energy and a 40 fs pulse duration. The hot electron dynamics on the surface was investigated with a spatial resolution of 20 nm [117]. Multiple photon transition is used to excite the photoelectron in tr-APRES, as the photon energy of a typical femtosecond laser (800 nm for Ti:sapphire laser as an example) is below the work function. Such a nonlinear process is very sensitive to the local electric field and band structure, thus leading to a contrast in the photoemission signal. Tr-PEEM has been utilized extensively in imaging the dynamics in surface plasmon [118, 119], carrier recombination [120] and transportation [121, 122] and relaxation, such as hot electron cooling [123] in semiconductors.
Similar to the development of tr-APRES, the advent of high harmonics and particularly attosecond pulses opened up new opportunities to probe surface dynamics with unprecedented temporal resolution, together with the spatial resolution of PEEM. In 2007, Stockman et al proposed an attosecond nanoplasmonic-field microscope [124], which was later termed as atto-PEEM. In the new scheme, the waveform-stabilized femtosecond pulse excites the surface plasmon, leading to localized fields on the nanometer scale, while the synchronized IAP probes the instantaneous nanoplasmonic field imprinted in the energy of photoemission. Ideally, ∼100 as temporal resolution and ∼20 nm spatial resolution could be achieved at the same time. It was also suggested to equip the PEEM with a TOF detector to get the energy resolution [125]. It also can be realized by hemispherical detector [126] or bandpass energy filter [127].
Mikkelsen et al presented the first atto-PEEM setup including 1 kHz, 35 fs, 800 nm IR pulses as the pump and XUV APTs as the probe [128]. The preliminary results are shown in figures 10(b)–(e). Chew et al reported an atto-PEEM with a few-cycle IR laser and XUV IAPs, equipped with a TOF detector [129] following their proposal [124]. The spatial resolution in these results was 200–300 nm, which was not very desirable in comparison to the prediction. The quality of imaging was severely limited by the chromatic aberration and the space charge effect, which was limited by the low repetition rate of the driving laser in both cases. Therefore, the development of atto-PEEM focused on the upgrade of the driving laser [130, 131]. The quality of the imaging was significantly improved and the exposure time was reduced by an order of magnitude by using 200 kHz repetition instead of 1 kHz. Recently, Zheng et al reported the XUV PEEM with a 50 nm spatial resolution and 300 meV energy resolution using a 10 kHz driving laser. The photon flux illuminated on the sample was found to be 2.3 × 109 photons s−1, corresponding to 2.9 × 1014 photons s−1 before the XUV optics and filters [127].
6. Conclusion
The generation of attosecond pulses at the dawn of the 21st century, particularly with the collaboration of attosecond science and condense matter physics in the past decade, has paved the way to temporal-resolved measurements combined with high energy-, momentum-, spatial- and angular- and spin-resolution in the intrinsic time scale of electron motion in various novel quantum materials and nanostructures. HHG in solids does not only serves as the source of coherent XUV attosecond pulses, but also involves complex electron–hole dynamics that are sensitive to the electronic properties of the bands. The understanding of the underlying physics of HHG in solids allows the all-optical measurement of band structure and the imaging of the valence charge density. Owing to the rapid development of high repetition rate, high average power lasers, HHG based IAP or APT has been employed in time-resolved photoemission spectroscopy such as tr-ARPES and tr-PEEM. The unique power to study the non-equilibrium state in the full Brillouin zone with potentially sub-femtosecond precision, though compromise between time and energy resolution has to be made, makes them very promising techniques to answer fundamental questions such as about the dynamics in superconductivity. Nevertheless, it is still an open question for scientists in both attosecond and material sciences to find an appropriate way to apply attosecond pulses, either in the spectral domain or temporal domain. Atto-ARPES might be a good example to adapt well-developed techniques from gas-phase attosecond physics to condensed matter. We also note that the combination of photoemission spectroscopy with attosecond pulses has been planned in a few attosecond user facilities, for instance in ELI-ALPS [132] and SECUF [133]. The free electron laser also emerges as the source for time-resolved photoemission spectroscopy [134]. The further collaboration triggered by the user facilities may help to transfer the opportunity for fundamental advances in the understanding of ultrafast dynamics in materials.
Acknowledgments
This study is supported by the National Natural Science Foundation of China (Grant Nos. 91850209, 12174435 and 12034020) and the National Key Research and Development Program of China (2017YFB0405202, 2018YFB1107200).