

Abstract
Serious challenges in energy and the environment require us to find solutions that use sustainable processes. There are many sustainable electrocatalytic processes that might provide the answers to the above-mentioned challenges, such as the oxygen reduction reaction (ORR), water splitting, the carbon dioxide reduction reaction (CO2RR), and the nitrogen reduction reaction (NRR). These reactions can enhance the value added by producing hydrogen energy through water splitting or convert useless CO2 and N2 into fuels and NH3. These electrocatalytic reactions can be driven by high-performance catalysts. Therefore, the exploration of novel electrocatalysts is one of the important electrocatalytic fields. In this paper, we aim to systematically discuss a variety of electrocatalysts used for sustainable processes and to give further insights into their status and associated challenges. We invited many famous research groups to write this roadmap with topics including platinum (Pt) and its alloys for ORR, oxides for ORR, chalcogenides for ORR, carbon-based hollow electrocatalysts for ORR, carbides for ORR, atomically dispersed Fe–N–C catalysts for ORR, metal-free catalysts for ORR, single-atom catalysts (SACs) for ORR, metal boride (MB) electrocatalysts for water splitting, transitional metal carbides (TMCs) for water splitting, transition metal (TM) phosphides for water splitting, oxides for water splitting, sulfides for water splitting, layered double hydroxides for water splitting, carbon-based electrocatalysts for water splitting, Ru-based electrocatalysts for water splitting, metal oxides for CO2RR, metal sulfides for CO2RR, metals for CO2RR, carbon for CO2RR, SACs for CO2RR, heterogeneous molecular catalysts for CO2RR, oxides for NRR, chalcogenides for NRR, C3N4 for NRR, SACs for NRR, etc. Their contributions enabled us to compile this 2020 roadmap on electrocatalysts for green catalytic processes and provide some suggestions for future researchers.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Jiandong Liu1, Jianmin Ma1 and Zhicheng Zhang2
1 School of Physics and Electronics, Hunan University
2 Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, School of Science, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin)
A potential energy crisis and increasing environmental problems drive us to explore sustainable energy that can replace fossil energy. Many interesting reactions inspire the study of sustainable processes, such as those of the hydrogen evolution reaction (HER), CO2RR, and the nitrogen reduction reaction (NRR) [1–4]. These sustainable reactions can provide H2 energy, gas, or liquid fuels and ammonia (NH3), and can be driven by solar energy, electricity or their combination (i.e. photoelectrocatalytic processes). Among these, electrocatalytic processes can be continuously carried out without insolation. Therefore, they are preferred from the perspective of the stability of reaction processes. Also, the oxygen evolution reaction (OER) is coupled with the HER to form so-called water splitting and is thus considered to be an important reaction [5]. The oxygen reduction reaction (ORR) is another reaction involving O2 [6], which is an important reaction for fuel cells and metal-air batteries.
Electrocatalysts play crucial roles in determining the performance of the sustainable processes mentioned above. Various strategies have been employed to improve the catalytic performance of electrocatalysts such as doping, defect design, hybridization, single-atom active centers, and their combinations [7–12]. Figure 1(a) presents the materials published up until 26 March 2020 that are relevant to different electrocatalytic processes. The numbers of papers reported are: 3016, 6495, 6573, 1132, 9927, 4147 for water splitting, OER, HER, CO2RR, ORR, and NRR, respectively. It can be seen that ORR is an extremely active field of study. We also studied the trend of electrocatalysts over the last decade. Figure 1(b) illustrates the number of papers relevant to electrocatalysts that were published each year from 2011 to 2020. The active trend is very clear: the number of papers published has risen from 524 in 2011 to 4777 in 2019, which is a nine-fold increase. From the start of 2020 to 26 March 2020, 1070 papers were published, which is near the total number for 2014 (1119). This also indicates that the field of electrocatalysis is active and strongly pursued by scientists.
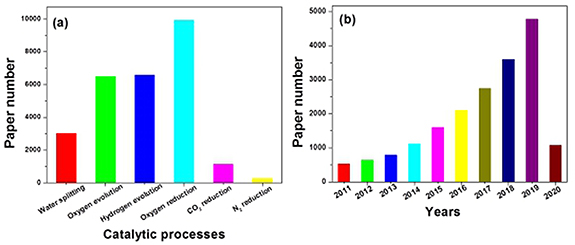
Figure 1. (a) The number of papers published relevant to different electrocatalytic processes up tountil 26 March 2020. Here, the papers were searched for on the Web of Science using the topic 'electrocatalyst' and the search results were refined by a further keyword termn (i.e. water splitting, oxygen evolution, hydrogen evolution, oxygen reduction, CO2 reduction, N2 reduction) for all document types on 26 March 2020. (b) The number of papers relevant to electrocatalysts undefinedpublished each yearundefined from 2011 to 2020; here, the papers were searched for on the Web of Science using the topic 'electrocatalyst' for all document types on 26 March 2020.
Download figure:
Standard image High-resolution imageAlso, we studied the different kinds of material used as electrocatalysts. Figure 2 shows the numbers of papers published up until 26 March 2020 that are relevant to different kinds of electrocatalyst. In those papers, some important kinds of electrocatalyst, such as oxides, sulfides, carbides, phosphides, carbons, platinum, ruthenium, and single atom, were analyzed. Metal-free carbon electrocatalysts were popular targets. Moreover, oxides and ruthenium have also been very actively studied.

Figure 2. The number of papers published relevant to different kinds of electrocatalyst up until 26 March 2020. Here, the were papers were searched for on the science Web of Science using the topic 'electrocatalyst' and further filtered using byusing a second term (i.e. oxide, sulfide, carbide, phosphide, carbon, platinum, ruthenium, single atom) for all document types on 26 March 2020.
Download figure:
Standard image High-resolution imageThis roadmap gives a comprehensive overview of the electrocatalysts used for ORR, water splitting, CO2RR and NRR. The main kinds of electrocatalyst shown in figure 2 will be comprehensively discussed in the following sections, which will include statuses, challenges, and concluding remarks. We invited some active researchers to give their views on different kinds of electrocatalyst. It is strongly expected that this roadmap will provide guidance for future research.
1. Pt and its alloys for ORR
Yuchen Qin
College of Sciences, Henan Agricultural University
Status
Platinum (Pt) is regarded as an excellent electrocatalyst for the ORR and represents a benchmark for ORR catalysts. The effects of the particle size, structure and shape of Pt nanocrystals (NCs) on ORR performance have been deeply explored. However, the unsatisfactory activity, high cost, and insufficient reserves of Pt limit its large-scale application. Therefore, minimizing the quantity of Pt required in catalysts and further improving the ORR's performance are essential for its extensive commercial deployment [13].
Initially, noble metals were generally employed in alloys with Pt, owing to their matching lattice parameters. Many Pt–noble metal alloys with various structures such as Pt–Au nanowires [14], Pt–Pd hollow nanostructures [15], Pt–Ag nanowires [16], etc, have successfully been prepared and have exhibited enhanced catalytic activity and stability in the ORR. This enhancement of catalysis can be attributed to the modification of Pt's electronic structure due to synergies between the metals. However, the improvement to the ORR's performance is limited. In particular, alloying Pt with noble metals cannot effectively reduce the cost. Therefore, it is desirable to alloy more suitable metals with Pt to further boost the ORR's performance and significantly reduce the cost.
Pt–transition metal (TM) alloys have attracted much attention, owing to the lower proportion of Pt required and their superior catalytic performance when they are used in the ORR. The enhanced catalytic property of Pt alloys originates from the TMs [17]. Thus, the type of TM plays a significant role in improving the ORR's performance. Alloys of Pt and late TMs have been widely studied for many years, and the main factors that may be responsible for the high ORR activity include the strain effect, the ligand effect and the surface structure [18, 19]. The strain and ligand effects can modify the d-band center or vacancy of Pt, resulting in a weakening of the adsorption of oxygen-containing species, which is crucial for the improvement of ORR performance [20]. Rough and Pt-skin surfaces are formed owing to the dissolution of TMs during ORR measurement in an acid environment or with initial acid treatment. More dissoluble TMs generally lead to higher ORR activity, but lower stability [21]. Thermal annealing is often carried out on Pt–late TM alloys to cause the segregation of Pt atoms on the surface, resulting in a Pt-skin surface and excellent ORR performance. The increased degree of ORR activity after thermal annealing is slightly different, following the sequence Pt < Pt3Ti < Pt3V < Pt3Fe < Pt3Ni < Pt3Co [22]. Apart from Pt–late TM alloys, alloys of Pt with early TMs and rare earths can also improve the activity and stability of the ORR, and are even superior to Pt–late TM alloys. Theoretical calculations demonstrate that the excellent catalytic properties of Pt–early TM alloys or Pt–rare earth alloys can be attributed to the ligand effect from the non-noble metals in the sublayers [23].
Since the start of the twenty-first century, many Pt intermetallic (ordered) alloys such as PtCo, PtFe, PtNi, PtZn, PtFeCo have successfully been synthesized through high-temperature annealing. These ordered Pt alloys usually deliver superior catalytic activity and stability, compared to disordered alloys, due to their stronger structural stability [24]. Even after ORR testing, the ordered structure was able to be maintained and only a few surface atomic layers became a Pt-like skin. According to theoretical calculations, the stronger Pt–metal covalent bond and more negative formation heat of ordered Pt alloys resulted in a superior ORR performance.
Another important development of Pt alloy catalysts is the fabrication of thin film structures. Distributing Pt alloy NCs on a nanostructured polymer thin film can not only effectively eliminate carbon corrosion and contact resistance between NCs and carbon but also reduce the proportion of low-coordination-number atoms. These factors all contribute to the much higher ORR activity of Pt alloy catalysts with a thin film structure, compared to that of carbon-supported Pt alloys.
Current and future challenges
Over the past decade, many notable advancements have been made in the development of Pt and Pt alloy ORR catalysts. The surface structure, electronic structure, strain, size, morphology and supporting material all play significant roles in improving the ORR's performance [25]. However, many challenges and opportunities still remain.
The D-band center plays an important role in the adsorption energies of intermediate species, and determines the catalytic activity of the ORR. Tuning the surface strain of Pt alloys can effectively influence the position of the d-band's center [26]. In Pt alloys, tensile strain can upshift the d-band's center, leading to stronger interaction with adsorbates, whereas compressive strain downshifts the d-band's center, weakening the adsorption energy (figure 3). Therefore, an effective and simple method for accurately controlling the surface strain of Pt alloys has shown great promise for catalytic enhancement.

Figure 3. A schematic diagram of the relationship between surface strain and the d-band center of Pt.
Download figure:
Standard image High-resolution imageDefect engineering is a hot spot in the field of catalysis and nanomaterials. The construction of defect sites (including vacancies), doping, and the amorphization of Pt alloys exhibit great potential for the enhancement of ORR activity. However, the preparation of Pt alloys with ordered defect sites is an enormous challenge. Porous Pt alloys can improve the catalytic property of ORR due to an increase in the numbers of active sites and boundary atoms. The dealloying method is the most common method for generating a porous structure. However, the resulting pores are disordered and cannot maximize the nanoconfinement effect required for the enhancement of catalytic performance. Therefore, the exploration of an effective strategy for preparing Pt alloys with an ordered porous structure is important for increasing the ORR's performance and decreasing the amount of Pt used.
The mechanistic investigation into ORR performance using Pt alloy catalysts at the atomic level has fallen behind synthetic studies. In this respect, advanced in-situ characterization techniques including in-situ Raman spectroscopy, in-situ Fourier-transform infrared spectroscopy (FTIR), in-situ transmission electron microscopy (TEM) and in-situ x-ray absorption spectroscopy (XAS) should be employed to detect transient states in the ORR process and directly reveal the reaction pathway and mechanism. Currently, the understanding of the relationship between the electronic structure and the ORR activity of Pt alloys is that it is a 'volcano-type' relationship [27]. Thus, balancing the adsorption energies of reactive intermediates and available reactive sites on the surface is essential for the improvement of ORR performance. Also, increasing the interaction between the catalyst's support and the Pt alloys is significant for realizing an effective distribution of Pt-based NCs on the support, which is beneficial for the enhancement of activity and stability.
According to the United States Department of Energy (DOE), the target for catalysts in proton-exchange membrane fuel cells (PEMFCs) is to reduce the total Pt loading to 0.125 mgPt cm−2 and increase the mass activity (MA) to 0.44 mA  at 0.9 V in an actual fuel cell device. Although the ORR performance of advanced Pt alloy catalysts reported previously was far higher than the DOE target, most of them were measured by a rotating disc electrode (RDE) system. There is a great difference between the catalytic property of Pt alloys as confirmed by RDE systems and practical performance achieved in an actual fuel cell device; therefore, a novel, fast, and low-cost catalyst evaluation technology should be explored to replace RDE systems. Until now, it has been rare for Pt alloy catalysts to exhibit such an excellent performance that they can be transferred into real fuel cell devices. Therefore, the design and preparation of Pt alloys with high activity and long-term stability that have been tested in real fuel cell devices remains a great challenge.
at 0.9 V in an actual fuel cell device. Although the ORR performance of advanced Pt alloy catalysts reported previously was far higher than the DOE target, most of them were measured by a rotating disc electrode (RDE) system. There is a great difference between the catalytic property of Pt alloys as confirmed by RDE systems and practical performance achieved in an actual fuel cell device; therefore, a novel, fast, and low-cost catalyst evaluation technology should be explored to replace RDE systems. Until now, it has been rare for Pt alloy catalysts to exhibit such an excellent performance that they can be transferred into real fuel cell devices. Therefore, the design and preparation of Pt alloys with high activity and long-term stability that have been tested in real fuel cell devices remains a great challenge.
Concluding remarks
This field has been regarded as a research hot spot for many years and has achieved significant advances. Although some scientific and technological problems remain, Pt alloy catalysts have exhibited great potential for widespread application.
Acknowledgments
This study was financially supported by the National Natural Science Foundation of China (21706074, 21972038), Natural Science Foundation of Henan Province (2023000410209), Key Research and Promotion Project of Henan Province (202102210261, 202102310267) and Top-notch Personnel Fund of Henan Agricultural University (30500682).
2. Pt-based electrocatalysts for ORR
Yan-Jie Wang
School of Materials Science and Engineering, Dongguan University of Technology
Status
With the exploration of clean and sustainable energy sources, the large-scale implementation of PEMFC technology that generates electricity from the electrochemical conversion of hydrogen and oxygen into water has been urgently demanded for use in commercial, residential, and transport applications. However, electrochemical conversion suffers greatly from certain issues, notably the sluggish speed of the ORR and the use of costly Pt-based catalysts at the cathode, which account for over 55% of the total PEMFC cost [17, 28]. Currently, Pt-based catalysts are undoubtedly the most active electrocatalysts for the ORR in PEMFCs, delivering the highest catalytic contribution to the ORR. Due to the scarcity and high price of Pt, reducing the Pt loading (particularly in the cathode catalyst layer), as well as improving its catalytic activity and stability to meet the requirements of PEMFC commercialization have become hot topics of widespread concern [29, 30].
In an effort to develop an optimum Pt-based ORR catalyst over the past few decades, significant progress has been made in the design and fabrication of nanostructured Pt-based catalysts using advanced materials science and nanotechnology, in which the size and morphology of the catalysts have been considered as critical factors for improving the catalytic activity and stability [17, 31]. For Pt-based nanostructures, it is well known that when the size of the catalysts is controlled in the nanoscale regime, especially over the small range of 3–5 nm, the attractive physical and/or chemical properties that do not emerge in the bulk state can afford excellent opportunities to obtain a large electrochemically active area and a high degree of catalytic activity. Meanwhile, for the structural morphology to enhance the catalytic performance, important insights into the nature of the active sites and their involvement in the ORR have shifted from traditional empirical trial-and-error methods to rational structural design and fabrication at the molecular or atomic levels, corresponding to the surface electronic structure of the catalysts and based on an understanding of the ORR catalytic mechanism. Both molecular catalysts (e.g. the molybdenum-doped hyperoctahedral Pt3Ni NCs with high-index facets) and atomic catalysts (e.g. single-atom Pt-based catalysts with good architectures) with well-designed geometric morphologies can favor high ORR activity and stability [32, 33]. Given the rapid development of advanced Pt-based ORR catalysts, a great need for updated design concepts has emphasized Pt-based nanostructures (figure 4) in three main trends: (a) Pt-based nanoparticles (NPs), (b) Pt-based nanotextures, and (c) Pt-based branched or anisotropic elongated nanostructures. Depending on their unique morphologies and nanoscale dimensions, these nanostructured Pt-based catalysts are preferred for precise control of structural reactivity, resulting in improved catalytic performance and electrochemical/physicochemical stability [31]. Also, insights into the relationship between structure and property, including the effects of Pt's facets, geometric architectures, compositional profile, and support, on the ORR's performance, have been significantly highlighted by the development and creation of advanced Pt-based nanostructured catalysts for practical PEMFC applications.

Figure 4. Various nanoscale Pt-based electrocatalysts.
Download figure:
Standard image High-resolution imageTo obtain low-Pt-content fuel cell electrodes with high Pt utilization efficiency, research advances in surface engineering strategies (figure 5) have been made using Pt-based nanostructured catalysts. Based on surface facet technology, coupled with composite engineering and a doping strategy, Mo-doped Pt3Ni (Mo–Pt3Ni) octahedral NPs [33] exhibit excellent ORR performance (i.e. an MA of 6.98 mA µgPt −1 and a specific activity (SA) of 10.3 mA cmPt −2 as measured by RDE at 0.9 V versus a reversible hydrogen electrode, RHE) compared to commercial Pt/C catalysts (i.e. 96 mA mgPt −1 and 0.127 mA cmPt −2, respectively). After 8000 potential cycles between 0.6 and 1.1 versus RHE at a sweep rate of 50 mV s−1, the Mo–Pt3Ni octahedral NPs display decreases of only 6.2% and 5.5% from the initial SA and MA, respectively, suggesting very good stability. Meanwhile, the combined surface strategy of composite engineering and defects demonstrates validity in the preparation of ultrafine jagged Pt nanowires (J Pt NWs) [34] with the best present-day ORR performance (i.e. an MA of 13.6 mA µgPt −1 and an SA of 11.5 mA cmPt −2 at 0.9 V versus RHE). The accelerated durability test (ADT) at a sweep rate of 100 mV s−1 between 0.6 V and 1.0 V showed that after 6000 potential cycles, the SA and MA only dropped by 5.5% and 12%, respectively, indicating the strong stability of J Pt NWs. Thanks to their excellent thermal and chemical stability, unique surface coordination states, and faster electron and mass transport, ultrathin NWs not only favor high catalytic activity but also cause less Pt dissolution and Ostwald ripening in a realistic fuel cell.

Figure 5. Surface engineering strategies for enhancing ORR performance.
Download figure:
Standard image High-resolution imageHowever, to avoid large crystallites in shaped Pt-based NPs with insufficient oxygen contact in the electrode during ORR, an ultralow concentration of Pt alloy supported on platinum-group-metal (PGM)-free materials (denoted by LP@PF-2) [36] was fabricated from engineered active sites with a combination of Pt and PGM-free catalytic sites, providing synergistic catalysis over the LP@PF-2 with improved activity (an MA of 12.36 mA µgPt −1 in the RDE test at 0.9 V versus RHE) and stability. The measured MEA at an internal resistance-corrected (iR-free) voltage of 0.9 V demonstrated that LP@PF-2 can achieve an MA of 1.77 mA µgPt −1, which is the best ORR performance of a membrane electrode assembly (MEA) measurement in the current technical landscape. In particular, after the standard durability test, the LP@PF-2 demonstrated stability comparable to the benchmark required by the DOE 2020 targets [37]. Moreover, at a very low Pt cathode loading of 0.033 mgPt cm−2, the rated power density reached 0.37 W cm−2, suggesting the excellent potential of LP@PF-2 for use in real fuel cells.
Current and future challenges
For Pt-based catalysts operating in PEMFCs, it is desirable to conduct the ORR through a four-electron pathway due to its direct energy efficiency. An experimental explanation of the electrochemical reaction mechanism in PEMFCs, required to clearly understand the details of the ORR, is challenging during the development of advanced Pt-based catalysts, because many intermediates of the ongoing ORR, depending on the nature of electrode material, catalyst, and electrolyte, are not easily probed via in situ characterization [38]. This often makes the ORR mechanism controversial, even though density functional theory (DFT) can provide theoretical help by modeling the electrochemical reactions of Pt-based catalysts. Moreover, in the recent single-atom Pt-based catalysts that provide enough atomic utilization and active sites to improve catalytic performance, isolated single atoms often prefer a two-electron pathway to catalyze ORR [39], which may result in difficulties in the theoretical understanding of the electrocatalytic mechanism, and thereby, in improving electrocatalytic activity.
Besides, other major technological challenges exist in the utilization of nanoscale Pt-based catalysts in real fuel cell operation: (a) as opposed to limited-scale laboratory fabrication, large-scale synthesis procedures for consistent, reliable, and reproducible NP quality need to be developed, in which the size and morphology should be controlled to maintain high activity and stability; (b) in the preparation of highly active Pt-based catalysts (e.g. facet-dependent Pt-alloy NCs), more reliable and controlled strategies are needed for the removal of surfactant(s) so that the size and morphology of NPs can be effectively controlled; (c) Pt-based nanostructured catalysts usually exhibit a large performance discrepancy between rotating disk electrode (RDE) screening and membrane electrode assembly (MEA) testing, because the polymer electrolyte membrane used, coupled with the electrode configuration, often results in an MEA with limited proton and oxygen transport as well as waterflooding within the cathodic electrode, which does not exist in the RDE. More systematic studies could be expected to clarify the correlation between changes in operating conditions and electrocatalytic properties; (d) for Pt-based nanostructured catalysts, in which the Pt or Pt-alloy NPs are generally deposited on conductive support materials (e.g. carbon) with high surface areas to prevent their agglomeration, more detailed analysis of the interaction between Pt and its support should be sufficiently addressed to clarify the influence of the support material on the activity and stability of Pt-based catalysts. More importantly, carbon corrosion at high potentials can make Pt-based NPs coalescent and detachable in long-term fuel cell operation, which requires more durable support candidates, such as modified carbon or noncarbon support materials with high surface areas and good conductivities.
Concluding remarks
Advances in surface engineering have created an important insight into improving the ORR activity and stability of Pt-based nanostructured catalysts in PEMFCs. Based on surface engineering strategies for building nanoscale structures and morphologies, there are new opportunities to make a big breakthrough and resolve the research bottleneck in the development of highly catalytic Pt-based ORR catalysts, while theoretical studies are still needed to uncover the cause of the catalyst's performance discrepancy when used with RDEs versus MEAs.
Acknowledgments
This work was supported by the Startup Grant of High-level Talents (GB200902-30, No. 196100041018) and the Foundation of Regular Research Team (TDYB2019007, No. 196100043028) at the Dongguan University of Technology.
3. Oxides for ORR
Yao Wang
State Key Laboratory of Heavy Oil Processing, College of Chemical Engineering and Environment, China University of Petroleum
Status
PEMFCs and metal–air batteries have been investigated for several decades due to their high efficiency, ease of operation, zero emission and most importantly, their unlimited renewable source of reactants. However, the sluggish kinetics of ORRs limit their large-scale commercial application [30, 40–42]. Transition-metal oxides (TMOs), such as cobalt oxides, nickel oxides, iron oxides, and manganese oxides are arousing enormous attention as promising alternatives to noble-metal-based electro-catalysts for ORRs, due to their intrinsic activity and sufficient stability in an oxidative electrochemical environment [43–48].
On one hand, extrinsic methods for enhancing the electrical conductivity of TMO catalysts to eventually achieve high ORR activity are based on combining them with other foreign components. Graphene, as the most promising conductive substrate, has been widely used to support TMOs to enhance ORR performance, due to its superior electrical conductivity [49]. This hybrid possesses enhanced electrical conductivity and fast electron transfer, thus accelerating the ORR's kinetics. Additionally, there is a synergistic effect between TMOs and graphene, which is beneficial to the ORR's performance. Furthermore, the doping of graphene with nitrogen inspires further development of the superior performance of TMO catalysts. In addition to increased activity, the introduction of graphene also enhances durability by anchoring the TMO components, which can be attributed to the strong effect of the interaction between the support and the TMOs. In general, the size of TMOs is always large, which is detrimental to rapid electron transfer. Micrifying the sizes of TMO particles not only greatly increases the contact interface between TMOs and conductive materials but also strongly promotes electron transfer.
On the other hand, the electrical conductivity of TMOs changes remarkably, from that of a semi-conductor to a metal-like conductor, through the introduction of other cations or the construction of desired oxygen vacancies. Firstly, doping another metal cation (Ni2+, Co2+, Fe2+ et al) into the lattice of TMOs can modify their electrical conductivity [50, 51]. However, the introduction of massive metal cations into bulk TMOs would probably form a new material. In this case, the crystal structure and composition of the pristine materials would be changed, thus affecting the adsorption/desorption energy of the intermediates. Secondly, constructing defects, especially oxygen vacancies, is regarded as the most effective method for altering the intrinsic conductivity of TMOs by strongly tuning their bandgaps. Some reports have shown that oxygen vacancies can be formed by a simple heat treatment. The existence of oxygen vacancies induces the bandgaps to become smaller, enhancing the intrinsic conductivity. Thus, rational control of the oxygen vacancies enhances the metallic characteristics of TMOs.
In a basic solution, the details of the ORR's performance are complex and are under investigation. It is generally accepted that O2 is reduced to OH− or HO2 − through the 4e− or 2e− pathways, respectively. In terms of TMO catalysts, it has been reported that the 2e− plus 2e− process is the dominant pathway for the alkaline ORR process. However, the adsorption energy between the catalyst's surface atoms and the reactant is not just related to the crystal facets, but also to the valance state of the surface metal atoms of TMOs. According to the associative pathway, the O–O bond will be be further cleaved to produce an O* intermediate after the OOH* intermediate is formed, and then an electron will transfer from the surface metal atoms to the adsorbed O* intermediate to form M(M+1)+–O2−. Based on the strategies discussed above, it is important to construct a tight relationship between TMO catalysts and ORR performance to guide the process of catalyst design (figure 6).

Figure 6. The advantages, challenges and fabrication strategies of TOM catalysts.
Download figure:
Standard image High-resolution imageApart from the previously discussed examples of TMO-type catalysts, other special materials, such as perovskite-type materials, spinel-type materials, and other ternary TMOs are widely used for ORRs. Firstly, the perovskite-type ORR catalysts usually consist of rare earth metals or alkaline earths (A) and TMs (B) in the form of ABO3. Implanting foreign cations into the bulk of perovskite to replace the A-site and B-site could greatly improve the ORR's catalytic performance. In the past, perovskite catalysts showed an enhanced ORR performance, which can be traced back to the 1970s. Matsumoto and co-workers documented that LaNiO3 perovskite material possessed an intrinsic activity, phase structure, and specific surface and electronic conductivity in the ORR [52]. It was reported that a so-called 'star' material based on perovskite oxide catalysts showed unprecedented ORR performance. In 2011 [35], Yang Shan-horn and coworkers reported that ORR activity for oxide catalysts was primarily correlated with σ*-orbital (eg) occupation and the extent of B-site transition-metal–oxygen covalency, which reflects the critical influences of the surface electron structure and metal–oxygen covalency on the competition between O2 2−/OH− displacement and OH− regeneration of surface transition-metal ions, demonstrating the importance of electron structure in controlling ORR catalytic performance, as shown in figure 7.
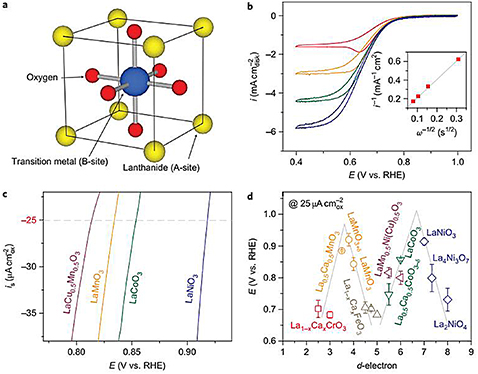
Figure 7. ORR activity of perovskite transition-metal-oxide catalysts. (a) ABO3 perovskite structure. (b) Oxygen-reducing activity of an LaCu0.5Mn0.5O3 electrode in O2-saturated 0.1 M KOH at a 10 mV s−1 scan rate and rotation rates of 100, 400, 900 and 1600 r.p.m. Koutecký–Levich analysis (inset) of the limiting currents (0.4 V) indicates a 4e− transfer reaction. (c) Specific activities of LaCu0.5Mn0.5O3, LaMnO3, LaCoO3, and LaNiO3. The potential at 25 μA cm2 ox is used as a benchmark for comparison. (d) Potentials at 25 μA cm2 ox of the perovskite oxides have an M-shaped relationship with the d-electron number. Data symbols vary with the type of B ion (Cr, red; Mn, orange; Fe, grey; Co, green; Ni, blue; mixed compounds, purple), where x ¼ 0 and 0.5 for Cr, and 0, 0.25 and 0.5 for Fe. Error bars represent standard deviations of at least three measurements [35].
Download figure:
Standard image High-resolution imageBesides, spinel-type ORR materials can be denoted by AB2O4 with specific proportions of A and B, where A stands for a divalent metal ion (such as Co, Ni, Mg, Zn etc) and B represents a trivalent metal ion (such as Co, Mn, Ni, Fe, Al etc). General methods for boosting the ORR performance of spinel-type materials include adjusting the composition and valence, designing the morphology and structure, and introducing defects and vacancies, the same as for ample TMO-type catalysts. Finally, some other ternary TMOs have also been explored, such as double perovskites, pyrochlore-type oxides, Ruddlesden–Popper-type oxides, LiCoO2-related oxides and Mn-based mullite oxides. It should be noted that the synergistic effect plays an important role in enhancing ORR activity due to the amount of intimate interfaces of different TMO components.
Current and future challenges
As is known, for the ORR, high-conductance materials are needed during the catalytic process, while the semiconductor characterizations of TMOs limit their ORR activity. Several strategies have been developed, such as combining TMOs with other components by in/ex situ methods, doping modifications and introducing defects and vacancies, and developing new materials and fabrication methods, among which, combining TMOs with other superior conductive support materials, such as pure metal NPs, carbon materials (graphene and carbon nanotubes (CNTs)), and other conductive polymers, is considered a high-efficiency method for improving the conductivity of bulk materials [35]. This strategy is based on the fact that introducing foreign components into TMOs by in-situ or ex-situ methods limits the margin of ORR performance enhancement. Another method is to optimize the intrinsic electrical conductivity of TMOs by atomic-level regulation, for example, by doping them with other metal atoms and developing oxygen defects [53]. This strategy can essentially convert TMO catalysts from semiconductors into metallic-type conductors, thus improving their intrinsic activity for the ORR.
Apart from the conductivity, the specific crystal structure and facets are other factors that affect ORR performance [54–56]. TOMs contain ordered but specific crystal and amorphous structures, which possess distinctly catalytic behaviors. Thus, exploring the detailed relationships between these structures and ORR performance is necessary for the rational design of the desired TMO catalysts. Also, TOMs with different facets exhibit different adsorption/desorption abilities and electrical conductivities. According to the Sabatier principle, the interactions between the catalyst surface atoms and the reactant should be neither too strong nor too weak, demonstrating the presence of maximum catalytic activity to some extent. The regulation of crystal facets of TOMs is an efficient strategy for improving ORR performance.
Concluding remarks
In recent years, research in the emerging fields of energy storage and conversions based on TMOs has been demonstrated at the atomic level. The development of the ORR in PEMFCs and metal–air batteries has been obtained by improvements in TMOs. Several strategies discussed in this paper have been elaborated to improve the activity and stability of TMO catalysts. Therefore, a deep exploration of the underlying relationships between TMOs and ORR performance is the key viewpoint for the rational design and construction of TMO catalysts in the future.
4. Chalcogenides for ORR
Rou Tan and Xiaochuan Duan
Pen-Tung Sah Institute of Micro-Nano Science and Technology, Xiamen University
Status
To solve the growing energy and environmental crisis caused by fossil fuels, much research effort has been devoted to clean energy systems, such as fuel cells and rechargeable metal–air batteries. ORR is a key fundamental electrochemical reaction involved in the energy systems mentioned above, which relies heavily on high-performance electrocatalysts to meet its practical applications. In general, the precious metal Pt and its complexes are the most common catalysts in commercial use for the ORR. Unfortunately, the high cost and scarcity of Pt-based catalysts severely limits their large-scale application. Therefore, there is an urgent need to develop alternative highly active low-cost ORR catalysts. To this end, more economical and competitive ORR catalysts that are not based on precious metals have been developed in succession [57–59]. In particular, layered transition-metal dichalcogenide (TMD)-based catalysts have attracted much interest because of their diverse and tunable catalytic properties [60, 61]. Previous studies have revealed that TMDs with well-defined structures exhibit superior catalytic activity for HER and OER, while their ORR performance is unsatisfactory due to their sluggish kinetics, including low conductivity and weak adsorption of ORR intermediates. Thus, the pursuit of high-performance TMD-based catalysts is conducive to the realization of trifunctional catalysts that are simultaneously efficient for HER, OER, and ORR, which can fulfill the needs of highly efficient renewable energy systems [62].
Current and future challenges
Layered TMDs possess a common chemical formula MX2 (M = transition metal, X = chalcogen) with a sandwich structure (X–M–X) held together by strong intralayer bonding (covalent bonding) and weak interlayer bonding (van der Waals interaction). According to the M–X arrangement, there are three known phases for MX2, namely one-layer tetragonal (1 T), two-layer hexagonal (2 H) and three-layer rhombohedral (3 R). Generally, in the 1 T phase, the TM atomic coordination is octahedral, while the 2 H and 3 R phases are trigonal prismatic (figure 8) [63]. The catalytic performances of layered MX2 nanosheets that expose the prismatic edges and basal planes can be tuned by reducing their size and phase transitions. On one hand, reducing the size of trilayer X–M–X to the atomic level endows MX2 ultrathin nanosheets with low-coordination step edges (metallic edge states) and corner atoms that are usually considered to be the active center in catalytic processes such as HER and ORR. On the other hand, the catalytic activity of MX2 can be adjusted by a phase transition that influences its electronic conductivity. For instance, the metallic 1 T phase of molybdenum disulfide (MoS2) exhibits higher activity compared with the semiconducting 2 H phase due to improved electron density, and thus favors fast electronic transport in the electrocatalytic process [64].

Figure 8. Illustration of TMDs and three typical 1 T, 2 H and 3 R phases with different metal coordinations and stacking sequences of MX2 structural unit cells. Reproduced with permission [63]. Copyright 2018, The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageCurrently, TMDs are used as a potential catalyst for the ORR based on the four-electron path. However, their sluggish kinetics restrict further application in clean energy-based devices. To improve the catalytic performances of TMDs for the ORR, various strategies have been applied: (a) atomically thin MX2 nanosheets manifest an ORR performance that is comparable to that of commercial Pt/C catalysts, especially in their methanol tolerance and stability. However, ultrathin MX2 nanosheets with high surface energy are inclined to restack together under the van der Waals attraction between their interlayers, thus reducing the density of exposed catalytic active sites. To improve the structural stability of ultrathin MX2 nanosheets and avoid agglomeration, highly conductive carbon-based frameworks such as CNTs, graphene or reduced graphene oxide (rGO), carbon fiber cloth, etc, have been used for hybridization with MX2 nanosheets. Moreover, considering the intrinsic ORR activity of carbon materials, a synergistic effect between the MX2 nanosheets and the abovementioned conductive carbon framework can provide more active sites for ORR and also facilitate electron transfer. Song et al reported a high-performance ORR catalyst consisting of ultrathin MoS2 nanosheets (∼5 nm) on carbon nanotube forest-covered graphite foam (GF-CNT@MoS2) made using the atomic layer deposition method (figure 9(a)) [65]. Benefiting from an integrated structural advantage, this composite catalyst exhibited high catalytic activity for both the ORR and the OER, and was successfully applied in a Li–O2 battery. (b) Given that the sluggish kinetics of TMDs in the ORR mainly originate from the unfavorable adsorption of oxygen-containing intermediates, especially in the case of the X-terminated layer of TMDs, the replacement of an X atom by other heteroatoms can significantly reduce the energy barrier for ORR intermediate adsorption on TMD surfaces, which is conducive to the reaction kinetics of the ORR. Singh et al systematically examined the effects of nitrogen (N) and phosphorus (P) doping on TMD catalysts for the ORR using first-principle calculations, and indicated that the substitution of X (S or Se) in the MX2 monolayer by N or P atoms induced a high spin density on the MX2 basal plane with improved chemical activity for O2 activation and adsorption. Thus, N-doped MX2 catalysts manifest efficient catalytic performance for the ORR based on the four-electron reduction mechanism, whereas over-strong binding by oxygen-containing intermediates in P-doped MX2 catalysts blocks the ORR (figure 9(b)) [66]. Also, the defect types and sites of heteroatom doping also greatly affect the ORR performance of TMDs. For pristine surfaces and surfaces with point defects in TMDs, large overpotentials (>1.0 V) are needed to activate the ORR, while the ORR overpotentials at edge defects are very low (0.66 V), corresponding to high activity, close to that of a prototypical Pt catalyst (overpotential ∼0.45 V) [67]. (c) Heterointerface tailoring has proven to be an efficient strategy for promoting the electrocatalytic properties of TMDs. The introduction of additional chalcogenides leads to an interfacial charge redistribution in the heterointerfaces that effectively reduces the adsorption energies of ORR intermediates, leading to the generation of more active catalytic sites and the promotion of electron transfer [68]. In the case of ultrathin Ni3S2/MoS2 heterostructured nanosheets (figure 9(c)), the presence of abundant heterointerfaces provides numerous highly active sites based on Mo edges and Mo–Ni–S sites [69]. Furthermore, the heterointerfaces in Ni3S2/MoS2 accelerate electron transport and enable the binding of more oxygen-containing intermediates for the ORR.

Figure 9. (a) Schematics of the preparation of GF–CNT@MoS2 catalysts. Reproduced with permission [65]. Copyright 2019, Wiley–VCH. (b) Scaling relation between adsorption-free energies of oxygen-containing intermediates on N-doped and P-doped MX2 monolayers. Reproduced with permission [66]. Copyright 2018, Wiley–VCH. (c) Schematics of the synthetic protocol for ultrathin Ni3S2/MoS2 nanosheets. Reproduced with permission [69]. Copyright 2019, The Royal Society of Chemistry.
Download figure:
Standard image High-resolution imageConcluding remarks
Layered TMDs have been considered as promising trifunctional electrocatalysts for the HER, OER, and ORR. However, the unsatisfactory ORR performance of TMDs that stems from their sluggish kinetics limits their further application in highly efficient clean energy systems. Thus, it is necessary to develop modification strategies to improve the ORR performance of TMD-based electrocatalysts. Comprehensive strategies including heteroatom doping, hybridization with conductive carbon materials, and heterointerface tailoring need to be considered to achieve optimal catalytic activity for the ORR. Although much progress has been made, the field of high-performance TMD-based catalysts for the ORR is still challenging. A deep understanding of the ORR mechanism of TMDs will be of great assistance in the realization of superior TMD electrocatalysts with high activity and stability. Through rational design and the fabrication of TMDs with well-defined structures, TMD-based multifunctional electrocatalysts hold great promise for the energy-related field in the near future.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (Grant No. 21601148) and the Natural Science Foundation of Fujian Province (Grant No. 2017J05090).
5. Carbon-based hollow electrocatalysts for ORR
Tong Zhen Tian, Cai Hong Zhang, Wen Wen Xie, Nian-Wu Li and Le Yu
State Key Lab of Organic-Inorganic Composites, Beijing University of Chemical Technology
Status
With the current surge in energy consumption, energy storage systems such as lithium-ion batteries are approaching their performance limits. New energy technologies, such as PEMFCs and metal–air batteries, with higher energy densities and lower production costs, have attracted tremendous research attention as promising candidates to address this problem [70–72]. The operation of both PEMFC and metal–air devices relies on a series of electrochemical processes at both electrodes. Amongst these, the most critical reaction is the ORR at the cathode, which determines the performance of the whole device [73]. The ORR can proceed through two pathways: one is the direct four-electron transfer pathway, and the other is the indirect four-electron pathway containing a two-step two-electron reaction route. In energy storage systems, the direct four-electron pathway is the first choice for realizing high efficiency [74–76]. Large-scale commercialization of the PEMFCs and metal–air batteries requires highly efficient, durable and low-cost ORR catalysts. In catalytic design, there are two main strategies for improving electrochemical performance. One is to increase the apparent activity through structural optimization, increasing the number of exposed active sites per gram and enhancing the utilization of the active sites by an improved mass transfer of electrolytes. The other is to improve the intrinsic catalytic activity per active site by the introduction of heteroatoms [77]. The heteroatoms change the oxygen-binding ability of the active site, altering the proton–electron transfer to diverse intermediates (OOH*, OH*, O*). Currently, the most widely used commercial ORR catalyst is Pt/C, which has excellent half-wave potential and limit current, showing its excellent intrinsic catalytic activity and apparent catalytic activity, respectively. However, the widespread commercial application of precious metals has been greatly hindered by the scarcity of reserves, high cost, carbon monoxide poisoning and methanol cross-reaction issues. Therefore, it is necessary to explore new low-cost and highly efficient catalysts to replace or reduce the use of noble metals. Carbon-based materials have been considered as promising alternates due to their abundance in nature, corrosion resistance, high conductivity and controllable surface properties [78]. Benefitting from large specific surface areas, defined active sites, limited voids and adjustable mass-transfer rates, hollow nanostructures have proved themselves in advanced electrocatalyst designs [79]. When applied to carbon materials, hollow structured carbon electrocatalysts can further optimize mass and electron transport, maximizing the usage of active sites. Here, we focus on recent research progress in the design of carbon-based hollow electrocatalysts for the ORR (figure 10). Based on their composition, hollow carbon-based catalysts are divided into three categories: metal-free catalysts, carbon with single-atom active sites, and carbon-based composites.

Figure 10. The design of carbon-based hollow electrocatalysts for ORR.
Download figure:
Standard image High-resolution imageCurrent and future challenges
For metal-free catalysts, the introduction of heteroatoms (such as N, B, S) is the approach most widely studied to effectively increase catalytic activity. The heteroatomic doping of carbon-based catalysts induces charge transfer between the doping site and the adjacent carbon atoms, so that the electron distribution can be rearranged. In particular, carbon atoms accompanied by more electronegative N or B atoms can improve the chemisorption of oxygen atoms. Also, N atoms bound in pyridinic sites or graphitic sites may enhance the electrochemical performance. B sites can also act as electron donors for the reduction reaction. Moreover, S doping can increase the spin density of graphene. The appearance of local states in the electron band structure enables the generation of active sites in the inert carbon atomic network, but there is still some doubt about the specific role of the doping species. Therefore, it is highly important to investigate the contribution of intrinsic carbon defects to the ORR. Conceptually, these intrinsic defects (vacancies, pentagon and/or heptagon formation, Thrower–Stone–Wales (TSW) defects, etc) are regarded as topological deviations that do not destroy the graphitic lattice. However, these structural changes can modify the atomic orbitals to create localized electronic states, which can induce improved ORR activity. For example, the hollow nanocages (figure 11(a)) designed by Hu and co-workers [80] had three kinds of defective carbon structure: (a) pentagon defects at the corners, (b) edge defects at the broken fringes of the shells, and (c) hole defects from micropores (figure 11(b)). DFT calculations further revealed that pentagon and zigzag edge defects were the catalytic active sites (figure 11(c)). Due to their defect engineering and unique hollow structures, these defective nanocages demonstrated an ORR performance comparable to that of N-doped carbon in an alkaline solution (figure 11(d)). A deep understanding of the inherent defects of carbon is helpful for the further defect engineering of carbon catalysts.

Figure 11. (a) High-resolution TEM (HRTEM) image of carbon nanocages. (b) Three typical defect locations within the carbon nanocages. (c) DFT calculations for the ORR activities of different defects. (d) ORR performances of carbon nanocages annealed at different temperatures. (e) Schematic illustration of the formation of hollow carbon spheres with single Co atoms. (f) Corresponding EXAFS fitting curves in R space. (g) ORR polarization curves. (h) Schematic illustration of the formation of a single-holed Co/NC hollow particle. (i) Field-emission scanning electron microscopy (FESEM) image of single-holed Co/NC hollow particles. (j) Tolerance test at 0.5 V in O2-saturated 0.1 m KOH solution with the addition of methanol after 2000 s. Panels (a)–(d) reproduced from [80] with permission. Copyright 2015, American Chemical Society. Panels (e)–(g) reproduced from [84] with permission. Copyright 2017, John Wiley and Sons. Panels (h)–(j) reproduced from [86] with permission. Copyright 2017, John Wiley and Sons.
Download figure:
Standard image High-resolution imageFor hollow carbon-based ORR catalysts containing metal species, the investigation of metal-based active sites at the atomic level on the carbon substrate is a hot research topic. Single-atom catalysts (SACs) have a theoretical 100% utilization of metal sites, thus significantly reducing the use of metal. Therefore, the combination of an SAC and a hollow carbon-based catalyst can largely control the production cost of ORR catalysts. As a typical example, Li et al [84] synthesized a catalyst with isolated single Co atomic sites fixed to hollow carbon spheres through a template-assisted pyrolytic (TAP) method (figure 11(e)). SiO2 and Co-TIPP were selected as the template and the cobalt source, respectively. As confirmed by the Fourier-transformed (FT) k3-weighted extended x-ray absorption fine structure (EXAFS) spectrum, the single Co atoms were atomically dispersed on the hollow carbon spheres with a coordination number of four (figure 11(f)). As an ORR catalyst, the hollow material displayed excellent catalytic activity, comparable to that of Pt/C in acidic media (figure 11(g)). The authors then compared this kind of hollow sphere with the same hollow sphere without an isolated single atom of Co and a solid ball with a single atom of dispersed Co. It was concluded that both the hollow structure and the single point gave the catalyst excellent ORR performance. A subsequent stability test showed that the hollow spherical catalyst had good cyclic stability due to the strong interaction between the single atomic Co site and the carbon substrate, and that the hollow spherical substrate was able to maximize the exposure of the active site and enhance the mass transfer of the catalyst.
Owing to the diversity and controllability of the structure and the composition of the precursors, hollow carbon-based composites obtained by the pyrolytic method using other organometallic complexes are another hot research direction. As an example, metal-organic frameworks (MOFs) are ideal precursors for the synthesis of many carbon-based functional materials with versatile morphologies and chemical compositions [85]. For example, Guan et al [86] synthesized a single-hole Co/N doped carbon hollow sphere from MOF precursors (figure 11(h)). Polystyrene (PS) spheres were selected as hard templates for the self-assembly of ZIF-67 NPs. The subsequent annealing treatment consumed the inner PS core and transformed the ZIF-67 into Co/N-doped carbon. The fast release of the gaseous product left a single hole in the sphere (figure 11(i)). An electrochemical test in 0.1 M KOH showed that the single-holed Co/N-doped hollow carbon spheres had a comparable ORR performance to that of Pt/C, with a half-wave potential of 0.87 V, and better tolerance for methanol (figure 11(j)). The reason for the excellent catalytic performance and stability is that the single-holed hollow structure greatly improves the mass transfer of the catalyst, while the active Co sites and N species are highly separated within the carbon shell.
Concluding remarks
The development of cheap and efficient ORR catalysts that can replace Pt/C is crucial to the commercialization of the related energy technologies. Carbon-based hollow materials have been widely investigated as promising ORR catalyst materials. In this review, we briefly discussed three potential research directions for the design of carbon-based hollow catalysts. However, there are still many problems with hollow carbon-based materials. The large consumption of templates is a big issue in the preparation of hollow structures. Besides, reaching a balance between the conductivity and the mechanical strength of carbon-based catalysts is critical, especially for hollow structures. Nevertheless, with progress in hollow materials and surface engineering, the performance of hollow materials will be improved, and the use of precious metals will be further reduced.
Acknowledgments
LY acknowledges the National Natural Science Foundation of China (Grant No. 51902016) and the financial support of the Fundamental Research Funds for the Central Universities (Grant No. buctrc201829). NWL acknowledges the financial support of the Fundamental Research Funds for the Central Universities (Grant No. buctrc201904).
6. Carbides for ORR
Chenhuai Yang and Zhicheng Zhang
Tianjin Key Laboratory of Molecular Optoelectronic Sciences, Department of Chemistry, School of Science, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin)
Status
Among green energy conversion technologies, fuel cells can directly and efficiently convert chemical energy into electrical energy, giving rise to water only, and not pollution. For fuel cells, the cathodic ORR is the rate-limiting reaction, due to its naturally complex and sluggish kinetics during electron transfer [87–89]. The mechanism of the ORR is rather complicated and mainly depends on the nature of the electrode and electrolyte, which has a crucial effect on the catalytic performance [90]. Hence, the rational design and synthesis of efficient ORR electrocatalysts with high onset potentials, low overpotentials, and high catalytic currents are important to the development of fuel cells. Currently, platinum (Pt) electrocatalysts still exhibit unparalleled catalytic performance for the ORR. However, scarcity of resources, high cost, sluggish ORR kinetics, and inferior operational stability due to deactivation by CO poisoning and the crossover effect hamper the commercial application of fuel cells. Therefore, it is desirable to replace Pt-based catalysts with new materials to make fuel cells useful for practical applications. Among various candidates, transition metal carbides (TMCs) have attracted intense interest for use as ORR electrocatalysts, which may be due to the following advantages: benefiting from an interstitial alloy structure, TMCs have advantageous properties such as high melting points, high electronic conductivity, excellent chemical stability and good corrosion resistance [91, 92]. Moreover, the group IVB-VIIIB TMs can alloy with carbon to form TMCs; these normally have formulas such as MC, M2C, and M3C and are made by controllable synthesis methods (e.g. carbothermal reduction, templates, MOF-assisted strategies, and so on), which greatly expand the number of TMCs [93].
Some bimetallic carbides such as Co6Mo6C and Fe2MoC have also been successfully prepared, enriching the range of TMCs available for use in ORRs [94]. Most importantly, their noble-metal-like electronic structures allow them to manifest a Pt-like nature in electrocatalysis, which ensures and even improves the catalytic efficiency of the ORR [95]. To improve their electrocatalytic activity and stability, TMCs have been encapsulated in carbon-based materials (e.g. CNTs, graphene). The introduction of these conductive supports is critical to improve the dispersity of downsized TMCs and strengthen the active sites via physicochemical coupling with TMCs. For instance, Wen et al reported a facile, scalable, cost-effective method to prepare core–shell structured N–Fe/Fe3C@C nanorods (Fe/Fe3C nanorods as the core and graphitic carbon as the shell) as electrocatalysts for ORR. The stability of the catalyst increased in neutral phosphate buffer solution (PBS) due to the complete enclosure of the (Fe/Fe3C) core by the graphitic carbon [96]. Moreover, the one-dimensional structure of the catalyst provided increased ORR active sites and exhibited a high ORR performance with a positive onset potential of 0.21 V. Yang et al reported a one-step soft-template-induced strategy for the controlled synthesis of bamboo-like carbon nanotube (b-CNT)/Fe3C nanoparticle hybrids (BCNFNHs) [97]. The BCNFNHs formed were highly active and stable catalysts for the ORR in both alkaline and acidic solutions. Specifically, the halfwave potential of the ORR using BCNFNHs reached 0.861 V vs. RHE in 0.10 M KOH, which is more positive than that of 20 wt% Pt/C (0.812 V vs. RHE). Furthermore, the BCNFNHs showed an ORR onset potential comparable to that of a Pt/C catalyst in an acidic solution. The BCNFNHs also exhibited outstanding stability and methanol tolerance, showing no ORR polarization curve shift and no cyclic voltammetric (CV) current changes in the presence of 1.0 M MeOH under both alkaline and acidic conditions.
Furthermore, TMCs can serve as versatile backbones/supports for other active materials, thereby forming multicomponent composites for the ORR. Based on first-principle DFT calculations, Zhou et al investigated the electrocatalytic properties of the heterostructures of N-doped graphene supported by TMCs (G/Ti2C, G/V2C, G/Nb2C and G/Mo2C monolayers) (figures 12(a)–(c)) [98]. Graphitic sheets hybridized with V2C and Mo2C monolayers showed enhanced ORR activity. Hollow-site C atoms located close to pyridinic N dopants were the most active sites and exhibited low overpotentials of 0.36 V (G/V2C) and 0.39 V (G/Mo2C) in the ORR (figure 12(d)). Considering their low cost, high stability, good activity, and Pt-like electronic structure, the use of TMCs instead of carbon to support Pt metal can effectively improve metallic stability and reduce Pt loading. Furthermore, because of the large binding energy of Pt-TMC, supporting Pt on TMCs hampers metal particle agglomeration. Xie et al reported that Pt combined with Ti3C2Tx was able to boost ORR activity with improved durability [81]. After 10 000 cycles, the half-wave potentials of the Pt/Ti3C2X2 did not visibly decrease, signifying the high electrochemical stability of the Pt/Ti3C2T2 catalyst.
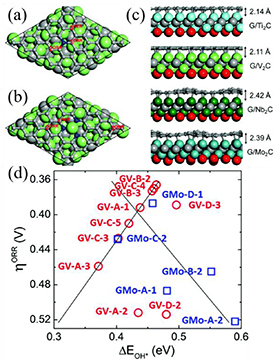
Figure 12. (a), (b) Top views of N-doped graphene supported by the V2C MXene monolayer, with N dopants in the graphitic and pyridinic forms, respectively. The red numbers indicate the active sites for the ORR and HER. (c) Side views of N-doped graphene on Ti2C, V2C, Nb2C and Mo2C MXene monolayers, respectively. The interlayer distances are shown for each system. The C, N, O, Ti, V, Nb, and Mo atoms are shown in grey, blue, red, cyan, green, dark green and turquoise colors, respectively. (d) Volcano plots of ORR overpotential vs. OH* binding energy [99]. 'GV' and 'GMo' are abbreviations for G/V2C and G/Mo2C, respectively.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Considering the significant advantages of TMCs, they will become a potential replacement for costly Pt-based catalysts. However, research into TMC-based electrocatalysts is in the early stages, and the future development of highly efficient TMCs still involves huge challenges, as follows.
- (a)Highly active TMCs with controlled/specific structures and crystal planes are normally obtained via solid-state reactions in a multistep process and using high-temperature treatments, which limit their large-scale controllable synthesis. Therefore, efficient, low-cost, and green methods should be explored for the facile synthesis of enhanced-performance TMCs.
- (b)For practical applications, it is vitally important to reduce the cost of TMC-based electrocatalysts and carbide loading and produce carbide under mild conditions. Hence, it is desirable to develop TMC-based electrocatalysts that have multiple functionalities and are highly compatible with the ORR in chemical environments with complicated species and highly variable parameters.
- (c)The surfaces of TMCs are easily oxidized, which limits their application in the ORR. Efficient strategies (e.g. metal functionalization) should be developed to enhance the stability of TMC-based electrocatalysts and improve OER performance by avoiding the oxidation of TMCs. Moreover, most of these emerging catalysts are completely inactive in acidic solutions. Extensive studies are required to understand the reason for this.
- (d)The electrochemical reaction mechanisms of ORRs based on TMCs are not well understood from the perspectives of thermodynamics and kinetics. The active sites of metal carbides are not clearly known. Hence, a methodology for the real-time precise monitoring of the dynamic variation of reaction intermediates on a catalyst's surface needs to be established, by combining advanced in situ operando techniques (such as in situ Raman spectroscopy, x-ray photoelectron spectroscopy (XPS), and synchrotron x-rays) to understand the mechanisms involved.
Concluding remarks
We described the current bottleneck of traditional Pt-based catalysts and highlighted the recent trends and great effort invested in TMC-based electrocatalysts for the ORR. We demonstrated that TMCs can be used as electrocatalysts or supports to improve the catalytic performance of the ORR. Despite the various challenges mentioned above, the field of carbide-based electrocatalysts for ORRs is a new frontier in research, with unlimited prospects and opportunities.
7. Atomically dispersed Fe–N–C catalysts for ORR
Yanyan Zhao
Rowland Institute at Harvard University
Status
The electrochemical ORR has attracted extensive attention due to its application in fuel cells [82]. The precious‐metal‐group (PMG) materials, such as Pt and Pd, are generally efficient electrocatalysts for ORR, however, their high prices and poor CO tolerance have hindered their large‐scale application. Therefore, it is necessary to replace the PMG materials with earth‐abundant catalysts but without sacrificing stability and activity. Currently, Fe–Nx –Cy catalysts are considered to be the most promising ORR catalysts due to their superior catalytic performance and are regarded as promising substitutes for PMG catalysts [100–102].
The sum of the existing knowledge inspires the researcher to recognize the importance of catalysts with well-defined structures. To meet this need, researchers first aimed to prepare an atomically dispersed catalyst with Fe metal centers within N or C frameworks. For instance, such a catalyst can be prepared by mixing the metal precursor and graphene-based materials using a sequential annealing process (figure 13) [83]. To get a clear picture of the prepared motif at the atomic level, microscopic and spectroscopic techniques are coordinated (figure 14). For the characterization of the Fe metal, the most direct evidence is obtained by high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) [103]. The bright spot highlighted in the background usually represents the metallic center. However, the microscopic data usually serves as a platform for the identification of the location. To further explore the local chemical information of the metal center, HAADF-STEM can also be combined with electron energy loss spectroscopy (EELS) to detect the coordination environment of target metal sites. For instance, Zelenay and co-workers applied high-resolution EELS to confirm the presence of adjacent N atoms around an Fe center [103]. Also, quantification of the Fe-to-N ratio from the EELS data helps to confirm an average composition. However, in some cases, the resulting catalysts might not exhibit adequate stability for high-resolution characterization. When studied by high-intensity experimental tools such as STEM, an atomically dispersed motif might undergo structural evolution, rendering structural information at the atomic level elusive. To partially address this challenge, spectroscopic studies such as X-ray absorption spetroscopy (XAS), including X-ray absorption near edge structure (XANES), and Extended X-ray absorption fine structure (EXAFS) spectra have also been developed to explore the average coordination environment of Fe [104]. XANES can help to identify the chemical valence state and spatial configuration, while EXAFS serves as a tool to identify the coordination numbers of Fe–N (first shell) and Fe–C (second shell). While Fe–Nx–Cy have been shown to be active in the ORR, there is still a debate about whether Fe2+ or Fe3+ is the active species. The answer to this will require XAS coordinated with Fe Mösbauer spectroscopy based on the recoil-free absorption of γ rays from Fe57 nuclei [82, 105]. A change in the coordination environment leads to a difference in isomer shift and quadrupole splitting. What is more, during the reaction, the valence state switch could also be observed by in-situ XAS and the atomic state could be monitored by in-situ STEM. Besides a detailed exploration of the coordination environment of the Fe center, the other key information is the chemical environment of the N. Similarly, when the researchers examined the performance of the ORR, the ratios of different kinds of N resulted in a divergence in terms of activity and selectivity. This observation begs the question: which N is necessary to achieve high performance? X-ray photoelectron spectroscopy (XPS) can be used to confirm the N species in the catalyst, such as pyridinic N, pyrrolic N, or graphitic-like N. Also, the dynamic state of N during the reaction can be monitored by an in-situ technique such as in-situ XPS or XAS. By combining these multiple pieces of information, a clear picture can be obtained of the detailed structure, oxidation state, and spin states of the Fe–Nx–Cy active sites.
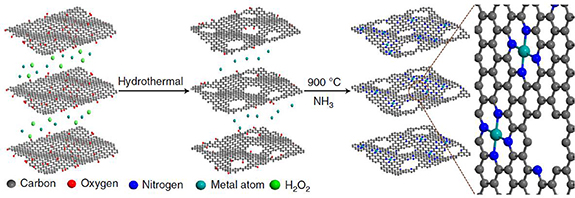
Figure 13. A typical method for preparing a well-defined MNx Cy (M=Fe, Co or Ni) atomic motif. A graphene oxide (GO) solution was mixed with H2O2 and an Fe precursor. The mixture was hydrothermally reacted to form graphene hydrogel. By sequent thermal annealing in an NH3 atmosphere, an atomically dispersed M–Nx–Cy motif was prepared. Reprinted and reused with the permission from [83], Copyright © 2018, Huilong Fei et, Springer Nature.
Download figure:
Standard image High-resolution image
Figure 14. A clear picture of the prepared motif at the atomic level, obtained by microscopic and spectroscopic techniques.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Fe–Nx–Cy catalysts have usually been prepared through the pyrolysis of molecular or polymeric precursors which contain metal centers, N– and C–containing ligands. Due to the heterogeneity of the prepared catalysts, the metal centers have a variety of chemical valences and C or N atoms are embedded in a diversified coordination environment [106]. The lack of a clear atomic picture of Fe–NX–Cy and the absence of an understanding of the ORR mechanism at the molecular level are the main challenges in expediting the development of this catalyst [107]. Although significant progress in terms of ORR performance has been reported in a large number of published works, these efforts face an important caveat: the active sites are not well defined. Because of this, enhancement of the performance mainly relies on trial and error. A variety of reasons are the cause of this issue [82, 83]:
- (a)The difficulty of preparing Fe–Nx–Cy catalysts free of metal nanoparticles, especially under annealing conditions.
- (b)Lack of comprehensive and accurate characterization tools for determining the structural details of the active sites, including both metal sites and N- or C-containing ligands.
- (c)The difficulty in distinguishing the individual role of N-doped C structures and metal sites. Moreover, the synergistic effect is not clearly understood at present.
Concluding remarks
Detailed structural information about active sites would make it much easier to capitalize on the molecular identities of catalysts to systematically study their mechanisms, which is critical to further performance optimizations. Moreover, knowledge about the dynamic nature of the active sites obtained by in-situ methods would help to unveil the whole catalytic process.
8. Metal-free catalysts for ORR
Hamna Zia and Farhat Nosheen
SA-CIRBS, International Islamic University Islamabad, Pakistan and Department of Chemistry, Division of Science and Technology, University of Education
Status
For important industrial processes, catalysts of metals and metal oxides are used for clean energy storage, energy generation, and material production. However, due to the high cost, poor durability, and low selectivity of these catalysts, certain problems must be solved. They also suffer from CO deactivation, are highly susceptible to gas poisoning and time-dependent drift, and have various harmful impacts on the environment. Furthermore, the worldwide commercial large-scale application of these renewable energy technologies has been hindered by the scarcity of precious metals [108–110]. Currently, fast and dynamic development is underway to replace metal-based catalysts with abundant, cheap, and high‐performance ORR electrocatalysts [111]. Up until now, various metal-free electrocatalysts have been synthesized, among which, carbon‐based metal-free catalysts have been reported to exhibit outstanding ORR electrocatalytic activity due to their high stability and good tolerance. Moreover, the constituents of these metal-free catalysts are highly abundant [99], and they also have the advantages of high energy conversion efficiency, low energy consumption, good corrosion resistance, and a wide application range. Therefore, they are considered as potential alternatives to metal-based catalysts [112].
Since the discovery of these metal-free catalysts, the overall cost has been dramatically reducedand the efficiency of renewable technologies has increased. These renewable technologies can alter our daily lives by providing us with very clean, renewable and efficient energy from water and sunlight [113]. Since the 1960s, research has been carried out to find effective alternatives to replace platinum catalysts [114]. Alloying platinum with some non-precious metals such as iron, cobalt, nickel, etc, was found to be a promising approach, but it was still not very effective for practical applications, due to the cost, scarcity, and limited reserves of platinum [78]. Stable, efficient, and low-cost catalysts are highly desirable to cater to large-scale applications. After intensive research efforts for over half a century to develop a metal-free catalyst for ORR, finally, the overall cost was dramatically reduced when vertically aligned nitrogen-doped carbon nanotubes (VA-CNTs) arrays were discovered in 2009 [6]. They exhibited a three-fold increase in electrocatalytic activity and high stability in comparison with commercially available platinum/C electrodes for ORRs in alkaline media and did not suffer from CO deactivation and methanol crossover. Later, several carbon-based metal-free heteroatom-doped catalysts were reported due to rapid development in the field of metal-free catalysis, for example: sulfur-doped graphene, boron-doped CNTs, phosphorus-doped graphite, edge-halogenated doped graphene nanoplatelets, iodine-doped graphene layers, phosphorous and nitrogen co-doped VA-CNTs, boron and nitrogen co-doped graphene, sulphur and nitrogen co-doped CNTs, nitrogen-doped ordered mesoporous graphitic arrays, quantum dots (QDs) supported by graphene nanoribbons, phosphorus-doped graphite layers, and nitrogen and sulfur co-doped graphene QDs [115].
Current and future challenges
Metal-free catalysts have seen great progress and development during the last decade. However, until now, the development of a carbon-based metal-free catalyst for ORR catalysis (defect-induced) is in the premature stage; therefore, further research is required to understand the overall mechanism. Metal-free catalysts will not be able to compete with metal-based catalysts until the following challenges have been resolved:
- (a)For alkaline solutions, some of the carbon-based metal-free ORR catalysts show similar or even better ORR activity compared to Pt/C, but in acidic media, most of these carbon-based catalysts show poor ORR activity compared to Pt/C.
- (b)The literature hardly explains the reduction of carbon dioxide by carbon-based metal-free catalysts. Up until now, no standard evaluation protocol has been applied using practical devices to check the long-term durability of metal-free catalysts, which can cause a hindrance for the commercial application of metal-free catalysts.
- (c)The multi-doping of sp2 carbon materials to achieve high ORR activity is a promising approach. To benefit from synergistic effects that are aroused by the multi-doping of carbon materials, the configuration of the dopants must be regulated and studied thoroughly by paying attention to all possible aspects. This may be helpful to the electrocatalytic performance of carbon materials in ORRs.
- (d)Surface active sites have a significant role in the reduction of current density and are the place where the ORR proceeds. There are huge numbers of topological defects present in active sites, giving a better onset potential, and thus making them highly active for the ORR. Moreover, a severe difficulty occurs for the ORR due to the low electron conductivity that is caused by large active sites. Hence, to optimize the performance of carbon‐based ORR electrocatalysts, a good balance between surface active sites and electron conductivity is necessary. Since the ORR involves multiple steps, and many intermediate species are formed, the handling of multiple defects and doping configurations becomes more complex and complicated.
- (e)The fabrication and design of catalytic materials and electrodes require structural evaluation and control at the molecular and macroscopic levels to form appropriate multi-scale hierarchical structures for optimized catalytic performance.
- (f)Novel doping or synthetic methods should be developed to control the location, distribution and content of dopants in heteroatom-doped carbon catalysts precisely.
- (g)The development of combined experimental and theoretical approaches is essential to understand the kinetics, mechanism and structure of the catalytic center.
- (h)The application of these catalysts in gas-phase reactions (for example, to the oxidative dehydrogenation of alkanes) has been very limited; their application in other new reaction systems must be expanded on an urgent basis.
- (i)Several basic factors are still unclear, for example, the exact role of the interface, the reaction mechanism, and the nature of the active sites during catalytic processes. A reliable identification method is needed that must identify functional groups and their structural defects and properties.
- (j)The impact of metal impurities on catalytic activity is still unknown. These metal impurities are present in trace amounts in the reaction mixture.
- (k)The development of facile, simple, and highly efficient strategies is required for large-scale industrial and commercial applications. Such methods are needed for the production of metal-free nanomaterials with well-defined structures and chemistries at a large scale. For large-scale commercial applications, many reaction conditions still need to be optimized and explored, which would help to enhance product purity and yield and thus minimize the overall cost.
Concluding remarks
Undoubtedly, extensive research in the field of metal-free catalysts will result in improved fuel economy and low harmful emissions, further reducing the overall cost of renewable generation of clean energy for industrial applications. Therefore, metal-free catalysts will undoubtedly receive more extensive attention in the fields of materials science, chemistry and catalysis.
9. Single-atom catalysts for ORR
Guangchao Zheng
School of Physics and Microelectronics, Zhengzhou University
Status
SACs are high-utilization heterogeneous catalysts, in which single metal atoms are uniformly dispersed on the surface of a support material [116]. Recently, research into SACs has received extensive attention and has offered new opportunities for numerous catalytic reactions, because SACs have an almost 100% atomic utilization rate, unique electronic states, strong interaction between the SAC and its support, quantum size effects and excellent catalytic performance compared to traditional catalysts, even nanocatalysts. SACs were first proposed by Zhang and his colleagues, who described the oxidation of CO catalyzed by a Pt SAC supported on FeOx , which was recognized in the field of catalysis and became a new frontier and focus [117–119]. In fact, before the emergence of the definition of SACs, several other concepts such as atomically dispersed heterogeneous catalysts, single-site heterogeneous catalysts, and site-isolated heterogeneous catalysts had already been mentioned in other reports.
So far, the strategies for preparing SACs include chemical vapor deposition, atom layer deposition (ALD), pyrolysis, the wet chemical method, the impregnation method, photochemical reactions, etc. As previously demonstrated, an SAC is the uniform distribution of individual atoms on a support. There is always a strong interaction between a single atom and the support. The strong interaction inhibits the aggregation or dissolution of the SAC, thereby enhancing its stability and durability. Also, it regulates the electronic structure of a single atom, thereby tuning the adsorption and desorption energies of intermediates in the catalytic process. Furthermore, it provides new active sites due to the non-metallic atoms. Therefore, the choice of support is also of great significance for the design, characterization, and theoretical models of SACs [120, 121]. To date, there are several kinds of support including carbon materials and their derivatives, 2D materials, porous materials (zeolites), and so on.
In particular, HADDF-STEM, EXAFS, and XANES technologies are widely used for the characterization of SACs. Researchers can evaluate individual metal atoms based on local structural features at the sub-nanometer level, coordination numbers, distances, evolution of oxidation states, and elemental distribution around active centers. The EXAFS spectrum reveals the coordination number of the metal-to-metal bonds, which is an index used to evaluate the quality of the SAC. In principle, metal-to-metal bonds should not exist at the active site. Therefore, if the EXAFS data does not show any information about metal-to-metal bonds, it can be concluded that there are only SACs. The structural information and catalytic mechanism can be further analyzed by infrared (IR) spectroscopy and DFT. Xiao et al have prepared uniform and high-efficiency Ir–N–C (Ir SAC) ORR electrocatalysts. In figure 15, the combination of HAADF-STEM with EXAFS and XAFS characterization confirms the presence of uniformly dispersed Ir SACs, which are coordinated with four N atoms and one O atom to form a stable Ir–N4–O structure. The turnover frequency (TOF) of the Ir SAC is 5.6 times higher than that of commercial Pt/C. The specific structure of Ir–N4 modifies the electronic structure of Ir and regulates the adsorption energy of the ORR's intermediate species, which mainly contributes to their excellent ORR catalytic activity and stability under acidic conditions [122].
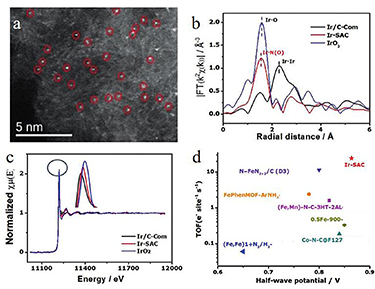
Figure 15. (a) HADDF-STEM image of Ir SAC on N-doped carbon. Monodispersed Ir SACs are indicated by red circles. (b) EXAFS and (c) normalized XANES spectra at the Ir L3 edge. (d) TOF and half-wave potential of the reported SAC. (Reproduced from [122]. Copyright 2019, Wiley–VCH.)
Download figure:
Standard image High-resolution imageAn SAC has been reported to be an efficient ORR electrocatalyst [123]. The ORR involves different pathways, mainly the direct pathway; the conversion of O2 by the four-electron reaction occurs directly to H2O (the four-electron path); the two electrons react to form products such as H2O2 intermediates (the two-electron path). The selectivity is generally determined by the catalyst's ability to break O–O bonds. The typical metal elements in SAC electrocatalysts include Pt-group noble metals which prefer a two-electron pathway, and transitional metals (such as Fe, Co, Cu, Mn, Ir, Zn, etc) which prefer the four-electron pathway. Low-cost transition-metal SACs, with high activity and selectivity, can replace Pt-group noble metals for ORR electrocatalysis, promoting the development of sustainable clean energy conversion and storage technologies. Electrochemical tests show that the half-wave potential of an Fe SAC electrocatalyst in the ORR reaction is as high as 0.91 V, which is 90 mV higher than that of conventional Pt/C catalysts. Fe-based SACs have better performance than Pt/C catalysts in the ORR under alkaline conditions, exhibiting smaller half-wave potentials.
In recent years, we have witnessed much significant research into the use of SACs in the ORR, as well as significant advances in the field of SACs. Zhang et al developed a universal route to fabricate a range of metal SACs by electrochemical deposition methods for both cathodes and anodes [124]. Studies have shown that the introduction of a second metal atom can further enhance the activity of SACs. Han et al reported a Co–Ni bimetallic SAC supported on nitrogen-doped hollow carbon nanocubes for dual-function use in the OER and the ORR. High SAC yields and bimetallic synergistic effects gave it lower overpotential, a high electron-transfer number, and good reversibility, which were superior to many reported OER/ORR performances of metal SACs [125]. Furthermore, engineering the supports with heteroatoms (N, P, S) is considered to be an effective method for improving the ORR performance of SACs. For example, metallic atoms supported on N-doped carbon materials have strong synergistic effects of high electrical conductivity and adjustable electronic states. Yao et al reported that an SAC supported on nitrogen-doped carbon (Mn, Fe, Co, Ni, Cu as an active site) was able to change the 1e− ∼ 4e− transfer process, thus strongly regulating the ORR pathway [126]. Chen et al proposed an atomic interface strategy to construct a Cu SAC supported on N/S-doped carbon materials, which had excellent ORR performance (E1/2 = 0.893 V vs. RHE) in alkaline media because of the construction of a Cu+–N4–C8S2 atomic interface, which was confirmed by x-ray absorption fine structure (XAFS) research based on synchrotron radiation and DFT calculations [127]. Recently, Zeng et al developed a general method for the synthesis of more than 30 different metal SACs with different electronic states deposited on the same cathodes and anodes, based on different reactions. The SACs deposited on cathodes showed high activity for the HER, while SACs deposited on anodes showed high activity for the OER [128]. Therefore, it is particularly important to design efficient, inexpensive and versatile electrocatalysts.
Current and future challenges
Despite some progress, there are still urgent challenges to be overcome to improve the catalytic performance and selectivity of the ORR. SAC has a certain significance in the field of catalysis: it has the greatest TOF because of the maximum utilization of atoms. However, HADDF-STEM and spherical aberration electron microscopy only provide local information; EXAFS and XANES are also averaged statistics. There may be a coexistence of clusters or small NPs and SACs in SAC systems, in which a few clusters may play an important role in catalytic performance. Therefore, it is still necessary for the current synthetic method to synthesize SACs and confirm that there is 100% single atom dispersion.
In past research, composition, low-coordination atoms and ligands played a big role in the properties of nanocatalysts. For example, a bifunctional SAC may have better catalytic performance compared to a monometallic SAC [129]. Besides, an understanding of the structure–activity relationship is also necessary for an in-depth understanding of the mechanisms of SACs. Ligands have been viewed as regulating the electronic structure of modified NPs, thus affecting the catalytic performance of NPs. For nanocatalysts, the modification of ligands will inevitably lead to more significant changes in the electronic structure, which will have a profound impact on catalytic performance. Research into composition, structures and low-coordination atoms would provide instructions for the rational design of SAC ORR electrocatalysts.
Because of the higher surface energy and thermodynamic instability of single atoms, the choice of support is of significant importance in preparation and practical application, to prohibit single atoms from aggregating into clusters or even NPs, thereby improving the catalytic efficiency, stability, durability, and selectivity. Also, the key to the challenge of preparing SACs is the contradiction between increasing the maximum loading amount and controlling agglomeration. In the process of preparing SACs, there is a limitation on the loading amount of SAC (generally 2% or less) to avoid the agglomeration of SACs. Lower metal loadings often lead to a lower TOF of the SACs, limiting their performance and application. Therefore, the preparation of high-efficiency, stable, and high-load SAC ORR electrocatalysts is still quite challenging.
Concluding remarks
SACs, with their extreme atomic utilization, unique electronic states, and quantum-scale effects have recently become a new frontier in the field of ORR electrocatalysis, because of their excellent performance and low cost. Advances in preparation methods, characterization technologies, and simulation model calculations indicate that SACs have great potential for use in ORR electrocatalysis.
Acknowledgment
This work was supported in part by the National Natural Science Foundation of China (Grant No. 21902148).
10. Metal borides for water-splitting
Suraj Gupta
School of Engineering, University of Liverpool
Status
Since H2, with its zero-carbon footprint, is on its way to being the new energy carrier, the development of associated technologies is at an all-time high. Electrocatalytic water-splitting is tipped to be the leading technology for the renewable production of H2 fuel. Current state-of-the-art water electrolyzers employ precious-metal-based (Pt, Ru, Ir) electrocatalysts that are expensive and have scarce reserves in the Earth's crust, limiting their use for large-scale implementation [130]. Amongst the low-cost alternatives, metal borides (MBs) have emerged as one of the most promising families of electrocatalysts for implementation in commercial systems [131, 132]. The first use of MBs for water splitting was reported in 1974 when Kuznetsova et al [133] tested some MBs for use in the HER. In the following years, there were a few more reports of the use of MBs for the HER and the OER, but they were not seen as potential electrode materials for industrial use. During this time (1980–2000), research into MBs was mainly focused on the use of commercial glasses containing borides or the use of energy-intensive methods (such as spin melting) to synthesize materials [132]. In 2009, Nocera et al [134] reported an in-situ-formed cobalt borate (Co–Bi) catalyst deposited by electrodeposition for near-neutral water-splitting. This was followed by reports of crystalline and amorphous MBs, based on Mo [135] and Co [136] respectively, that re-triggered the interest in these materials amongst the research community. From 2015 onwards, a plethora of different material combinations has been reported within the MB family [131, 132]. Figure 16(a) presents some of the best MBs in alkaline conditions for OER. The graph indicates that overpotentials as low as 237 mV have been achieved for the OER using an optimum loading of Fe–Ni–B catalyst. Likewise, for the HER, figure 16(b) depicts that the performance of MBs is on par with that of industrial Pt electrodes. The performance and stability of MBs surpass those of Pt in an alkaline medium. In contrast to the case of the OER, MBs are active and stable for the HER in acidic and neutral solutions, with the lowest overpotentials of 45 mV and 54 mV shown by NiBx catalysts in acidic and neutral electrolytes, respectively [137]. These graphs suggest that MBs are amongst the best-performing electrode materials, with bi-functional activity (the capability to catalyze both the HER and the OER) in an alkaline medium, rendering them best suited for alkaline water electrolyzers.

Figure 16. (a) A Chart comparing the OER performances of some of the best MB/borate electrocatalysts with respect to their mass loading. All catalysts are reported at Aa pH of 14 except for metal borates, which are reported at a pH of 9.2. The graph also indicates the best catalysts from other families of non-precious-metal catalysts as well as precious-metal-based catalysts. (Acronyms used—C: Vulcan carbon; CC: carbon cloth; CP: carbon paper; NF: Ni foam; Ni: Ni foil/plate; Ti: Ti mesh/foil.) (b) Comparison chart representing the HER performance for the best-performing MB electrocatalysts in Pt solutions of different pHs as well as other non-precious-metal catalysts. (Acronyms used—C: Vulcan carbon; NC: N-doped carbon; GNR: graphene nanoribbons; CP: carbon paper; NF: Ni foam; Ni: Ni foil/plate; Ti: Ti mesh/foil; NCF: nanoparticles dispersed on carbon microflowers, NSs: nanosheets.) Reproduced from [132].
Download figure:
Standard image High-resolution imageOver the last five years, research into MBs has mainly focused on trying new material combinations and strategies to improve their performance and stability. The most prominent material engineering strategies that have been employed to boost the performance of MBs include the formation of ternary/quaternary alloys, the incorporation of P to form phospho-borides, the use of porous conducting supports to support MBs, and the development of unique nanostructures. Of these strategies, the most successful results have been obtained with ternary and quaternary alloys. For instance, tri-metallic boride Fe–Co–Ni–B yields a low overpotential of 274 mV and achieves 10 mA cm−2 in the OER, with a minimal catalyst loading of 0.3 mg cm−2, without using any substrates. The strategy of using porous substrates is also highly effective, but the issues related to an accurate determination of the geometric and electrochemical surface areas are difficult in such systems, and hence, they must be adopted with caution.
Despite these reports, it should be noted that TMBs are the youngest family of inexpensive electrocatalysts and hence have significant potential for further advances. Although they show good promise for the HER in acidic and neutral electrolytes, their exceptional bi-functional performance and stability in alkaline solutions render them the most ideal candidates for implementation in extreme alkaline solutions. The other characteristic that gives MBs an edge over other non-precious families of electrocatalysts, such as metal sulfides and phosphides, is the availability of non-toxic, low-cost, and room-temperature synthesis strategies. Additionally, the bi-functional characteristic helps in reducing the cost of manufacturing different sets of electrode materials. With further advances in this family of materials, it is expected that MBs will replace both anode and cathode coatings, primarily in commercial alkaline water electrolyzers.
Current and future challenges
Though MBs have a very short history of use in electrocatalytic water-splitting, they have been used for other catalytic applications for over a century. In particular, amorphous MBs of Fe, Co and Ni have been of particular interest in various catalytic applications such as hydrogenation and H2 generation from metal borohydrides [138]. The chemistry of such amorphous MBs has been a topic of debate for a long time and this continues to be the case. In a simple amorphous mono-metallic boride (for example, Co–B, Ni–B, etc), B exhibits higher electronegativity than its metal counterparts. A higher value of Pauling's electronegativity suggests a higher affinity towards electrons and hence, B is expected to be the electron-rich site, taking electrons from the bonded metals. However, in these amorphous systems, this phenomenon does not hold good. It is usually observed that the metal atom is enriched with electrons, while the B site is electron deficient. This 'reverse electron transfer' phenomenon has consistently been observed by several researchers, especially in MBs containing Co and Ni [132, 138]. My group demonstrated the experimental and computational evidence of this phenomenon for amorphous cobalt–boride (Co–B) [136]. We provided concrete computational data to confirm that the crystalline structures of Co–B exhibited electron-enriched B-sites, which reversed when an amorphous cluster containing the same number of atoms was simulated. As per Sanchez et al [138], the transfer of electrons in amorphous MBs is governed by the ratio of the concentrations of B and metal (M) in the compound. In an MBx system, reverse electron transfer (from B to M) is observed only in metal-rich borides when x ⩽ 2. So far, there has not been a single experimental report that validates these theories comprehensively. This is one of the primary fundamental challenges in understanding the chemistry of MBs.
In the context of understanding the reaction mechanisms of the OER/HER in MBs, very little work has been done, particularly for the OER. From the various reports, the most common theory for the OER over MBs is the formation of active metal oxy-hydroxy (M–OOH) species on the surface of the catalyst. In our recent article [139], we presented experimental evidence for the formation of a Co–OOH layer on the surface of a Co–B film after the OER. Similar reports have been made by other researchers using Ni- and Fe-based borides [132]. However, incisive reports explaining the favorability of formation of this surface species on MBs, compared to bare metals, do not exist. Such experiments need to be accomplished using in-situ/operando techniques capable of probing the chemical states of the elemental sites during the OER. Since it is a light element, it is difficult to probe B sites using conventional in-situ spectroscopic methods, which presents a real technical challenge for researchers. Another theory explains that the B in an MB system gets leached out during the OER, leaving pores in the catalyst's surface, eventually leading to an increase in the electrochemical surface area and hence, a better OER [140]. However, this theory needs to be validated by consistent reports examining the content of dissolved elements in used electrolytes. In addition to these experimental challenges, the computational simulation of amorphous systems that replicates the exact experimental conditions is also a challenging task, which is often ignored. The development of advanced computational methods to support this objective will present exciting prospects for unravelling the mystery of amorphous MBs. Most of the above discussion is focused on amorphous MBs, but their crystalline counterparts are equally exciting and easier to explore, owing to their known structures and stoichiometries. For crystalline MBs, determination of the most active planes and subsequent tailoring of these materials to expose more of these planes must be adopted, to push the present boundaries of electrochemical performance.
The last big challenge with MBs lies in the synthesis of pure-phase crystalline Mo-based borides. Mo-based borides are the acid-stable members of the family of MBs and demand special attention to expedite the use of MBs in proton-exchange membrane (PEM)-based electrolyzers. New and greener synthesis strategies need to be developed to obtain pure-phase Mo-based borides with different nanostructures, preferably in the form of 2D layered structures.
Concluding remarks
Undoubtedly, research into MBs holds the key to finding the most appropriate low-cost electrocatalyst, especially for use in alkaline water electrolyzers. So far, research into these materials has been widely scattered; this roadmap will serve as a guide to streamlining the effort to achieve the final goal of commercializing MBs.
Acknowledgments
S Gupta acknowledges a research grant from the UK Commonwealth Scholarship Commission through the Commonwealth Rutherford Fellowship (INRF-2017-139).
11. Transition-metal chalcogenides for water splitting
Xianhong Wu, Zhiyu Wang and Jieshan Qiu
State Key Lab of Fine Chemicals, Liaoning Key Lab for Energy Materials and Chemical Engineering, Dalian University of Technology
Status
The development and efficient utilization of clean sustainable energy are eagerly desired, to overcome the crisis arising from the fast depletion of fossil fuels and increasing environmental pollution, accompanied by ever-growing energy consumption. Hydrogen perfectly meets the demand of clean energy for modern society is not only a critical raw material in the clean energy industry but also perfectly meets this demand due to its substantial merits, including, but not limited to: (a) it is a nearly infinite energy resource, making up 75% of the universe; (b) the by-product of its use, water, is totally environmentally friendly, safe, and fully recyclable; (c) its calorific value (142.351 kJ kg−1) is, in fact, three times more potent than gasoline and much superior to other fossil fuels; (d) it is storable and transportable on a large scale; (e) it provides an incomparable rate of energy supply to energy-consuming facilities (e.g. electrical automobiles), compared to batteries. With these advantages, hydrogen looks promising as one of the leading and cost-effective options for storing energy from renewables and strengthening global energy security.
Water electrolysis represents one of the most efficient and clean ways of producing high-purity hydrogen, especially when coupled with renewable solar PV, wind and hydropower, compared to the traditional approach of steam–methane reforming. However, it suffers from a huge overpotential (about 570–770 mV) originating from the high thermodynamic barrier of the OER and HER (compared to the theoretical minimum value of 1.23 V), which causes high energy consumption and thus processing cost. Electrocatalysts based on noble metals (commonly Pt or Ru/Ir groups) perform well in enhancing the efficiency of energy utilization and hydrogen production, but their large-scale implementation is prohibited by their unaffordable cost and great scarcity in the Earth [141]. Currently, the extreme lack of highly efficient non-precious-metal electrocatalysts that can satisfy the strong industrial demand has become a critical bottleneck in the widespread application and development of hydrogen economics around the world.
Among various candidates, TMCs have shown great potential as alternatives to noble metal catalysts. With noble-metal-like electronic structures, they actively perform hydrogen adsorption and activation, manifesting a Pt-like electrocatalytic behavior [142]. Benefiting from the high electronic conductivity associated with their interstitial alloy structure, TMCs are innately superior to semiconducting catalysts such as metal chalcogenides, phosphides, and nitrides in terms of charge transport kinetics [143]. Moreover, their good chemical stability and wide pH applicability also make them versatile for practical use in various applications, such as PEM-based acidic electrolysis, neutral seawater electrolysis, and alkaline electrolysis. Since the pioneering work by Vrubel and Hu in 2012 [144], research into TMC-based electrocatalysts has experienced a surge (figure 17(a)). Specific attention has been paid to the design of TMC nanostructures (M=Mo, W, V, Ta, Fe, Co, Ni, etc) with controlled compositions, structures, electronic properties, and chemical states for reducing overpotential and accelerating the kinetics of the HER and OER. Various strategies, including nanocrystallization, crystallography engineering, morphology tailoring, heteroatom doping, and defect and hetero-interface engineering have been utilized to boost the intrinsic activity, accelerate the charge transport kinetics, and optimize the interaction of TMCs with the reactant, intermediate species, and gas products [143]. To further improve their electrocatalytic activity and stability, TMCs have been further coupled with conductive supports such as soft carbon materials (e.g. CNTs, graphene) and MXenes [145]. The introduction of these conductive supports is critical for improving the dispersion of downsized TMCs and strengthening active sites via physicochemical coupling with TMCs. More importantly, it offers prominent interfacial electronic and structural coupling to reduce the water dissociation barrier under proton-lacking conditions and optimize the hydrogen binding energy (HBE) (a primary indicator of HER activity), to a near-zero value, to accelerate the HER without suppressing H2 release. On this basis, a free-standing 3D electrode configuration has been also adopted to achieve fast mass transport, which significantly extended the liquid–solid–gas interface and allowed the rapid release of gaseous products from the electrode's interior during vigorous electrocatalytic reactions. These advantages are vitally important to realize industrial-level current densities (>500–1000 mA cm−2) [135]. For binder-free 3D electrodes, they have the benefit of avoiding clogging due to an insulating binder (e.g. Nafion) that occurs in traditional cast electrodes, thereby minimizing the deterioration of the intrinsic activity of TMCs and electrode polarization. These strategies also work well in improving the electrocatalytic performance of non-metal carbides such as graphitic C3N4 [146]. Due to great efforts, the overpotential for acquiring a current density of 10 mA cm−2 (ηj = 10) and the HBE have rapidly approached the levels achieved by Pt in the HER (figure 17(b)). For the OER, the state-of-the-art TMC-based electrocatalysts exhibit a relatively high ηj = 10 of more than 200–300 mV, which still cannot compete with their phosphide/sulfide/hydroxide counterparts. Moreover, since TMCs generally suffer from fast oxidation in the OER, and due to the lack of stable OER electrocatalysts that are efficient under acidic conditions, the development of TMC-based HER electrocatalysts that are efficient under alkaline conditions promises to drive the practical use of water electrolysis, when coupled with efficient noble-metal-free OER catalysts such as Ni-based layered double hydroxides (LDHs) and perovskite-type oxides.

Figure 17. (a) The numbers of publications and relevant citations on the topic of TMCs for electrocatalysis during 2012–2017. (b) Rapid reduction of the overpotential required to acquire a current density of 10 mA cm−2 for the HER using typical TMC-based electrocatalysts during this period. The above figures were summarized by Tang et al [143]. Copyright. 2019, Wiley–VCH.
Download figure:
Standard image High-resolution imageRecently, a new, yet huge, family of 2D TMCs based on early transitional metals (MXene) has attracted considerable attention for engineering high-performance water-splitting electrocatalysts [147]. They have a good combination of high conductivity (104–105 S cm−1) associated with a high electron density of states near the Fermi level, excellent hydrophily, and rich surface chemistry offered by rich surficial chemical groups. A wide chemical and structural variety, ultralow work functions, and electronegative surfaces further provide the feasibility of precisely regulating the electrophilicity of active sites and catalytic properties. So far, there is still plenty of room for improving the HER activity of MXenes so that the shielding effect of their surficial groups on active sites and ionic diffusion can effectively be overcome. Nevertheless, recent works have demonstrated the great potential of MXene for engineering highly active and multifunctional collaborative catalytic interfaces in hybrid electrocatalysts [148]. It offers overall enhancement of electrical conductivity, exposure of reactive sites, water dissociation kinetics, H+/H2O adsorption and the HBE due to the synergy of intrinsic merits of MXene and guest active phases such as LDH, MOFs, TMCs and noble metals. Accordingly, a series of multifunctional electrocatalysts have been developed to satisfy the complicated criteria of water splitting in a highly variable chemical environment, which exhibits excellent performance over a wide pH range and even with saline seawater.
Significant progress has been also been made in understanding the chemical nature and mechanisms involved in the HER/OER by a combination of ex-situ analysis and operando techniques such as scanning electrochemical microscopy (SECM), electrochemical quartz crystal microbalance (EQCM), operando microscopy, synchrotron radiation and theoretical calculation based on density functional theory and molecular dynamic computation [149]. These powerful tools provide an atomic/molecular insight into the relationship between structure, composition, electrode configuration, and electrocatalytic properties, thereby uncovering the mechanisms of water-splitting reactions over TMC-based electrocatalysts while providing guidelines for the exploration of novel non-noble-metal electrocatalysts. Nevertheless, the relevant field is still at an early stage relative to the booming research into the design and fabrication of catalysts, and many issues (e.g. the variation of intermediate species on the catalyst's surface, the rate-limiting step, the accurate identification of the active sites of TMCs in the OER) are still confusing and under debate.
Current and future challenges
Despite significant progress and impressive performance, research into TMC-based electrocatalysts is still in the early stages and their application in large-scale hydrogen production from water electrolysis still has a long way to go. There are still several main challenges to the further development of TMC-based electrocatalysts for water splitting, including, but not limited to:
- (a)TMCs are usually fabricated via solid-state reactions at high temperatures, which limits the large-scale production of high-purity TMCs. More efficient methods for the facile synthesis of TMCs with precisely tailored crystallography, crystal facets, and chemical states should be explored
- (b)For practical applications, it is vitally important to cut the cost of TMC-based electrocatalysts by such methods as synthesis from cheap raw materials such as biomass, the reduction of carbide loading, production under mild conditions, etc.
- (c)To meet the complicated demands of practical use, it is necessary to develop TMC-based electrocatalysts with multiple functionalities and good compatibility for water splitting in chemical environments with complicated species and highly variable parameters. This not only helps to reduce processing cost and system complexity but is also is helpful for an understanding of the electrocatalytic mechanism of water splitting reactions in different chemical states.
- (d)The surfaces of TMCs are easily oxidized, which limits their application in the OER. An efficient strategy should be developed for enhancing the stability of TMC-based OER electrocatalysts or boosting the OER performance by controlled use of the oxidation of TMCs.
- (e)The methodology of real-time precise monitoring of the dynamic variation of reaction intermediates on the catalyst's surface needs to be established by combining the advanced operando techniques for understanding the mechanisms involved in water-splitting reactions, for example, the chemical configuration and strength of H-binding on TMCs, the structural evolution of TMCs during the OER and its effect on the chemical state of real active sites and the OER species on them, etc.
Concluding remarks
We highlighted the recent trends and great efforts made to engineer water-splitting electrocatalysts based on TMCs. The great promise of TMC-based electrocatalysts for the HER in alkaline electrolyzers was highlighted. The challenges in the development of efficient TMC-based water-splitting electrocatalysts were discussed in terms of scalable and low-cost production, functionality tailoring, and the new perspectives on the analysis methodology for TMC-based electrocatalysts, as well as the development of TMC-based OER catalysts. We hope that this contribution will guide the scientific community devoted to the field of carbide-based catalysts for electrocatalysis, a burgeoning and cutting-edge research arena with boundless prospects and opportunities. The gap between the innovative development of carbide electrocatalysts and practical use in energy-driven applications will eventually be bridged by flourishing development and intense multidisciplinary efforts in the future.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC, Nos. 51522203, 51772040), Fok Ying Tung Education Foundation (No. 151047) and the Fundamental Research Funds for the Central Universities (No. DUT18LAB19).
12. Transition-metal phosphides for water splitting
Guangyao Zhou and Lin Xu
Jiangsu Key Laboratory of New Power Batteries, School of Chemistry and Materials Science, Nanjing Normal University
Status
The ever-growing global energy demand and severe environmental pollution have motivated extensive exploration of eco-friendly and sustainable energy sources. H2, with a high gravimetric energy density and environmental friendliness, is considered to be a promising energy carrier [150, 151]. Electrochemical overall water splitting (OWS), including the HER and the OER, has been recognized as one of the most competitive approaches for H2 production. Unfortunately, the sluggish reaction kinetics at both electrodes result in a higher cell voltage than the theoretical value (1.23 V) [152, 153]. The current state-of-the-art electrocatalysts for HER and OER are Pt and RuO2/IrO2, respectively. Nevertheless, the scarcity and mono-functionality of these benchmark catalysts significantly impede their widespread application. Therefore, enormous efforts have been devoted to developing earth-abundant electrocatalysts for the OWS.
Recently, transition-metal phosphides (TMPs) have been demonstrated to be effective electrocatalysts for the OWS due to their unique electronic structure. In 2005, Liu described the HER process on a Ni2P (011) surface by DFT calculation, predicting that Ni2P could be a promising HER catalyst [154]. Up until 2013, Zhang and Raymond experimentally proved the high HER performance of TMPs in succession [155, 156]. Following their work, a myriad of nanostructured TMPs was developed as efficient HER electrocatalysts. Besides, it was found that phosphides can be transformed in-situ into surface oxy/hydroxides and phosphates at a high overpotential, which can serve as active sites for the OER [157]. Therefore, TMPs can usually act as bifunctional electrocatalysts to catalyze the HER and the OER simultaneously. The abovementioned findings have stimulated the overwhelming development of TMPs as bifunctional electrocatalysts for use in the OWS. Although a range of TMPs has been reported as electrocatalysts for the OWS over the past years, their catalytic activities are still far from satisfactory due to their low intrinsic activity. To address these issues, the rational design of high-efficiency TMP-based catalysts requires the overall consideration of both thermodynamic and kinetic aspects. Currently, compositional regulation and architectural engineering provide reliable strategies to improve the OWS performance.
For compositional regulation, heteroatom doping and hybridization are usually employed to modulate the electronic structure, thus significantly boosting the intrinsic activity. The introduction of foreign elements (such as cations and/or anions, etc) with different electronegativities and electron configurations into a TMP lattice can modify the electronic structure and alter the adsorption free energies of reaction intermediates, dramatically enhancing the OWS performance. For example, Peng et al found that the incorporation of appropriate Fe into Ni2P was able to change the local atomic and electronic structure of Ni2P and optimize the Gibbs free energy of reaction intermediates during both the HER and OER on Ni2P, leading to favorable electrocatalytic activity with overpotentials of 208 and 33 mV at 10 mA cm−2 for the OER and the HER, respectively, and a cell voltage of 1.57 V at 100 mA cm−2 for OWS [158]. Our group recently reported that introducing a moderate amount of O into CoP nanosheets remarkably regulated the electronic structure and reduced the charge transfer resistance of CoP, giving rise to a significantly enhanced OWS performance with a cell voltage of 1.6 V at 10 mA cm−2 [159]. Kim et al found that the synergetic effect of a multi-element dopant made a great contribution to improving the activity of Co–Ni–S–P/graphene hybrids [160]. Apart from heteroatom doping, interfacial engineering by coupling TMPs with other components (such as sulfides, oxides, carbides, etc) also represents a powerful strategy for improving the OWS performance. The hybridization of different components results in the formation of heterojunctions, which may modulate electronic structures, increase the number of active sites, and accelerate electron transfer. For instance, Jiao's group constructed V-doped CoP@a–CeO2 hybrid nanoarrays grown on a carbon cloth substrate as a high-efficiency electrocatalyst for OWS. Due to the presence of V as an electron donator and the charge redistribution caused by CeO2, the resulting catalyst exhibited favorable Gibbs free energy of hydrogen adsorption and lower free energies for water absorption/dissociation, thus displaying outstanding OWS activity, with cell voltages of 1.56 and 1.71 V to at 10 and 100 mA cm−2, respectively [161].
Architecturally, nanostructural engineering also plays a significant role in promoting the OWS performance of TMPs. Unique nanostructures with large specific surface areas and plentiful pores can increase the number of active sites and the electrolyte–electrode interfacial contact area, and facilitate mass transport, thus expediting reaction kinetics. For instance, Lou et al synthesized Ni–Co–P hollow nanobricks (HNBs) using Ag2WO4 cuboids as reaction templates. Benefiting from a hollow structure, the Ni–Co–P HNBs exhibited excellent OWS activity compared to their Ni–Co–P nanosheet counterparts [162]. On the other hand, the direct growth of TMPs on conductive substrates (e.g. carbon cloth/paper, Ni/Cu foam, Ti mesh, etc) to form hierarchical structures is an effective approach for increasing electron transfer efficiency, ensuring a sufficient permeation of the electrolyte, and preventing the detachment of TMPs, hence improving electrocatalytic activity and stability. Moreover, the substrate covered by the active component can be directly used as a self-supporting and binder-free working electrode, thus avoiding the conventional post-coating of powdery electrocatalysts on a working electrode with the assistance of polymeric binder additives. For instance, Shen et al prepared self-supported MoP nanoflake arrays on a Ni foam (MoP/NF); the prepared MoP/NF working electrode required overpotentials of 265 and 114 mV for the OER and the HER, respectively, and a cell voltage of 1.62 V for OWS at 10 mA cm−2 [163].
Current and future challenges
Despite these significant advances, several major challenges remain. (a) Due to the complicated elementary steps involved in water splitting under different pH conditions, the mechanistic and fundamental workings of TMPs for OWS are still not well understood at the molecular level. (b) Since surface reconstruction such as the reduction, oxidation, and amorphization of TMPs may occur during electrolysis, it is highly desirable to develop in-situ/operando characterization technologies to probe the actual active catalytic sites and identify the interfacial structure during the electrochemical process. (c) Although theoretical calculation represents a powerful tool to predict electrocatalytic performance and disclose the possible reaction mechanisms for TMPs, more efforts should be devoted to the construction of reasonable models that are closer to real reaction systems. (d) For heteroatom doping and interfacial engineering, the 'synergistic effect' is frequently mentioned to explain the boost in catalytic activity. However, the exact synergistic effect in different systems is not yet fully understood and should be further explored. (e) Although many TMP-based catalysts show exceptional OWS performance at the laboratory level, the scalable and cost-effective synthesis of catalysts and their practical industrial application remain greatly challenging [164, 165].
Concluding remarks
In conclusion, we have briefly summarized several popular strategies for designing high-performance TMP-based bifunctional electrocatalysts for electrochemical water splitting. Moreover, the current challenges and future directions of TMP-based electrocatalysts have also been tentatively proposed. We believe that the flourishing development of TMP-based electrocatalysts for OWS will light up the future of the hydrogen economy.
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (21972068).
13. Oxides for water splitting
Kang Liu, Junwei Fu and Min Liu
School of Physics and Electronics, Central South University
Status
To cope with increasing energy consumption and climate change, the development of sustainable and clean energy is very important for future life. The conversion of renewable and residual energy to chemical energy by electrocatalytic water splitting offers a promising strategy for fuel production [77, 166–169].
In the water-splitting process, the thermodynamic potential is E0 = −1.23 V between the two half reactions of the HER and the OER (scheme 1). The reaction equation is closely related to electrolyte pH. Owing to the sluggish four proton-electron process, the OER usually has a high overpotential and this is considered to be the key factor that restricts water splitting.
Oxides such as iridium oxide (IrO2) and ruthenium dioxide (RuO2) are commonly used as the OER electrocatalysts in water splitting. However, due to their high cost, these high-efficiency OER catalysts are difficult to use in the mass production of water splitting. At present, various TMOs (including rutile, perovskite, spinel metal oxides, etc) have attracted wide attention as the alternative candidates for the OER, due to their low cost and abundance. Although the activity of metal oxides in OER has largely been improved through defect engineering, doping, and heterojunction construction, etc, they still suffer from poor activity, stability, and durability, especially in acidic conditions. Also, TMOs are often unstable in the HER, because they are easily reduced. These problems hinder the application of oxides in water splitting.
Current and future challenges
The ideal catalyst should have high activity, stability, duality, and conductivity and also low cost. However, there is still a big gap between existing and ideal catalysts. It is highly desirable to combine experiment, characterization and theory to deepen the understanding of oxides and the water-splitting reaction.
Improving the activity and stability of metal oxides for experimental water splitting. Because the surface active sites directly affect the reaction, catalytic activity can effectively be regulated by controlling the morphologies and the compositions of metal oxides. In an alkaline solution, Su et al found that exposing a Co3O4 (111) surface showed higher OER activity than the (100) surface [170]. Similarly, mesoporous Co3O4 also displayed high activity, due to numerous surface defects [170]. Regarding the composition effect, Zhang et al reported that the addition of Fe and W into CoOOH regulated Co–OH interaction and decreased the overpotential to 190 mV for the OER [171]. Other metal oxides, such as Ba0.5Sr0.5Co0.8Fe0.2O3−δ, Nix Fe3−x O4, Ni1−x Fex OOH, etc, also displayed excellent OER performances, compared to single metal oxides [127, 128]. Besides, highly conductive materials, i.e. Ni, Au, and carbon materials are often used as substrates in combination with metal oxides. The synergistic effect between the metal oxides and the substrate is an efficient way of promoting catalytic activity. For example, NiFe layer double hydroxides loaded onto graphene oxide (GO) showed a low overpotential of 195 mV for the OER [171]. Co3O4 displayed better properties on a nickel substrate than on other substrates [170].
On the other hand, the reaction equation for water splitting shows that the OER requires a larger electrode potential in an acidic solution (scheme 1). Until now, the OER catalysts used in acidic solutions have mainly been RuO2- and IrO2-based materials. The exploration of new metal oxides that can reduce the amount of noble metal required for the acidic OER is a hot topic. Many studies have reported that the OER activity of RuO2 can be enhanced by doping and mixing it with other metallic elements (such as Co, Ni, Ce, etc) [171]. For IrO2, high levels of activity are directly linked to IrO6's octahedral structure. Increasing the ratio of IrO6 octahedra is a direct way of improving performance. Impressively, owing to its self-healing process, MnOx can also be adapted to perform the OER under acidic conditions [172].
Furthermore, their poor stability and complex mechanisms limit the application of metal oxides in the HER, due to the easy reduction of oxides on the cathode. The exploration of novel metal oxides for the HER is a key factor for OWS. Li's group found that a RuIrOx nano-netcage catalyst exhibited remarkable activity and durability in OWS [173]. Besides, dynamically self-optimized (DSO) NiFe LDH nanosheets also showed a lower overpotential in the HER and the OER in an alkaline solution [174]. In short, the search for efficient, stable oxide catalysts for water splitting is still a work in progress.
In situ techniques for monitoring the evolution of catalytic structure, active sites, and intermediate species in electrocatalytic conditions. To study the origin of the activity of metal oxides in the OER, obtaining operando insight into the catalytic structure during catalysis has become a hot topic. Based on operando XAS measurements, Su et al found that Ni(OH)2 deprotonated to form NiOOH2−x (containing Ni4+ ions) under an applied potential (figure 18(a)) [175]. Similarly, using operando ambient-pressure XPS, Favaro et al revealed that Co(OH)2 transformed to CoOOH and that highly active Co4+ centers were the active sites under catalytic conditions (figure 18(b)) [176]. Thus, most people think that the OER catalytic activity of metal oxides originates in high-valency M-Oxo centers.
Scheme 1. Water-splitting half reactions in acidic and alkaline solutions.
| Acidic solution | E0 (V) | Alkaline solution | E0 (V) | |
|---|---|---|---|---|
| HER | 4 H+ + 4e− → 2 H2(g) | 0.00 | 4 H2O(l) + 4e− → 4OH− + 2 H2(g) | –0.83 |
| OER | 2 H2O(l) → O2(g) + 4H+ + 4e− | –1.23 | 4OH− → O2(g) + 2H2O(l) | –0.40 |
| Overall | 2H2O(l) → O2(g) + 2H2(g) | –1.23 | 2H2O(l) → O2(g) + 2H2(g) | –1.23 |
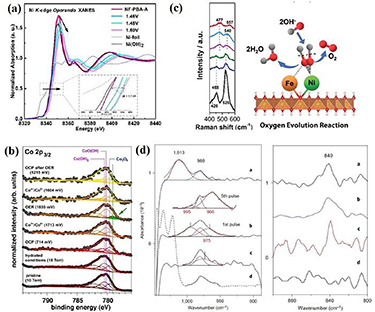
Figure 18. (a) Operando Ni K-edge XAS spectra of NF-PBA-A at different potentials. Reproduced from from [175] with permission. Copyright © 2018, American Chemical Society. (b) Operando ambient-pressure XPS evolution of Co 2p3/2 under hydrated conditions and as a function of the applied potential. Reproduced from [176] wi with permission. Copyright © 2017, American Chemical Society. (c) In situ Raman spectra collected from a large wavenumber region of NiFe LDH during the OER process in 1 M KOH at various overpotentials vs. RHE [174]. (d) Rapid-scan FTIR spectra of water oxidation catalysis. Reproduced from [177] with permission. Copyright © 2014, Springer Nature.
Download figure:
Standard image High-resolution imageMeanwhile, tracking the active sites and intermediate species is essential to explore the reaction mechanism of OER. Qiu et al revealed through in situ Raman techniques that the Ni and Fe sites of NiFe LDH interacted with OH* species differently under different potentials (figure 18(c)) [174]. Under reaction conditions, Zhang et al found that a Co3O4 surface had two intermediate states, a surface superoxide and an oxo Co(IV) site, using time-resolved Fourier-transform infrared spectroscopy (figure 18(d)) [177]. At present, the biggest challenge is building the 'structure–activity' relationships, which requires multiple in situ spectroscopic measurements to simultaneously monitor the catalyst's structure, the active sites, and the intermediate species.
Obtaining a universal descriptor of catalytic activity and a deep theoretical understanding of the OER reaction mechanism. Obtaining a universal descriptor of catalytic activity is key to understanding the nature of OER catalysts, and would facilitate the design, prediction and screening of novel oxide catalysts. The adsorption energies of intermediates have been widely used as a successful descriptor in the design of novel metal oxides. Combining experimental and theoretical metal oxide data, Nørskov's group constructed a volcano plot for metal oxides using GO–GOH as the descriptor (figure 19(a)) [77]. They found that the trend of experimental data matched the theoretical data in the volcano map. They also gave a reasonable explanation of the high activity of IrO2 in acid, by theoretically analyzing the binding energies of intermediate species [77]. Suntivich et al reported that the use of eg electrons as descriptors described the activity of perovskite samples containing TMs (figure 19(b)) [178]. However, the results of theoretical prediction often deviate greatly from reality, due to the complexity of the catalyst's surface. In recent times, the prediction of efficient catalysts and the identification of potential descriptors using artificial intelligence have become hot topics.

Figure 19. (a) OER volcano plot for metal oxides. Reproduced from [77] with permission. Copyright © 2017, American Association for the Advancement of Science. (b) The correlation between the applied potential (E) and eg electrons for each perovskite compound at a current density of 50 mA cm2. Reproduced from [178] with permission. Copyright © 2011, American Association for the Advancement of Science.
Download figure:
Standard image High-resolution imageTo understand the influence of oxide surface structures on water splitting, it is very important to study the reaction mechanism theoretically. The OER reaction intermediate species (including OH*, O*, OOH*, O2*) are closely related to the active sites, surface defects, and so on. For example, the reactant of OER is OH− in an alkaline solution (H2O in acid solution). In an alkaline solution, the typical reaction path consists of four elementary steps as follows: (a) OH− adsorbs at the active sites to form OH* species, which subsequently interact with another OH–, forming O* species at the active site and releasing H2O; (b) the O* species continue to react with another OH–, generating OOH* species at the active site; (c) the OOH* species combine with another OH–, creating O2* species at the active site and releasing H2O; (d) the O2* species desorb from the active site and are replaced by OH− at the active site. On many oxides, the OER occurs through this typical reaction pathway [172]. Also, other studies have reported that surface lattice oxygen also plays an important role in the OER [172]. The surface lattice oxygen species (hydroxide radicals) combine with adsorbed O* species, forming OOH* species and leaving surface oxygen defects. Subsequently, the OH− continues to interact with OOH* species to generate the O2 and H2O, and another OH− fills the defect site. Owing to the complex structure of oxide surfaces, clarifying the reaction mechanism is one of the current challenges.
Concluding remarks
Although the performance of oxides is improving, they are not ideal materials and they are far from practical application. Metal oxides still have great potential for electrocatalytic water splitting. At present, the catalytic activity and stability of metal oxides remain the current focuses in water splitting. To construct efficient oxide catalysts, the urgent concerns at this stage are to deepen insights into oxides and to build structure–activity relationships using advanced equipment and theoretical analysis. Moreover, the conductivity and duality of metal oxides should be investigated in the future. Also, it would be beneficial to the development of water splitting to design novel metal oxides and understand their reaction mechanisms through theoretical calculation. Furthermore, to adapt to various conditions, the exploration of multifunctional metal oxides for efficient pH-universal OWS will offer new opportunities.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21872174 and U1932148), the International S&T Cooperation Program of China (2017YFE0127800), the Hunan Provincial Science and Technology Program (No. 2017XK2026), the Project of Innovation-Driven Plan in Central South University (20180018050001), the State Key Laboratory of Powder Metallurgy, Shenzhen Science and Technology Innovation Project (JCYJ20180307151313532), the China Postdoc Innovation Talent Support Program, China Postdoctoral Science Foundation (2018M640759), the Thousand Youth Talents Plan of China and the Hundred Youth Talents Program of Hunan.
14. Sulfides for water splitting
Sang-Il Choi
Department of Chemistry and Green-Nano Materials Research Center, Kyungpook National University
Status
Since H2 gas is considered to be a zero-carbon energy source, splitting water into H2 and O2 using nanocatalyst-based systems is of great interest to the environmental and sustainable energy communities. Pt and Ir are well-known electrocatalysts used in the HER and the OER, respectively. However, due to their rarity in the earth's crust and their high costs, transition-metal sulfide (TMS)-based nanoscale catalysts have emerged as economical and earth-abundant alternatives for water splitting systems [179–181]. The main strategies for developing TMS catalysts with the performance to match and even beat noble-metal catalysts are to adjust their composition and morphology.
In terms of composition, the main purpose of selecting a TM candidate is to adjust the electronic structures of the surface TM and S sites [182]. In the acidic HER, the Gibbs free energy of H adsorption (ΔGH) at the S site is considered a descriptor, which is a significant negative value affected by many TMs. However, only a few TMSs such as FeS2, CoS, CoS2, Co9S8, Ni3S2, MoS2, and WS2, have shown an optimal ΔGH that favors H2 evolution. The story for the catalytically active sites of TMSs when used for the alkaline HER and the acidic/alkaline OERs differs from the acidic HER trend described above; specifically, the electronic structure of the TM is considered to be a major factor in the reactions. In alkaline media, TMSs form S–TM–H2O networks, where the binding of the TM and the hydrate is altered by the S atom to promote the HER. In the case of both electrolytes of the OER, the key route for improving reaction kinetics is the pre-formation of TM–OOH intermediates. Due to electronegative S, the electron-lacking TM sites of TMS start to accelerate the oxidation of TM–OH to TM–OOH, facilitating the O2 evolution kinetics.
By controlling the morphology of the TMS catalysts, more promising catalytic sites for water splitting have been unveiled (figure 20) [183]. One well-known strategy has been to prepare TMS catalysts of high porosity, sub-nanometre size, and with 1- and 2D structures, so as to have larger surface areas that are HER and OER active. It has also been shown that exposing specific crystal facets to the surface of nanocatalysts enhances the kinetics of the adsorption/desorption of reaction intermediates. In particular, facet-controlled nanostructures, enclosed by low-coordination numbers of metal and sulfur, have demonstrated that their unique sites become more active for water splitting catalysis than structures without facet control. More recently, advanced synthetic methods using surface etching and Kirkendall effects have suggested that surface defects and novel structures have been exposed as new active sites. TMSs with hollow morphologies are one of the expected products of etching and Kirkendall-effect processes. These types of material possess a larger number and density of active sites than their solid counterparts. However, although the TMS catalysts developed have outperformed the activity of Pt and Ir, their stability remains an issue for long-term operation in industrial use.
Current and future challenges
The goal of TMS research is to apply advanced catalysts in practical electrolyzers over the long term. Electrolyzers operate at current densities of >0.2 and >0.6 A cm−2 in alkaline and acidic media, respectively, at cell voltages of >1.8 V [184]. Under these severe conditions, surface-atom dissolution, morphological deformation, and particle agglomeration have been revealed to be major issues that degrade the catalytic performances of TMSs during electrolytic operations. Therefore, the development of active and stable TMS catalysts suitable to the reaction and pH conditions, as discussed below, continues to be a current and future challenge.
For the HER under acidic conditions, the dissolution of TMSs is significant, as their surfaces can be corroded by the transformation of S atoms to thiosulfates. Recent reports have introduced P-decorated TMSs as an approach for improving catalytic stability in acids [185]. By replacing S with P, the bond strengths of Co and S/P anions become stronger, which can suppress surface atom corrosion. As an example, a surface treatment of P on CoS2 maintains its current density for 20 h, while pristine CoS2 loses 70% of its activity after only 30 min. Some similar P treatment methods offered a new strategy for improving catalyst stability, but the limited elemental choices were not able to not offer extensive research opportunities for catalyst design and thus few cases are under investigation.

Figure 20. Representative schematic diagram of synthetic methodologies for morphologically controlled nanostructures [183].
Download figure:
Standard image High-resolution imageThe design of a TMS catalyst for the acidic OER is more demanding and has not succeeded in long-term operation. Under high operating potentials, the formation of TM–O, TM–OH, and TM–OOH intermediates and the dissolution of the surface S occur simultaneously before O2 evolution takes place. Thus, strong TM–O bonds lead to TM dissolution during the OER. Recently, the hierarchical structure of CoMoNiS–Ni foams has been used to demonstrate the complete coverage of Ni foams by Co9S8, MoS2, and Ni3S2, preventing corrosion and enhancing OER stability compared to other compositional options [186]. This complex composite used the synergistic effect of several TMSs to promote catalytic stability, but after 80 min of operation, the current density decreased by 20%, and therefore, this composite was still unable to meet industrial requirements. On the other hand, TMS can be used as a template to improve the catalytic stability of corrosion-resistant Ir and Ru during the acidic OER. As an example, an IrRu alloy anchored to a Cu sulfide template maintained its morphology and OER performance during 100 h of continuous operation in 0.1 M HClO4, whereas IrRu alloy alone was deactivated after 10 h [187].
In alkaline solutions, the corrosion and agglomeration of TMSs are milder than in acids. However, since the catalytic performance is weaker in alkaline conditions than in acids, enhancing the activity causes stability problems or vice versa. There is, as yet, no representative result that improves both catalytic activity and stability, but some trials have been introduced to optimize both. For example, CoSx , which suffers from Co dissolution, is less stable (about 40 times) but has more active surface defects that function as catalytic sites for the HER than MoSx [188]. By interconnecting CoSx and MoSx , CoMoSx has become a new HER catalyst model, providing stabilized catalysts with highly active structures (figure 21). In the case of the OER, TMSs have been well-accepted since the surface phase was transformed to OER-active hydroxide species. For example, Ni3S2 grown on Ni foam showed recorded OER performances for more than 50 h of operational time after Ni–Fe–OH was formed on its surface [189].
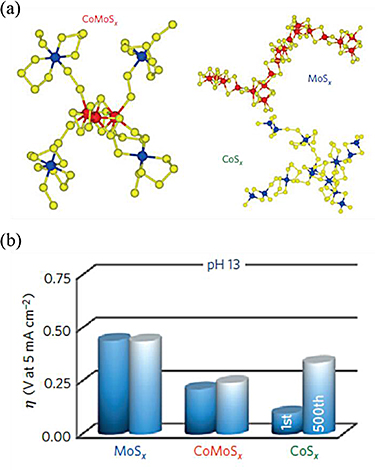
Figure 21. (a) CoMoSx chalcogel consists of Mo3S13 clusters interconnected by CoS6 octahedra that are capped by polysulphide oligomers in a compact and robust structure. Yellow, red, and blue spheres represent S, Mo, and Co atoms, respectively. (b) Initial overpotentials and after 500 potential sweeps for MoSx , CoS2, CoMoSx chalcogels.
Download figure:
Standard image High-resolution imageThere may be more issues, one of which is to understand the real catalytic sites involved during the water-splitting operation. Large elemental TMS dissolution can create unstable but vigorous new sites. However, it is highly challenging to identify these actual catalytic sites under alkaline/acid conditions. Therefore, the development of in-situ/operando measurement techniques, which allow the viewing of real-time changes in such active surface structures during reactions, is still required for future work in this and related fields [190]. Although in-situ/operando tools using Raman spectroscopy, XPS, ICP-MS, and XAS have recently been introduced, atomic-resolution TEM-EDS to capture elemental dissolution will be highly challenging in future technologies.
Concluding remarks
As discussed in this chapter, TMSs have been investigated as promising non-precious-metal catalysts for water splitting. However, the design of TMSs to improve long-term catalytic stability in acidic and alkaline media has been limited by many problems, such as the corrosion, deformation, and agglomeration of the catalysts. Therefore, great attention should be paid to the future design and exploration of advanced catalysts to inhibit the degradation of catalyst stability while improving activity under the surface elemental and structural transformations occurring during reactions.
Acknowledgment
This work was supported by the National Research Foundation of Korea (NRF-2018R1C1B6004272).
15. Layered double hydroxides for water splitting
Junfeng Xie
College of Chemistry, Chemical Engineering and Materials Science, Shandong Normal University
Status
Electrochemical water splitting has been considered as one of the most promising pathways to produce clean hydrogen energy economically and efficiently. Aimed at reducing the required overpotential of the half reactions, namely, the HER and the OER, tremendous efforts have been devoted to exploring highly efficient electrocatalysts for improved water splitting. Among a large variety of Earth-abundant water-splitting electrocatalysts, TM-based LDHs are highly attractive candidates for the alkaline OER and have received substantial research interest. Typically, an LDH possesses a layered structure with a mixed M2+ and M3+/4+ hydroxide matrix and intercalated anions between layers, which is easy to form into a two-dimensional (2D) nanosheet morphology with a large surface area [191]. By simply adjusting the composition of the metal ions, the synergistic effect between metals can easily be modulated for potential catalysts. Numerous works focused on this method have been reported that have pursued improved OER activity by adjusting the composition of LDHs, e.g. by tuning the metal ratios or adopting elemental doping in the LDH lattice, while structure-oriented enrichment of the catalytically active (or pre-oxidatively active) sites via structural design (i.e. construction of nanopores, amorphous structures, or hierarchical structures) could lead to further optimization of the OER activity. With the aid of those strategies, LDH-based materials could be promising candidates for use as electrocatalysts for electrochemical water splitting.
Current and future challenges
To optimize OER activity, enlarging the surface area via simple liquid exfoliation is the most convenient method, which endows the exfoliated LDH nanosheets with highly exposed surface sites for potential ion/gas interactions. Hu's group reported that liquid-exfoliated NiFe and NiCo LDH nanosheets outperformed a commercial RuO2 catalyst in both OER activity and stability [192]. By further optimization of the composition to form ternary LDH nanosheets, the OER activity can show remarkable enhancement compared to unary hydroxide and binary LDH. For example, Bao et al proposed a series of ternary LDH-based catalysts (Co–Mo–V and Fe–Ni–V LDHs), which exhibited high activity in the OER and even for OWS [193, 194]. The synergy between ternary metal sites introduced an optimized electronic structure, which was responsible for the facilitated OER activity. Similar regulation can also be confirmed in LDH-structured α-Ni(OH)2 hierarchical nanosheet arrays with controllable Cu, Ce and Fe incorporation, in which the electronic structure can effectively be optimized, leading to the facile accumulation of high-valence Ni species that are highly active for electro-oxidation reactions. Also, the unique wire-on-sheet morphology of the hierarchical nanosheet arrays offers more active sites for the pre-oxidation reaction, therefore realizing efficient performance for water splitting with the synergy of the optimized electronic structure [195–198]. It is worth noting that OER activity usually exhibits a strong relationship to the contents of metals in LDHs, which can be regarded as a result of the modulation of the electronic structures to meet an optimal *OH adsorption energy, and subsequently, optimized reaction kinetics. By simply adjusting the concentrations of dopants or modulating the contents of multi-metals, the OER activitiy shows obvious variation between the above materials. For example, 1% is optimal for Fe-doped Ni(OH)2 nanoarrays, while lower or higher concentrations can lead to reduced OER activity [196]. Hence, apart from compositional concerns, the ratio/concentration of each component also needs to be considered.
Besides, it is worth noting that although LDH nanosheets possess a large surface area, the basal planes are mainly terminated by hydroxyl groups which are relatively inert in participating in the pre-oxidation reaction to form catalytically active high-valence metal species. Based on this recognition, our group proposed a facile etching intralayered Ostwald ripening approach to fabricate highly porous NiFe LDH nanomesh by removing Zn ions from ternary ZnNiFe LDH nanosheets [199]. The nanopores in the LDH nanomesh catalyst were able to provide abundant edge sites for the pre-oxidation reaction, and the single-crystalline feature further ensured facile charge transport behavior along the 2D nanomesh, thereby leading to remarkably enhanced OER activity, compared to nonporous nanosheets with the same composition. More importantly, by conducting post-catalytic investigations, enrichment of the catalytically active species around the nanopores was confirmed, which proved that the edges were more active in undergoing the pre-oxidation reaction that produced the catalytically active species (figure 22). Besides, the presence of abundant nanopores in the 2D nanomesh catalyst was able to provide sufficient buffering space for potential volume changes during the electrochemical processes, thus guaranteeing the excellent operational stability of the NiFe LDH nanomesh catalyst.
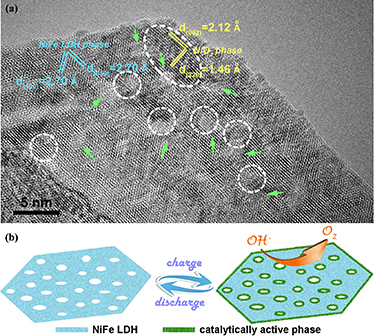
Figure 22. (a) Post-catalytic HRTEM image shows the emergence of an α-NiOOH phase around the nanopores. (b) Schematic to illustrate the generation of high-valence species around the nanopores [199]. Copyright: Elsevier 2018.
Download figure:
Standard image High-resolution imageBesides, to promote the pre-oxidation reaction in the LDH-based OER electrocatalysts, the construction of a partially amorphous structure is also highly effective. By simply adjusting the reaction temperature, a NiFe LDH nanosheet array catalyst with edge amorphization can be obtained, which exhibits a considerable concentration of local Ni3+ ions that are active for OER. Compared with highly crystalline and fully amorphous counterparts, the partially amorphous NiFe LDH nanoarray catalyst displays significantly enhanced OER and urea oxidation performance (figure 23) [200]. That is, the edge amorphous structure can provide more under-coordinated metal sites, which not only stabilize the high-valence metal ions but also promote Ni2+–to–Ni3+ conversion, thereby leading to an improvement in electro-oxidation reactions with the synergistic effect of the binary composition. Alternatively, a partially amorphous structure can also be achieved by controllable edge sulfurization. Using hydrothermal sulfurization, an edge sulfurized CuNiCo LDH catalyst can be obtained, which exhibits enhanced electro-oxidation performance for the OER and hydrazine oxidation [201]. It is worth noting that although the fully amorphous structure appears to be rich in the coordinately unsaturated metal ions, poor in-plane conductivity becomes the limiting factor in further realizing the electric connection of such metal sites, thereby exhibiting poor activity for OER catalysis.
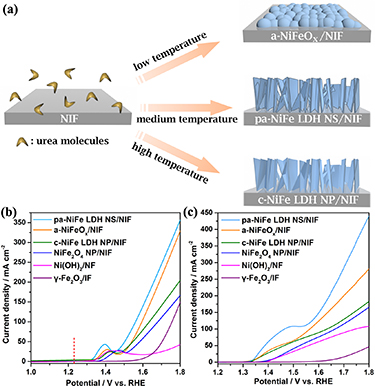
Figure 23. (a) Modulating the crystallinity by varying the reaction temperature. (b), (c) OER and urea oxidation performance [200]. Copyright: RSC, 2018.
Download figure:
Standard image High-resolution imageConcluding remarks
TM-based LDHs have been investigated as efficient water-splitting catalysts during the past decade. To improve the OER performance of LDH-based catalysts, boosting the generation of high-valence species during the pre-oxidation reaction should be given special attention, and optimizing the electronic structure is another effective strategy for regulating electrocatalytic behavior. Through the construction of a highly porous morphology or partially amorphous structure, the pre-oxidation reaction can effectively be boosted, thus resulting in accumulated high-valence species for enhanced OER activity. For future optimization of LDH-based electrocatalysts, a synergistic strategy based on the abovementioned methods would be highly promising in pursuing improved water splitting performance.
Acknowledgments
This work was supported by the Natural Science Foundation of Shandong Province (ZR2018JL009) and the Key Research and Development Program of Shandong Province (2019GGX103051).
16. Carbon-based electrocatalysts for water splitting
Xinwen Peng and Tingzhen Li
State Key Laboratory of Pulp and Paper Engineering, South China University of Technology
Status
Hydrogen, due to its numerous advantages such as high energy density (3400 Kcal Kg−1), high utilization rate, wide application, cleanliness, and zero-carbon emissions has been considered an ideal energy carrier to replace fossil fuels [150, 202]. Among various hydrogen evolution technologies, electrocatalytic water splitting has attracted great attention due to its environmental friendliness, high efficiency, and high hydrogen purity. State-of-the-art electrocatalytic water splitting technology is highly dependent on the efficiency of precious metal catalysts, which dominate the reaction rate of the HER and the OER [109, 203, 204]. Typical Pt-based and Ru/Ir-based catalysts exhibit benchmark HER and OER activity, and can effectively reduce the overpotential of the OER and the HER at the electrodes, thus realizing high-current hydrogen production at low cell voltages [205]. However, the limited stability, scarce reserves and high cost of these noble-metal-based catalysts severely impede their commercialization in water-splitting electrolyzers [206]. Therefore, great effort has been devoted to developing non-precious metal electrocatalysts with high activity and low cost. Among various alternative electrocatalysts, carbon-based electrocatalysts with good electrical conductivity, low price, acid-alkali corrosion resistance and high stability have been attracting great attention. Carbon-based electrocatalysts for water splitting can be traced back to the initial period of the United States space program. Up until now, they have also received increasing research efforts aimed at large-scale commercialization [203]. On one hand, carbon-based electrocatalysts can be obtained from various precursors, and the porous structure, large specific surface area, and ion transport channels of the prepared catalysts can significantly improve their electrochemical performance. On the other hand, the electronic structure of carbon-based electrocatalysts can easily be regulated by TM atom modification or heteroatom doping to produce more catalytically active sites or enhance the inherent activity of the active sites, thus significantly improving the efficiency of water splitting [206]. Therefore, carbon-based electrocatalysts are considered to be promising alternatives to noble-metal-based catalysts for water splitting. Although they have excellent physicochemical properties and economic potential, the large-scale commercialization of carbon-based electrocatalysts for water splitting is still a challenge due to the agglomeration/dissolution of TM atoms in strongly acidic/alkaline electrolytes and their easy oxidation in air [207]. Therefore, the development of carbon-based electrocatalysts for water splitting is quite significant and full of opportunities and challenges.
Current and future challenges
Although great efforts have been made in recent years to develop carbon-based electrocatalysts for water splitting, their large-scale application still suffers from numerous challenges. On one hand, hydrogen production from water splitting using carbon-based electrocatalysts comes with the cost of high mass loading or a high surface area, since the active site density of carbon-based electrocatalysts is relatively low [203]. In the past decade, carbon-based electrocatalysts have been proven to exhibit high levels of HER or OER catalytic activity that are very close to those of precious metals, because of the surface phenomena obtained by increasing the mass loading of catalysts or increasing the surface area to compensate for the low intrinsic catalytic activity [203]. However, increasing the mass loading of catalysts will make its cost equivalent to that of precious-metal catalysts [208]. On the other hand, although carbon-based electrocatalysts for water splitting can meet various harsh requirements, most of the carbon-based bifunctional catalysts usually only show good activity for one half reaction at the expense of the activity in the other half-reaction, leading to a moderate performance for OWS. At the same time, if different catalyst materials are used in the two electrodes, this may result in inconvenience in the preparation of catalysts and the assembly of the electrolyzer [209]. Furthermore, the mechanisms of bifunctional active sites are not clear and should be given more attention to guide the rational design of high-performance carbon-based bifunctional electrocatalysts [210]. Last but not least, considering the catalytic activity of the carbon materials themselves (poor or no performance), the development of practical and viable carbon-based electrocatalysts for water splitting is still a challenge [211].
To address the above issues, some promising approaches have been proposed and are under investigation (figure 24). Carbon-based single-atom metal catalysts aimed at improving the active site density of carbon-based electrocatalysts have received more and more attention. Using atomically dispersed metal for catalytic active sites has the huge advantages of maximum atom-utilization efficiency and atomic economy, which are expected to address the low density of inherent active sites in carbon-based catalysts [212]. Besides, integrating active OER and HER catalysts as heterostructures will provide an avenue towards a robust OWS catalyst, which can compensate for the inconvenience of fabricating different catalyst materials for the two electrodes [209]. The interface formed between the two types of active materials can form more active centers than a single component, because of strong chemical bonding, electronic interaction, or synergism, and the presence of both HER and OER active components in heterostructures will render good levels of activity when used as both anode and cathode catalysts [213]. Furthermore, carbon-based electrocatalysts with specific electron-donor characteristics are the ideal low-cost alternatives to noble metals, so it is necessary to produce active sites and improve catalytic performance by the surface chemical modification of carbon-based catalytic materials, using, for example, surface functionalization, recombination, doping, defect engineering, and other activation with active species [207]. For instance, heteroatom (e.g. N, P, S, and so on) doping can break the electrical neutrality of C atoms, adjust the electronic structure of carbon, and produce positively charged active sites, thus enhancing electrocatalytic activity [210]. Additionally, the protection of the carbon matrix can protect active metal-compound NPs from dissolution and aggregation, which is helpful for improving the stability and acid/alkali corrosion resistance of carbon-based catalysts.
Figure 24. Carbon-based electrocatalysts for water splitting and their design strategies.
Download figure:
Standard image High-resolution imageConcluding remarks
The various advantages, such as high electrical conductivity, structure and shape tunability, acid/alkali corrosion resistance and cost merits of carbon-based electrocatalysts make them highly attractive for water splitting. Although remarkable progress has been made in recent years, there are still unresolved challenges to their commercialization, such as their inherent weak catalytic activity and complex processes of synthesis and regulation of active sites. To further improve the performance of carbon-based electrocatalysts for water splitting and realize their widespread application, more efforts focused on the following aspects are required: (a) The development of a highly efficient synthesis strategy and a facile preparation process; (b) the synthesis of electrocatalysts with bifunctional and enhanced electrochemical performance by recombination, doping, defect engineering, and structural adjustment; (c) further investigation and a deep understanding of the mechanisms of the catalytic process. In conclusion, carbon-based electrocatalysts for water splitting are extremely important for clean energy, and more efforts should be made to improve their electrochemical performance and realize their commercial application.
Acknowledgments
X W Peng thanks for the National Natural Science Foundation of China (31971614), Guangdong Natural Science Funds for Distinguished Young Scholar (2016A030306027), State Key Laboratory of Pulp and Paper Engineering and Fundamental Research Funds for the Central Universities.
17. Ru-based electrocatalysts for water splitting
Gaoxin Lin and Jiacheng Wang
Shanghai Institute of Ceramics, Chinese Academy of Sciences
Status
Electrochemical water splitting is a promising way to produce high purity H2 and O2. To be competitive in practical applications, it is necessary to develop catalysts that are efficient, robust, inexpensive and green, which can improve the sluggish kinetics of the two half reactions (the HER and the OER) of electrochemical water splitting [214–216]. The state-of-the-art catalysts for HER and OER are platinum (Pt) and iridium (Ir) based materials, respectively. However, their high cost and low stability hinder their large-scale application.
Compared with Pt and Ir, relatively cost-effective ruthenium (Ru) has been widely used in selective hydrogenation, CO oxidation, and water splitting [217, 218]. In 2016, Bhowmik et al reported a 1D–RuO2–CNX composite material for both the HER and the OER in electrolytes of all pHs. It exhibited low overpotentials of 93 (for the HER) and 250 mV (for the OER) at 10 mA cm−2 [219]. Later, Ru metal was discovered to have excellent HER activity; a free-standing 2D combination of Ru//RuO2 used for water splitting possessed an overpotential of about 330 mV [166]. It was proved that the strength of the Ru–H bond (∼65 kcal mol−1) was similar to that of Pt–H, which resulted in the fast kinetics of Ru metal in the HER [220].
The further development of Ru-based catalysts for water splitting mainly aims to increase the intrinsic activity and maximize the exposure of active sites of the catalysts to achieve high efficiency. Normally, the main strategies include: (a) the design of porous or nanosized structures; (b) the modulation of the electronic structure. Both of these strategies can also be optimized simultaneously. Ru-based catalysts, including a three-dimensional hierarchical RuNi heterostructure [221], Ru/nitrogen-doped carbon [222], and Co-doped RuO2 nanowires [223] have exhibited promising prospects. The activity of the above catalysts was enhanced by modifying the electronic structure of Ru to reduce the water adsorption energy and the water cleavage barrier. However, the performance still needs to be improved, especially at a large current density, to meet the requirements of practical applications.
Current and future challenges
Current challenges are related to optimizing the activity and stability of catalysts in acid electrolytes. As highlighted above, the intrinsic activity of the catalyst was enhanced by modifying its electronic structure through alloying with other metals or by being loaded on a polar substrate. However, the effects of the above methods were more limited in acid conditions. For instance, the channel structure of RuCu nanosheets was reported to alleviate the t2g−eg splitting of Ru and thus minimized the surface Coulomb repulsion of the catalyst, which led to improved water-splitting efficiency [218]. The catalyst was stable for more than 30 h at a current density of 10 mA cm−2 in an alkaline electrolyte, outperforming most of the reported catalysts (figure 25(a)). However, the period of stability was halved in an acid electrolyte (figure 25(b)). According to reports, the reduced stability of the catalyst in an acidic electrolyte was due to the electrochemical surface area loss, which resulted from the particle aggregation in the HER and lattice oxygen evolution in the OER. Recently, an RuIrOx nano-cage was reported for high activity and durability in water electrolysis [173]. The introduced Ir was able to simultaneously optimize the rate-determining steps in the HER and the OER, and stabilize Ru species at a low oxidation state during the cycles. During the HER, the RuIrOx nano-cage was reduced to an Ru2Ir alloy and exhibit enhanced activity. Thus, it demonstrated stability in acid for more than 24 h and worked irrespectively of the positive/negative terminals of the battery (figure 25(c)).
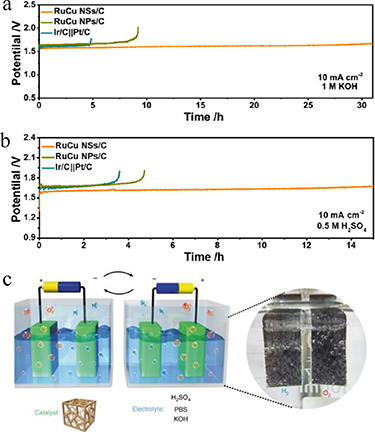
Figure 25. (a) Water-splitting chronoamperometry test of the optimized RuCu NSs/C, RuCu NPs/C and Ir/C||Pt/C at 10 mA cm−2 in 1 M KOH. (b) Water-splitting chronoamperometry test of the optimized RuCu NSs/C, RuCu NPs/C and Ir/C||Pt/C at 10 mA cm−2 in 0.5 M H2SO4. Copyright 2019 by Wiley. (c) Schematic illustration of the water splitting setup using an AA battery, showing that the RuIrOx nano-net cages can work irrespective of the positive/negative terminals of the battery; and a digital photograph of the evolution of H2 and O2 from the electrodes during electrolysis. Copyright 2019 by Springer Nature.
Download figure:
Standard image High-resolution imageMoving forward, we can conceive that a combination of a hollow nanostructure and control of the electronic structure may promote high activity and stability. A polysynthetic theoretical prediction of the rational electronic structure may accelerate the catalytic design cycle.
Concluding remarks
Recently, the continuing interest in the Ru-based electrocatalysts for water splitting has been fuelled due to their intrinsic activity and stability. Further development is intended to design efficient, robust, inexpensive and green catalysts for industrial applications. However, the current main work is still to solve the problems of activity and stability under acidic conditions. Understanding the real reactivity mechanism and the quantitative relationship between performance and electronic structure may guide the design of novel Ru-based catalysts for large-scale applications.
Acknowledgments
JW is grateful for financial support from National Natural Science Foundation of China (52072389) and the Program of Shanghai Academic Research Leader (20XD1424300).
18. Metal oxides for CO2 reduction
Jingrui Han and Hongyan Liang
School of Materials Science and Engineering, Tianjin University
Status
The greenhouse effect caused by excessive CO2 emission from the depletion of fossil fuels has attracted global concern. Since the Industrial Revolution, the concentration of CO2 in the atmosphere has increased from 278 ppm to more than 400 ppm. The global annual CO2 emission is 39.5 Gt, resulting from the consumption of 12 billion tons of oil, and it will increase to 75 Gt when the energy demand doubles by 2050. Great efforts have been expended to explore technologies for CO2 capture and utilization. Among them, the electrochemical CO2 reduction reaction (CO2RR) is considered to one of the most promising and environmentally friendly. At the same time, the CO2RR provides a potential energy transformation route to store intermittently generated renewable power, such as that produced by wind turbines and solar cells (figure 26) [224, 225]. However, during the CO2RR process, multiple electron/proton transfer steps result in a large reaction energy barrier (large overpotentials), and the similar thermodynamic potential differences between various products lead to a broad product distribution (low Faradaic efficiency (FE) of the targeted products) [224, 226]. Moreover, the competing HER is more kinetically favorable and makes H2 as its major byproduct [224, 227]. Therefore, it is imperative but challenging to explore electrocatalysts for efficient CO2RRs to decrease the active energy barrier, favor the selectivity of targeted products, and improve the resistance to deactivation.

Figure 26. The process of the artificial carbon cycle.
Download figure:
Standard image High-resolution imageAs earth-abundant catalysts, metal oxides play a critical role in the CO2RR, due to their low cost, excellent activity, and notable stability (figure 27). The d-orbital electrons of metal oxides can overlap the p-orbital electrons of the CO2 molecule to form *CO2 δ −, which can lower the energy barrier for CO2 activation. O2− anions terminated on the surface of a metal oxide have a larger size than that of metallic Mn+ cations, which can lower the coordination of smaller Mn+ cations with the bulk, leading to an abundance of undercoordinated metallic atoms as active catalytic sites [224, 227, 228]. Moreover, metal oxides contain a high density of defects (kinks, steps, terraces) and vacancy sites (oxygen vacancies, metallic vacancies), which can enhance catalytic activity. When an unsaturated surface oxygen reacts with water, the hydroxyl groups formed can favor the adsorption of CO2 molecules.
Figure 27. Overview of metal oxide electrocatalysts for CO2RR.
Download figure:
Standard image High-resolution imageSome research efforts have focussed on improving the performance of metal oxide catalysts by optimizing the sizes, introducing crystal facets, controlling oxidation, enhancing conductivity, and so on. NP catalysts have been demonstrated with enhanced activities compared to the bulk material, due to their high surface to volume ratios and their abundance of exposed active sites. New procedures have been developed to prepare and activate metal oxide catalysts at nanoscale sizes. To address their agglomeration issues, strategies such as surface modification, in situ electrochemical activation, and synergistic activation have been studied [227]. In some cases, supports have been used to stabilize NP catalysts with high dispersion, which also introduced further advantages such as increased charge transfer and synergistic effects. Recently, great efforts have been made to solve the poor electronic conductivity. The fabrication of one-dimensional (1D) nanowires instead of nanospheres has been proven to enhance the electron transportation in an SnO2 nanofiber, shorten ionic diffusion length and enlarge electrolyte–electrode contact, resulting in high current density [224]. Moreover, the preparation of a metal/metal oxide core/shell structure is another remedy for the conductivity issue. A Sn–Cu alloy/Sn core was used to simultaneously improve electrical conductivity and mass transformation, while an in situ reconstructed amorphous SnOx shell was responsible for catalyzing the CO2RR, leading to enhanced CO2RR activity [229]. Also, some studies have been devoted to the design of hybrid catalysts or the construction of an effective interface, which were able to enhance charge transfer and boost CO2RR performance. For example, a Zn2SnO4/SnO2 heterostructure on a porous microcube was able to construct uncoordinated sites, which simultaneously favored CO2 adsorption, promoted interfacial charge transfer, and optimized the reaction kinetic barriers; the ensemble effect benefited the electrochemical CO2RR performance [230]. Fractal β–Bi2O3 (f–Bi2O3) with roughened edges was uniformly deposited on porous fibrous substrates, which allowed facile control of the crystal size, increased the electron density of Bi2O3, and achieved an efficient CO2RR [231]. Moreover, various approaches have been proposed for the CO2RR, but no unified solution is available since the reaction pathway is complicated and still under investigation.
Current and future challenges
Looking back across the last decade, one can state that material and theoretical breakthroughs have driven the development of the CO2RR and that the improvement of electrocatalytic activity is highly dependent on chemical composition, morphology and crystal structure. The choice of metallic elements for catalysts is determined by the factors governing the activity and product selectivity for the CO2RR. Experimental investigations have revealed that Cu2O can facilitate CO2 activation and enhance the selectivity towards ethene (C2H4), ethanol (C2H5OH), or C2+ products because the under-coordinated Cu atoms or residual oxygen can promote C–C coupling [225, 227]. A SnOx catalyst has been confirmed to stabilize the CO2 •− intermediate and enhance the generation of formic acid (HCOOH), methanol (CH3OH) or carbon monoxide (CO) [224, 232]. Other metal oxides, such as ZnO, Co3O4 and TiO2 only show high selectivity towards the gas product CO [233]. Surface oxygen vacancies (Ov) are another key factor for the performance of metal oxide catalysts. Introducing Ov can form an electron-rich surface, which further increases the charge density around the valence band maximum, promoting CO2 adsorption and facilitating CO2 activation [233, 234]. Ov can be reduced to hydroxyl species on the surface of metal oxide through a protonation process, which plays a vital role in both the CO2RR and the HER. By controlling the coverage of hydroxyl groups on the surface, competition between the CO2RR and the HER can be modified, and the optimized OH coverage can suppress the HER and boost the generation of carbon products [230]. Structural features such as grain boundaries and facets can further enhance the activity of the CO2RR. A high density of grain boundaries in SnO2 can break local spatial symmetry and tune the binding energy of the reaction intermediates to accelerate the transformation of CO2 [224, 229]. Different crystal facets of Cu2O NPs have different catalytic activities; after comparing three facets, the mixture of [235] and [38] facets showed the highest selectivity for C2H4 [236]. Other factors, such as oxidized states, local pH gradient, local field effects, and ensemble effects have also been demonstrated to affect the activity of metal oxide catalysts [227].
Although different strategies have been used to boost the catalytic activity of metal oxides, there are some issues hindering their performance. Firstly, the highly active sites and grain boundaries are related to the small size of NPs, but agglomeration easily happens spontaneously during the operational process, leading to an enlargement of particle size and the disappearance of high-index facets, resulting in degraded performance [227]. Secondly, the poor conductivity of metal oxides impedes the efficiency of charge transformation, and limited current density leads to low productivity which cannot fulfill the industrial requirement for large-scale production. Thirdly, metallic Mn+ cations are reduced to M0 under negative potentials; the change of oxidation state is generally accompanied by morphological deformation and crystal structural transformation as well as the loss of surface oxygen; eventually, the advantages of metal oxides are weakened [227].
As well as the catalytic challenges, there are several obstacles to the commercial application of the CO2RR. For example, the dissolution of CO2 in solution is low, and the low concentration of CO2 in electrolytes hinders mass transport, limiting current densities and the reaction rate. An understanding of the electrocatalytic reaction pathway at the solid–liquid–gaseous interfaces is highly desirable but still under debate. Carbon dimerization only happens with copper, and no other element has been known to catalyze the conversion of CO2 to multi-carbon products; thus, the exploration of elements is still ongoing.
Concluding remarks
There is a need for catalysts that reduce CO2 at low overpotentials, efficiently and continuously producing desirable products without the formation of unwanted byproducts. The combination of advanced synthesis methods, surface, bulk and in situ characterization techniques, DFT modeling, and advances in the system engineering of reactors have enhanced the understanding and boosted the efficiency of the CO2RR through the improvement of catalysts' activity, selectivity, and stability. The acceleration of material discoveries should continue in the future with contributions from a variety of scientific and industrial fields.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (NSFC) (No. 51771132), Recruitment Program of Global Youth Experts of China.
19. Metal sulfides for CO2 reduction
Shuyu Li and Xiaotao Zhang
Tianjin Key Laboratory of Molecular Optoelectronic Science, Department of Chemistry, School of Science, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin)
Status
The overconcentration of CO2 in the atmosphere due to the excessive consumption of fossil fuels has contributed to serious consequences such as global warming, ocean acidification, and so on [237]. The electrochemical CO2RR stands out among the methods developed to address this crisis and can be powered by renewable energies (solar, wind, and tide, etc) to reduce CO2 into value-added chemicals and fuels under mild conditions [238–240]. However, it should be noted that CO2 is thermodynamically stable and chemically inert due to the high bonding energy (750 kJ mol−1) of the C=O double bond, therefore the reduction of CO2 is an energy-demanding and kinetically sluggish process. At the same time, the similar thermodynamic potential differences of the various products and competition from the HER lead to challenges in the high FE of the targeted product. That is to say, highly selective, energy efficient, and readily available electrocatalysts are urgently wanted.
Metal sulfides (MSs) have emerged as an economical, earth-abundant and special category of CO2RR electrocatalysts because the specific biological CO dehydrogenases contain a cluster of inorganic MSs as active sites for reducing CO2 to CO and HCOO−. Among these, two-dimensional MoS2 has received attention, due to its considerable activity and low cost [241]. However, the practical applications of MoS2 in the electrochemical CO2RR have been hampered by its high catalytic activity in the HER and poor conductivity, which means that an MoS2 electrocatalyst for the CO2RR with a high FE and low overpotential is a big challenge. To suppress the HER and improve its performance in the CO2RR, alloying and metal doping of MoS2 were developed. For instance, Abbasi et al synthesized niobium (Nb)-doped MoS2(VA–Mo0.95 Nb0.05 S2) catalysts that reduced CO2 to CO with an FE of up to 82% and a low onset overpotential of 90 mV [242]. Inspired by N-doped carbon materials as promising electrocatalysts for CO2RR, Lv et al first proved that doping elemental N into MoS2 was able to improve its CO2RR performance through theoretical and experimental studies [243]. They fabricated a combination of N-doped MoS2 nanosheets and N-doped carbon dots (N–MoS2@NCDs) by a 'one-pot' solvothermal reaction, which was able to selectively catalyze the conversion of CO2 into CO. In particular N–MoS2 @NCDs-180 with a high N doping content of 8.35 at% displayed a high FE of up to 90.2% and a low onset overpotential (130 mV). They attributed the outstanding performance to the fact that NCDs on the surface of N–MoS2 were not only able to enhance the material's conductivity, but also lower the onset overpotential. Moreover, DFT indicated that N doping of MoS2 was able to decrease the formation energy of the COOH* intermediate and weaken the adsorption strength of CO* due to the Mo atom on N–MoS2, which facilitated the electrical reduction of CO2 to CO. According to the DFT calculation by Li et al, the edge sites of MoS2 can facilitate the formation of the key intermediate *COOH in the process of the conversion from CO2 to CO, which means that exposing as many MoS2 edges as possible is an important design principle for electrocatalysts [244]. Besides, they found that electron transfer from electron-rich N-doped carbon (NC) to MoS2 edges during the CO2RR further decreased the Gibbs free energy of the intermediate *COOH. Following these two design principles, Li et al synthesized a hierarchical hollow electrocatalyst comprising edge-exposed 2H MoS2 hybridized with NC (NCMSH) for the CO2RR. The NCMSH displayed remarkably high CO production with an FE of 92.68% and negligible degradation over 24 h at an overpotential of 590 mV. Sn-based sulfides are promising for large-scale utilization due to their low cost, environmental friendliness, and high HCOOH selectivity. Chen et al enhanced SnS nanosheet catalytic activity for CO2 electroreduction by engineering its electronic structure to accelerate the mass transfer and charge transfer rate [245]. They modified hollow nanotubes composed of stannous sulfide (SnS) nanosheets with amino-functionalized carbon layers (SnS/Aminated-C) for the electrocatalytic reduction of CO2 to HCOOH with a high FE of HCOOH of 92.6% and a remarkable current density of 41.1 mA cm−2 at −0.9 V versus RHE as well as good stability for up to 15 h. The hollow structure increased the electrochemically active area of the catalyst and provided abundant active sites. Besides, DFT suggested that the abundant charge of the Aminated-C was transferred to, and accumulated on the Sn atoms' surfaces, which was conducive to the increase of the charge transfer rate at the interface between the SnS and the Aminated-C. At the same time, it was found that the key intermediates, OCHO* and CO2*, adsorbed strongly on the surface of SnS/Aminated-C, which facilitated HCOOH formation.
Current and future challenges
Although considerable progress has been made in the synthesis of MSs, research into applying these materials with different morphologies to the electrocatalytic reduction of CO2 has been relatively limited, compared to the widely reported photocatalysts. As with other electrocatalysts, great challenges in achieving industrialization remain. Therefore, it is of interest to suppress the activity for the HER and enhance the catalytic performance for the CO2RR based on MS electrocatalysts.
Concluding remarks
MSs have been investigated as promising electrocatalysts for CO2RR. In this chapter, we discussed some principles of the design of high-performance MSs, including engineering their electronic structure through doping, hybridization and so on. Also, researchers have made efforts to adjust the morphologies of the catalyst to expose more active sites. Even so, great attention should be paid to the future design and exploration of advanced electrocatalysts to inhibit the HER while improving activity during the CO2RR.
20. Metals for CO2 reduction
Yating Zhu and Zhicheng Zhang
Tianjin Key Laboratory of Molecular Optoelectronic Science, Department of Chemistry, School of Science, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin)
Status
With the large amounts of fossil fuels used in human activities, CO2 gas emissions are constantly increasing, leading to global warming and even threatening human or animal survival. Electrocatalysts for CO2 reduction can effectively convert CO2 into other carbon-containing compounds, thereby decreasing the CO2 amounts in the air. Metals have a huge promise for application in the electrochemical CO2RR due to their excellent catalytic performance; thus, they have attracted extensive interest. The reaction pathways are diverse, depending on the employed electrode metal, modifying the final CO2RR product. According to the different relative binding strengths of CO2RR intermediates such as *COOH, *OCOH and *CO, electrocatalysts based on metals can generally be divided into three categories [246–249]. The three categories of metallic electrocatalysts are shown in figure 28. The first kind is late TMs (e.g. Au, Ag, Zn, Pd), which tend to thermodynamically adsorb *COOH to produce carbon monoxide (CO), possessing excellent selectivity. The second is mostly main-group metals (e.g. Sn, Pb, Hg, In, Bi), which tend to thermodynamically adsorb *OCOH to produce formate (HCOO−). The third kind is Cu, which can stabilize both *CO and *COOH and can optimally bind with *CO as compared with other metals, thereby converting CO2 into various products [250, 251].
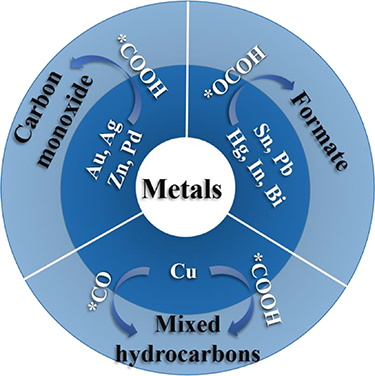
Figure 28. CO2RR products generated on various metal electrocatalysts [246].
Download figure:
Standard image High-resolution imageNotably, metal-based tandem catalysts have been investigated since 2016 for the electrochemical CO2RR [252, 253]. The tandem mechanism, CO spillover and the effects of metal active sites have been explored in the literature. Up until now, tandem catalysts have mainly been bimetallic nanostructures containing copper. This has provided some new thoughts regarding the enhancement of selectivity for the desired products.
The question of how to improve electrocatalytic performance has been investigated in recent years. As compared to other nanostructures, alloy catalysts can not only efficiently reduce the kinetic overpotential, but also adjust the selectivity towards the desired products. This is because the electronic structure, geometric strain, and chemical bifunctionality all change, thereby modifying the chemical binding strength of the intermediates on the alloy's surface [254]. Besides, SACs can also be synthesized to serve as CO2RR electrocatalysts with good electrocatalytic performance. Single noble-metal atoms can be effectively utilized in the electrochemical CO2RR.
Current and future challenges
Based on previous studies of metallic electrocatalysts for the CO2RR, the CO2 reduction reaction steps have been uncovered. Different metallic electrocatalysts have different overpotentials, current densities and Faradic efficiencies, thereby determining the activity and selectivity. However, there are several challenges to be solved in the future to realize practical application.
- (a)Noble metals, such as Ag and Au, have been extensively studied due to their outstanding catalytic activity and selectivity towards CO formation for the electrochemical CO2RR. However, their high cost impedes development and practical application. As a result, it is essential to improve noble metal utilization, reduce the consumption of noble metals or synthesize non-noble metallic electrocatalysts with high activity and selectivity to replace noble metals. Fortunately, up until now, non-noble metals have been widely investigated to efficiently convert CO2 into CO, thus replacing noble metals [255].
- (b)CO2RR selectivity for specific products is limited, as multiple proton/electron transfer steps require high overpotentials to drive the CO2RR [10]. This is more obvious when using Cu, compared to other metals. The product distribution of Cu-based catalysts largely depends on the nature of the copper surface. In 2019, a study explored the effects of different Cu facets on product distribution. According to the coordination number, the Cu facet structures were analyzed to uniquely identify the active sites (figure 29). This contributes to exploring the selective formation of desired products by identifying active sites in the future. Also, the reaction pathway of Cu catalysts is not just determined by the nature of the reaction's active sites, but by scaling relations between intermediates [256].
- (c)The CO2RR electrochemistry of metallic catalysts, such as its mechanism and intermediates needs to be more deeply explored to enhance selectivity [258]. Theoretical calculations and spectroscopic characterizations are expected to provide more evidence about catalytic mechanisms and reaction pathways.
- (d)There is always a competitive side reaction, i.e. the HER, which is kinetically favored over the CO2RR [259]. The competition result depends on the chemisorption strength of the two key intermediates during these two reactions [254]. A study demonstrated that a high coverage of CO intermediate can enhance the CO2 reduction rate and reduce the activity of the HER [259]. Further study is still required regarding the way to synthesize appropriate metallic catalysts with high activity and selectivity in the CO2RR by suppressing or reducing the activity of the HER.
- (e)Also, the effect of the metal active sites in controlling activity, selectivity, and stability in the electrochemical CO2RR still needs to be clarified. The question of whether active sites of the same metal play the same role in different catalysts needs to be explained.
- (f)Electrocatalysts with good stability need to be constantly investigated to realize the commercialization of CO2RR in the future. In general, the service life of CO2RR electrocatalysts prepared in the laboratory only reaches several tens of hours at most, which is far from the time demanded by practical applications [246].
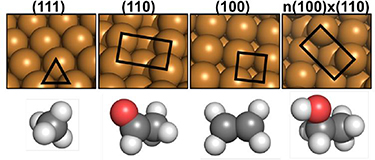
Figure 29. Identification of the active sites of the Cu catalyst. Cu(111) generates methane, Cu(110) generates acetaldehyde, Cu(100) generates ethylene, and the n(100) × (110) step generates ethanol. Reprinted with permission from [257], Copyright 2019 by the American Chemical Society.
Download figure:
Standard image High-resolution imageConcluding remarks
Although previous studies have provided much experience of the CO2RR based on metals, more effort is essential to a clear understanding of the mechanisms, pathways, active sites, intermediates and rate-determining steps. This will contribute to achieving high selectivity, activity, and stability, establishing a good basis for practical applications in the future.
21. Carbon for CO2 reduction
Ting He
Tianjin Key Laboratory of Molecular Optoelectronic Science, Department of Chemistry, School of Science, Tianjin University and Collaborative Innovation Center of Chemical Science and Engineering (Tianjin)
Status
The intensifying energy shortage and global warming issue have been two global challenges over the last few decades, which resulted from the overuse of fossil fuels due to the rapid development of human society. CO2 is the primary component of greenhouse gas derived from the combustion of fossil fuels. The increasing concentration of CO2 has captured solar energy within the atmosphere of the Earth, thus leading to an almost 4 °C temperature increase, compared to pre-industrial levels [260]. To meet the requirements of sustainable development, novel technologies such as CO2 capture, separation, and conversion are in great demand to reduce CO2 emissions [261–263].
The electrochemical CO2RR, which converts CO2 into value-added products, has attracted tremendous interest, not only for laboratory research but also for industrial application, due to its unique advantages [264]. (figure 30) Firstly, the utilization of CO2 feedstock decreases the concentration of CO2 in the atmosphere. Secondly, the production of CO2RR products, which so far are still mainly derived from fossil resources, will reduce the global demand for fossil fuels and the emission of CO2. Thirdly, the CO2RR process is controllable and moderate when operated under ambient conditions. Last but not least, the CO2RR can easily be scaled to facilitate the storage of other renewable energy sources such as solar power, wind power and so on.
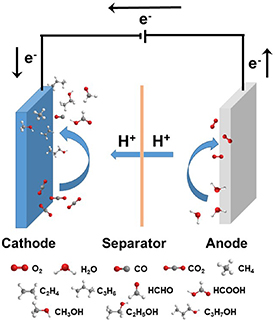
Figure 30. Illustration of the electrochemical CO2 reduction process.
Download figure:
Standard image High-resolution imageThe process of the CO2RR presents high thermodynamic and kinetic barriers due to the multiple electron transfers that take place during the reaction. Up to 16 different products result from the CO2RR, based on the number of electrons transferred. Two-electron products including carbon monoxide (CO) and formic acid (HCOOH) can be produced with high selectivity, while multi-electron products such as methanol, ethanol, methane (CH4), ethylene and so on have been synthesized with lower selectivity so far. Moreover, a competing reaction, the HER, can also proceed during the electrochemical process. Hence, it is highly desirable to develop efficient electrocatalysts to improve the kinetically sluggish process and improve the selectivity of products. There are four parameters for evaluating the performance of electrocatalysts: (a) FE, which demonstrates the selectivity of the current for a desired product; (b) current density, which reflects the rate of reaction; (c) overpotential, which represents the onset potential and the potential required to reach the maximum FE, and (d) stability.
Various CO2RR catalysts have been extensively explored during the past several decades [265]. Carbon-based electrocatalysts have attracted significant attention owing to their unique advantageous characteristics, compared to other species [266]. Firstly, the carbon matrixes of these catalysts are chemically inert, which can inhibit reconstruction and aggregation. Secondly, the electronic structures of carbon-based catalysts can easily be tuned by doping them with heteroatoms. Moreover, due to the lack of a d-band electronic structure, carbon-based materials can naturally break the scaling relations between the intermediates of CO2RR and the d-bands of metal surfaces. This leads to a reduction in the overpotential of the reaction and adjusts the selectivity of the product. Thirdly, carbon-based catalysts are cheap and can be prepared by various methods with different precursors on a large scale. Hence, carbon-based catalysts have been considered as potential alternatives to replace state-of-the-art metal catalysts when considering current density, energy efficiency, selectivity and stability.
Current and future challenges
The first example of a carbon-based catalyst for the CO2RR was nitrogen-doped multiwall carbon nanotubes (NCNTs) [267]. The NCNTs exhibited a maximum FE of CO of up to 80% at an overpotential of −0.26 V compared to their CNT counterpart. Moreover, multi-carbon products were also produced using a carbon-based CO2RR catalyst. Nitrogen-doped graphene quantum dots (NGQDs) were synthesized by exfoliating a GO precursor [268]. The FE of C2 hydrocarbons and C2–C3 oxygenates reached up to 55% at an overpotential of −0.74 V. Although many carbon-based CO2RR catalysts have been synthesized, and some of them have presented superior performance compared to other kinds of CO2RR catalyst, there are still several challenges that need to be solved to meet industrial requirements (figure 31).
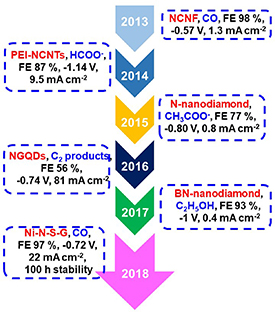
Figure 31. Roadmap presenting milestones in the development of carbon-based CO2RR catalysts.
Download figure:
Standard image High-resolution image(a) Competition between the CO2RR and the HER. The electrochemical hydrogen evolution is usually carried out using H2O as a proton source. The CO2RR is a multi-proton coupled process, and different products are produced, based on different numbers of transferred electrons. The overpotentials of all final products are close to the HER, hence, it is highly desirable to prepare catalysts with high selectivity and low overpotential, which are efficient toward the CO2RR while sluggish for the HER.
(b) High-efficiency and selectivity to multi-carbon products. So far, the two-electron-transfer product CO has been produced with a high FE and a low overpotential when catalyzed by carbon-based CO2RR materials. Nevertheless, the preparation of higher-order products involving more than a two-electron transfer still needs a high overpotential for the subsequent hydrogenation of the *CO species. Furthermore, the formation of multi-carbon products via carbon–carbon bond coupling by the dimerization of *CO and its hydrogenated derivatives is a kinetically sluggish process, which generally results in low energy efficiency and high power consumption. Many difficulties remain on the path to the selective production of desired multi-carbon products with higher commercial values.
(c) Techno-economic and industrial requirements. To achieve techno-economic and industrial targets, techno-economic analysis has demonstrated that the key performance parameters of the CO2RR have to reach an FE level larger than 90%, a cell voltage smaller than 1.8 V, a current density larger than 300 mA cm−a, and a stability of more than 80 000 h for economically compelling target products [269]. Meanwhile, the activity and stability of catalysts are an intrinsic trade-off relationship. Catalysts with the highest activity always possess the least long-term durability.
(d) Mechanistic understanding. Although a plausible mechanism for the CO2RR process has been proposed, the origin of active sites in carbon-based catalysts is still unclear. The definitive mechanism of the carbon–carbon bond used for coupling to multi-carbon products is still absent. The rational design of carbon-based CO2RR catalysts is highly desirable, compared to the current trial-and-error method. The establishment of a fundamental theory would benefit the design of catalysts with high efficiency and high selectivity, especially for high-value multi-carbon products.
Concluding remarks
In conclusion, the production of value-added carbonic products through the CO2RR process has the potential to solve the energy shortage and global warming issues. Until now, tremendous effort has been expended to reach that goal. There are still critical challenges, including enhanced activity, high-value product selectivity, and superior stability, which need to be further overcome. The development of carbon-based CO2RR catalysts that can produce higher-value commercial products is strongly expected in the future, for both commercial and environmental reasons.
22. Single-atom catalysts for CO2 reduction
Xiaoya Cui
Center for Programmable Materials, School of Materials Science and Engineering, Nanyang Technological University
Status
The excessive consumption of fossil fuels has caused an increasing accumulation of CO2, which consequently resulted in climate change and global warming. The CO2RR is one of the most promising strategies for reducing the greenhouse effect and converting excessive emitted CO2 into value-added chemicals using renewable electricity [270–272]. To date, enormous efforts have been devoted to improving CO2RR performance, to achieve, for example, high selectivity, low overpotential, good stability, and large current density. Numerous nanostructured electrocatalysts have been developed, such as metals, metal oxides, carbon-based compounds, MOFs, MS, etc, which exhibit enhanced catalytic performance, as compared to their bulk counterparts, due to the abundant active sites on their nanostructures. However, an investigation of the electrochemical CO2RR mechanism, especially the reaction pathway, in terms of the relationship between structures and their related catalytic properties, is not sufficient. SACs, featuring low coordinate states, homogeneous active sites, well-defined structures, and 100% metal utilization provide a potential platform for an investigation into the mechanisms of the CO2RR [273]. It is worth noting that the active sites on SACs can be facilely modified at the atomic level and that the local structure of nanostructured catalysts is relatively simple and uniform. Also, the electronic structures of SACs, which can be altered via strong SAC–support interactions, are dramatically distinct from their bulk counterparts. Several synthetic strategies have been proposed for the synthesis of SACs, including mass-selective soft landing, ALD, co-precipitation, impregnation methods, and atomic trapping at high temperatures. For the prepared SACs, the strong interaction between single metal atoms and the support guarantees a charge transfer between the metal and the associated interface, as well as the dispersion of isolated metal atoms on the surface of the support which prevents the aggregation of metal atoms. Recently, SACs made with Ni, Co, Fe, Mn, Zn, Sn, and Cu have been demonstrated to be active for the electrochemical CO2RR, providing high reduction-current densities and FEs [274]. The electrochemical CO2RR of SACs can mainly be optimized in three respects: tuning the coordination number of single atoms with N, constructing multimetallic SACs, and increasing the loading mass of metal atoms. Firstly, by establishing Ni–SACs with low coordination numbers, such as NiN2, and NiN3, the activity of Ni SACs for the CO2RR is optimized [275–277]. NiN2 exhibits a high CO FE of 92.0%–98.0% from −0.53 to 1.03 V. A DFT calculation indicated that the free energies of *COOH on NiN2 and NiN3 were much lower than that on NiN4. Similarly, by tuning the coordination number of Co atoms, the catalytic activity and selectivity for the CO2RR of Co SACs can also be modified (figure 32(a)) [9]. Therefore, the electronic structures of active sites can be altered by tuning the coordination numbers of metal atoms, and thus the electrocatalytic performance is enhanced. Secondly, multimetallic SACs have been proven to exhibit superior catalytic performance for the CO2RR, as compared to single metal-based SACs, due to the synergistic effects from the interactions between different metal components. For instance, the isolated diatomic Ni–Fe metal-nitrogen sites developed by Zhao and coworkers showed a high CO FE of more than 90% over a wide potential window from −0.5 to 0.9 V and improved stability over 30 h [278]. Also, by anchoring Fe‐N sites with cobalt phthalocyanine (CoPc), Lin et al synthesized CoPc@Fe‐N‐C which showed a high CO FE of more than 90% at a potential window of 0.71 V [279]. It is proposed that multimetallic SACs are beneficial for the C–C coupling and stabilization of C2 intermediates, due to the synergistic effect among various single-atom sites (figure 32(b)). Thirdly, the loading mass of active sites is another key factor that improves the catalytic performance of SACs. Impressively, Zhang et al fabricated TM-based SACs (Cr, Mn, Fe, Co, Ni, Cu, Zn, Ru, Pt or their combinations) with mass loadings of up to 1.8 wt% (figure 32(c)) [280]. In particular, Ni SACs with the highest mass loading (5.32 wt%), exhibited excellent activity for the electrochemical CO2RR, with a CO FE of 98.9% at −1.2 V (figure 32(d)). Moreover, Cu SACs can not only maximize the efficiency of Cu utilization but also maintain catalytic activity for hydrocarbon production, and thus obtain value-added products (e.g. C2H5OH, C2H6, CH4 and C2H4) [281]. Notably, Cu–CeO2 SACs show high catalytic activity and selectivity for CH4 with an FE of 58% during the CO2RR (figure 32(e)).
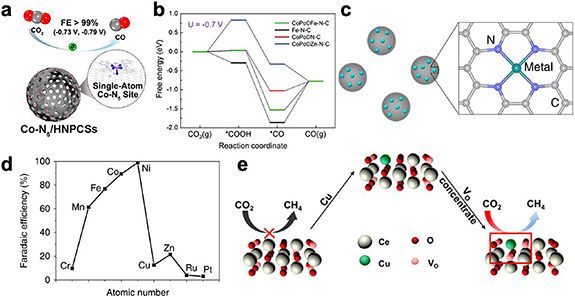
Figure 32. (a) Schematic illustration of CoN5 SACs with optimized catalytic activity and selectivity for CO2RR. Reproduced from [9] with permission. Copyright 2018, American Chemical Society. (b) Free energy diagram for CO production at −0.7 VRHE on Fe site in CoPc@Fe–N–C. Reproduced from [279]. with permission. Copyright 2018, Wiley–VCH. (c) Schematic illustration of TM-based SACs. (d) CO FE for metal-SACs at −1.2 VRHE. Reproduced from [280] with permission. Copyright 2019, Nature Publishing Group. (e) Schematic illustration of Cu SACs for the CO2RR to CH4. Reproduced from [281] with permission. Copyright 2018, American Chemical Society.
Download figure:
Standard image High-resolution imageAdvanced characterization techniques and synthetic strategies for precisely controlling nanostructures at an atomic scale are believed to be crucial for guiding the design of efficient electrocatalysts and the study of catalytic mechanisms. From the point of view of synthesis strategies, the modification of support materials (e.g. the construction of porous nanostructures, the introducion of defects, vacancies and heteroatom doping), optimization of the coordination environment of single atoms, and tuning the synergy between single atoms and supports should be regarded as effective methods for increasing the metal loading mass. Besides, advanced characterization techniques, such as atomic-resolution transition electron microscopy (TEM) (figures 33(a) and (b)), scanning tunneling microscopy (STM), and XAS, are urgently required to confirm the local atomic structures and the electronic structures of SACs [282]. In particular, STM can provide atomic-resolution images of the surface structure as well as the adsorbed molecules or atoms. Notably, XAS shows great advances in confirming the characteristics of local structures, e.g. oxidation state, coordination number, bond length, and atomic species (figures 33(c) and (d)) [281]. A synergistic combination of the above advanced techniques should be taken into account to accurately confirm the overall structures. Also, in situ characterization techniques should be considered for accurate verification of the dynamic changes and revelation of the nature of SACs. To boost engineering of SACs at the atomic level, theoretical simulations and experimental studies should be comprehensively developed and systematically combined to design electrocatalytic structures with outstanding activity and selectivity.
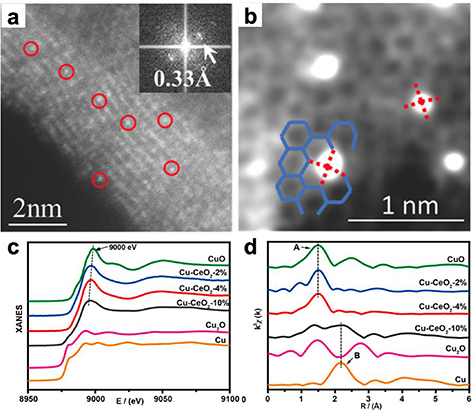
Figure 33. (a) High-angle annular dark-field imaging scanning TEM (HAADF-STEM) image of Ni SACs (marked with red circles) embedded in a CNT. (b) HAADF-STEM image of Ni single atoms showing the structured environment. Reproduced from [282] with permission. Copyright 2018, Wiley–VCH. (c) X-ray absorption near-edge structure (XANES) spectra at the Cu–K edge. (d) FT of extended EXAFS spectra of Cu–CeO2 SACs. Reproduced from [281] with permission. Copyright 2018, American Chemical Society.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Although SACs have demonstrated attractive performance for the CO2RR, there are still many challenges to be addressed. Firstly, it is important to guarantee a loading mass high enough for practical applications while maintaining the reaction centers as single atom sites during the CO2RR. Secondly, owing to the challenges of precise synthesis, SACs often comprise mixed structures and different coordination environments, which greatly hinder the development of high-performance SACs and the systematic investigation of the CO2RR mechanism using SACs. Thirdly, it remains a great challenge to design and synthesize multimetallic SACs featuring well-defined compositions and precisely controlled local structures. Multimetallic SACs can exhibit excellent activity and selectivity for multicarbon products, which is beneficial for the establishment of structure–activity relationships at the atomic scale and the study of catalytic mechanisms. Lastly, the combination of advanced characterization technologies and theoretical simulations is necessary to properly express the electrocatalytic process of SACs and illustrate the reaction pathways.
Concluding remarks
The development of SACs has greatly expanded research into electrocatalysis and shows promising potential for the electrocatalytic CO2RR. SACs provide more opportunities to tune active sites and optimize the activity, selectivity, and stability of electrocatalysts, establish the structure–property relationship, and further guide the design of highly efficient SACs and other catalysts for the CO2RR. The synergistic combination of novel synthetic strategies, advanced characterization technologies, and theoretical modeling represented by SACs is believed to deepen our understanding of CO2RR mechanisms and pathways.
23. Heterogeneous molecular catalysts for CO2 reduction
Haiqing Wang
Collaborative Innovation Center of Technology and Equipment for Biological Diagnosis and Therapy in Universities of Shandong, Institute for Advanced Interdisciplinary Research (iAIR), University of Jinan
Status
Currently, the excessive consumption of fossil fuels has resulted in a CO2 concentration of up to 414.11 ppm in the global atmosphere, which is higher than the safe upper limit of 350 ppm [2, 10, 283]. The resulting extreme climate and environmental changes, such as ocean acidification, climate warming, and the thawing of glaciers, have seriously threatened the survival and development of human beings. To steer the carbon cycle, various strategies have been developed for CO2 conversion, mainly consisting of chemical reforming, thermochemical, biochemical, mineralization, photochemical, and electrochemical methods, etc. With the rapid development of renewable electricity storage techniques, electrochemical CO2 reduction (ECR) has been recognized as a promising approach for directly converting carbon emissions and storing renewable electricity as fuels and value-added chemicals. Owing to the inherent stability and the low solubility of CO2, the thermodynamics and kinetics of ECR are always slow, and need a very negative redox potential (−1.9 V vs. SHE) to activate CO2 into the rate-determining CO2 •− radical species via the first electron transfer. At that operating potential, the HER is a competing side reaction and severely suppresses the FE. The ECR usually consumes different numbers of electrons and protons and goes through a complex stepwise reduction process. When 2/4/6/8/12 electrons are consumed, the production varies from CO, HCOOH or HCOO−, and H2C2O4/CH2O/CH3OH/CH4/C2H4 and C2H5OH, respectively [284]. The difference in reaction pathways depends on the types of intermediates generated on the surface of the catalyst. The final activity and selectivity of the catalyst are highly relevant to the tuning and stabilization of reaction intermediates. Therefore, the design of ECR electrocatalysts is of great importance to optimize reaction pathways for highly selective hydrocarbon production. Until now, metal-based heterogeneous electrocatalysts have demonstrated high performance for ECR, with relatively low overpotentials and various production distributions from C1 and C2 to C3. By contrast, molecular catalysts normally work in a homogeneous and indirect form for ECR [285]. The well-defined active metal sites are endowed with the accurate manipulation of ligand structures, improving mechanism-based activity. The redox cycling of the active metal centers is incapable of catalyzing more interesting products than the two-electron pathway. Although exciting advances have been made, the development of ECR is still suffering from poor product selectivity, low FE, and high overpotential. Besides the inherent chemical inertness of the CO2 molecule, the electrosynthesis pathways in single organic or inorganic catalysts still lack the modulation to tune the stability of intermediates and further improve the catalytic performance [264]. Very recently, heterogeneous molecular catalysts based on organic–inorganic hybrid nanomaterials have exhibited superior catalytic performance and interesting reaction processes for ECR, as compared to single organic or inorganic material, due to the integration of the advantages of both heterogeneous and homogeneous catalytic processes.
At present, the well-developed heterogeneous molecular catalysts can be generally classified into hybrids of molecular catalysts/carbon materials (metal phthalocyanine/porphyrins/polypyridine complex/carbon materials), organic functionalization of metal catalysts (Cu/Au/Ag/Pd/alloys catalysts modified by organic ligands), MOFs and their composites (Cu/Fe/Co/Zn/noble metal-based MOFs and their composites), and other organic-inorganic hybrid materials (hybrids based on COFs/TMS/g–C3N4). In particular, enzymes can be categorized as a special heterogeneous molecular catalyst due to their distinguished structure and activities (figure 34) [270, 286, 287]. MOFs and their composites are classical crystalline inorganic–organic hybrid nanomaterials consisting of metal nodes and organic linkers, which have been widely investigated for ECR. Their tunable porosity and shape can concentrate CO2 at the interface between the electrode and the electrolyte. Exposed unsaturated central metal sites and ligands can synergistically enhance the activity, selectivity, and stability of a catalyst. Moreover, the mixing of carbon materials with MOFs can overcome their inherent weakness of poor electronic conductivity. Hybrids of MOFs with active ECR nanostructures can potentially construct an effective heterogeneous interface to facilitate electron transfer for the activation of the CO2 molecule and the adsorption of reaction intermediates. Unfortunately, the low conductivity and stability of MOFs severely hinder the development of high-performance MOF-based electrocatalysts, which remains a significant challenge [288].
Figure 34. Classification of heterogeneous molecular electrocatalysts. Reproduced from [270]. Copyrighted by Springer Nature Ltd.
Download figure:
Standard image High-resolution imageIt is highly desirable to synthesize catalysts chemically or physically via a combination of methods including the hydrothermal method, CVD, PVD, laser and so on, to promote activity and selectivity by stabilizing targeted intermediates. The functional group of ligands, such as thiol, amine, carboxyl, phosphine, and hydroxyl, should specifically depend on the kind of inorganic substrate [289]. Many concepts should inspire the design of catalysts for generating C⩾2 products; the main concepts include enzyme-like second coordination sphere interaction for proximity between the catalyst and the intermediate (see figure 35(a)), tandem catalysis for highly valued productions (see figure 35(b)), control of the proton inventory for pH-dependent equilibrium (see figure 35(c)), selective inhibition of undesired reactions (see figure 35(d)), and stabilization of the electrode microstructure for continuous targeted reactions (see figure 35(e)). Advanced operando spectroscopic techniques and theoretical calculations are still very necessary to achieve a deep understanding of the interactions or bindings in heterogeneous molecular catalysts and the reaction pathways [290]. The electron transfer kinetics and mass transport at the solid/liquid interface should be taken into account when investigating critical surface limitations. The new PEM cell, liquid-flow cells, and waterproof GDE should be comprehensively developed to improve CO2 conversion to take place at a higher rate, i.e. at a commercial level, with a current density of >200 mA cm−c.

Figure 35. Opportunities for heterogeneous molecular electrocatalysts. Reproduced from [209]. Copyrighted by Springer Nature Ltd.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Current and future challenges mainly lie in the design of advanced catalysts to improve and optimize catalytic activity, selectivity, long-term stability and heterogenization techniques for practical applications. Molecular electrocatalysts per se suffer from poor conductivity and apparent aggregation, while the scaling relations in inorganic catalysts hinder multiple-electron production. The presence of additional binding interactions between molecular and inorganic components in heterogeneous molecular catalysts may tailor the stability of reaction intermediates and the stability and conductivity of catalysts [291]. The types of interactions should be designed well by mediating the ligands of the molecule to address the above problems. Novel cell configurations are needed to promote the diffusion of reactants, and the separation and purification of products. Heterogeneous molecular catalysts capable of reducing CO2 to C⩾2 products are still limited. Despite the development of advanced operando techniques and theoretical calculations, the ECR mechanisms are still unclear, and this extends to the identification of intermediate species, the structural evolution of molecules, the formation of C–C/H bonds and the interface between the electrolyte and the catalyst. Moreover, the poorly defined structure of heterogeneous molecular catalysts affects the accuracy of the final structure–activity relationship.
Concluding remarks
ECR has developed as a very promising route for dealing with greenhouse gases. It also opens up a multitude of possibilities for generating clean fuels and chemicals by converting renewable energy. The potential to apply heterogeneous molecular catalysts has been highlighted for the precise control of active sites to support high ECR performance. With concerted research into, and development of, catalyst optimization, advanced characterization techniques, theoretical methods, and cell configuration, it is believed that we will overcome the challenges and achieve a high-efficiency closed-loop anthropogenic carbon cycle for the health and safety of humanity.
Acknowledgment
This work was supported by the Provincial Natural Science Foundation (ZR2019BB025).
24. Oxides for N2 reduction
Zengxi Wei1, Quanhui Liu1 and Jianmin Ma1
!School of Physics and Electronics, Hunan University
Status
The Haber–Bosch process is the primary industrial method of producing ammonia (NH3), but its rigorous production conditions (high temperature and high pressure), excessive use of non-renewable energy sources, and vast released carbon dioxide have motivated considerable efforts to search for alternative methods of NH3 production [292–294]. Nowadays, the environmentally friendly electrochemical nitrogen reduction reaction (NRR) provides a promising opportunity to replace the Haber–Bosch process for NH3 production. Large-scale research has been devoted to designing effective electrocatalysts for NH3 production; however, it is still difficult to prevent low selectivity and Faraday efficiency from impeding the development of the electrochemical NRR. Thus, the development of efficient electrocatalysts to improve the selectivity and Faraday efficiency is the foremost issue in enhancing NH3 production.
Recently, metal oxides have been widely investigated for the electrochemical NRR, such as Fe2O3, TiO2, InO2, NbO2, SnO2, etc [295]. It was demonstrated that metal oxides could be used as effective electrocatalysts for NRR, due to the reduction of the competing HER. Liu et al first reported nano Fe2O3 as an electrocatalyst for the NRR. Nguyen et al adopted DFT calculations demonstrating that Fe2O3(0001) surfaces could be a promising active center for NH3 production with an applied bias of −1.1 V [296]. A more negative applied bias caused the NH3 selectivity to decrease because of the competing HER. Thus, screening other metal oxides with a low applied bias and a high NH3 production rate for applicability to the NRR has captured the attention of many researchers.
Current and future challenges
Metal oxides have been proposed as electrocatalysts for the NRR, but their low Faraday efficiency and production rate impeded the development of NH3 production. Thus, it is vital to design functional metal oxides for the electrochemical NRR with high activity and selectivity.
Fe-based oxides have been widely used as electrocatalysts for the NRR. Centi et al reported an catalyst consisting of Fe supported on CNTs for the NRR at room temperature and atmospheric pressure in a flow electrochemical cell operating in a gas phase. Under a −2.0 V applied bias, the NH3 production rate reached 2.2 × 10−3 gNH3m−2 h−1 in a flow of N2 [297]. Park et al demonstrated that the synthesized nanosized γ-Fe2O3 electrocatalysts were able to reach a FE of 1.9% and a weight-normalized activity of 12.5 nmol h−1 mg−1 at 0.0 VRHE (figure 36)300. The weight-normalized activity was dramatically increased to 55.9 nmol h−1 mg−1 after the γ-Fe2O3 was coated onto porous carbon paper to form an electrode. Sun et al also found that spinel Fe3O4 nanorods on a Ti mesh (Fe3O4/Ti) acted as an efficient electrocatalyst for the NRR [298]. At an applied bias of −0.4 V, the NH3 production rate reached 5.6 × 10−11 mol s−1 cm−2 and a high FE of 2.6% in 0.1 M Na2SO4. Also, they reported that a β-FeOOH nanorod was able to act as an efficient electrochemical NRR. After fluorine doping, the NRR activity was greatly improved, with an optimal NH3 production rate of 42.38 μg h−1 mgcat. −1 and an FE of 9.02%. DFT calculations demonstrated that the fluorine-doped β-FeOOH was able to decrease the reaction energy barrier from 0.59 eV to 0.24 eV, suggesting the enhanced NH3 production activity of the modified β-FeOOH catalyst. Additionally, multi-oxides have also been studied to find a possible electrocatalyst for the NRR. Zhao et al demonstrated that a CoFe2O4 nanocluster anchored on rGO was able to accelerate the N2 transformation to NH3. This catalyst was able to achieve a high NH3 production rate of 4.2 × 10−11 mol s−1 cm−2 and an FE of 6.2% in 0.1 M Na2SO4 [299].

Figure 36. Schematic of the electrochemical NRR. With permission from ACS [300].
Download figure:
Standard image High-resolution imageBesides Fe-based oxides, other oxides have been studied and used as NRR catalysts. Zheng et al demonstrated that a Zr4+-doped anatase TiO2 exhibited excellent electrochemical NRR performance with an NH3 production rate of 8.9 μg h−1 cm−2 and an FE of 17.3% at −0.45 V vs. RHE under ambient aqueous conditions [301]. DFT calculations revealed that the adjacent bi–Ti3+ pairs formed on anatase TiO2 were able to act as the most active centers in the NRR. As shown in figure 37, the A(101)-Vo (Vo on anatase (101) planes) presented adjacent bi–Ti3+ pairs that were effectively able to induce the chemisorption of N2 in a side-on configuration. The calculated free energy barrier for the bi–Ti2+ was 0.24 eV, which is lower than that of a single Ti2+ site of 0.5 eV. Mao et al reported Cr-doped CeO2 nanorods for effective NH3 production. The introduced Cr atom was able to act as an inducer for the oxygen vacancies, leading to a high FE of 3.84% and a large NH3 production rate of 16.82 μg h−1 mgcat. −1 in a 0.1 M Na2SO4 solution. Zhang et al demonstrated that SnO2 QDs supported on rGO were able to act as an efficient electrocatalyst for the NRR [302]. The ultrasmall SnO2 QDs were able to provide abundant activity sites for N2 adsorption and lower the reaction energy barrier for the rate-determining step of the NRR process.
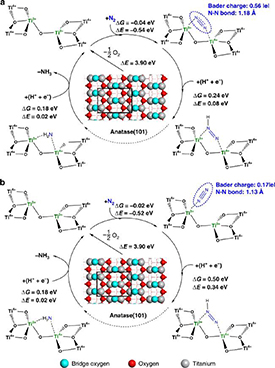
Figure 37. The free energies of the NRR at the (a) bi–Ti2+ activity site and (b) the single Ti2+ activity site, respectively. With permission from Nature publishing [301].
Download figure:
Standard image High-resolution imageAlthough metal oxides have been widely applied to catalyze N2 reduction, the NH3 production rate and FE are still ultralow, limiting industrial use. Also, it is difficult to confirm the activity sites of metal oxides, which obscures the mechanism of the NRR.
Concluding remarks
As discussed in this section, metal oxides can act as the proposed electrocatalysts for NRR. However, massive efforts should be devoted to designing ideal catalysts to improve the NH3 production rate and FE.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 51302079, 51702138), the Natural Science Foundation of Hunan Province (Grant No. 2017JJ1008), and the Key Research and Development Program of Hunan Province of China (No. 2018GK2031).
25. Chalcogenides for N2 reduction
Guangyin Fan1,2, Qian Liu1 and Xuping Sun1
1 Institute of Fundamental and Frontier Sciences, University of Electronic Science and Technology of China
2 College of Chemistry and Materials Science, Sichuan Normal University
Status
NH3 is an important chemical for the production of fertilizers, medications, and plastics. At present, value-added NH3 is mainly produced using the traditional Haber–Bosch process under rigorous conditions with the use of large amounts of energy and the generation of greenhouse gases. Alternatively, electrocatalytic N2 reduction (NRR) has been regarded as a perfect strategy for converting N2 to NH3 under ambient conditions with water as a hydrogen source, thereby supplying an appealing route for alleviating the energy and environmental issues caused by the Haber–Bosch process. Owing to the high bond energy and non-polarization features of N2, however, an efficient catalyst is required for N2 dissociation to produce NH3 via the NRR. Most importantly, the competitive HER exists at the same potential range, which considerably reduces the FE of the NRR process and hence greatly impedes the practical applications of this strategy [303]. Some meaningful achievements toward suppressing the HER have been made, as follows: (a) modulating the available H+ ions at the catalyst-to-electrolyte interface; (b) repelling H2O molecules in the NRR by constructing a porous hydrophobic nanolayer; (c) increasing the mass- and charge-transfer barriers of the applied electrolytes. As for electrocatalysts, noble-metal-based and non-noble-metal-based materials have previously been explored for their ability to produce NH3 by the electrocatalytic NRR. Because of fundamental interests and industrial applications, however, it is highly desirable to develop efficient non-noble-metal-based electrocatalysts that offer the potential for large-scale NH3 production via the green and sustainable electrocatalytic NRR under ambient conditions.
Current and future challenges
Currently, two basic reaction pathways, including disassociative and associative mechanisms, are proposed for the electrocatalytic NRR catalyzed by a heterogenous catalyst [303]. To accelerate the reaction kinetics of the NRR, tremendous effort has been exerted to design and synthesize novel nanostructures that can be used to promote the electrocatalytic performance of the NRR and prohibit the competitive HER. Among the non-noble catalysts developed, chalcogenide catalysts have sparked increasing research interest in the electrocatalytic NRR to NH3. Notably, 2D MoS2, which typically served as an efficient HER catalyst, has been demonstrated as an active NRR catalyst. For instance, Sun et al [304] first discovered that the 2D–MoS2 material favored for the HER was capable of catalyzing NRR to NH3, since the MoS2 edge sites were beneficial for N2 adsorption and activation (figure 38). The significant importance of this study lies in the discovery of an efficient MoS2 HER catalyst that can also be applied for NRR catalysis. In pursuit of advanced NRR performance for MoS2, several well-defined modification approaches have been explored, including defect modulation, heteroatom functionalization and increasing the catalytically active sites. The current research into the defect engineering of MoS2 has been demonstrated as an essential strategy to promote the NRR's performance. For example, Sun et al further indicated that the electrocatalytic performance for NRR was able to be enhanced using defect-rich MoS2 nanosheets in the same electrolyte [305]. Moreover, heteroatom-doping is regarded as another efficient way to achieve enhanced NRR electrocatalysts. Various metals have been introduced to MoS2 with different species, such as NPs, clusters, and atoms, among which Fe species have been widely investigated [306]. Recently, Zhao et al developed an in-operando-assisted strong Li–S interaction on S-abundant MoS2 to enhance the electrocatalytic NRR to NH3 [307]. DFT calculation demonstrated that the in-operando-driven strong Li–S interaction that happened at the S-edge sites of MoS2 was able to efficiently impede HER catalysis by decreasing ΔGH* on S-edge sites and enhancing the ΔGH* on Mo-edge sites, thereby promoting N2 adsorption and activation and leading to boosted electrocatalytic NRR performance. To further increase the active sites, MoS2 has been combined with various carbon matrixes (e.g. graphite oxides) to form hybrid nanostructures [308]. Except for the abovementioned MoS2-based materials, other metal (e.g. Fe, Re, Co, Nb) sulfides and their hybrids have also been synthesized for NRR catalysis [309–312]. Despite the tremendous efforts that have been committed so far, confirmation of the actual active sites of catalysts for the NRR remains elusive and needs further investigation. Also, the application of advanced in-situ analysis techniques and theoretical calculations can be of benefit for a better understanding of the NRR mechanism, helping scientific researchers to synthesize high-performance chalcogenide catalysts.
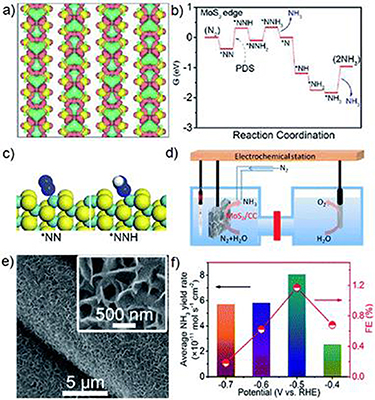
Figure 38. (a) Isosurface of deformation charge density viewed from above. Red and green represent charge accumulation and loss, respectively. The isosurface is 0.0025 a.u. (b) Free energy profiles for NRR at an MoS2 edge site. An asterisk (*) denotes the adsorption site. (c) Structures of key intermediates of the PDS. (d) A schematic diagram to illustrate the electrochemical setup for NRR tests. (e) Scanning electron microscopy (SEM) images for MoS2/CC. (f) Average NH3 yields and FEs of MoS2/CC at different potentials in 0.1 M Na2SO4. Reproduced [304] with permission from Wiley–VCH, copyright 2018.
Download figure:
Standard image High-resolution imageConcluding remarks
Herein, a brief review of the chalcogenide-based nanostructures for NRR was summarized. To improve the catalytic performances of chalcogenide catalysts, the origins of the activity enhancement need to be precisely determined and hence the rational design and modification of catalysts for NRR can be performed. Also, further solid evidence is required to verify the structure–performance relationship of chalcogenide catalysts, thus leading to a more suitable construction of components and a precise adjustment of interface chemistry to achieve a fast NRR kinetic. On the other hand, the reaction process of the NRR should be further optimized in combination with the electrode materials, electrolytes and reactor configuration. Based on the above discussion, the construction and modulation of chalcogenide catalysts with well-defined and enhanced catalytically active sites are highly desirable in terms of developing efficient NRR catalysts in future work.
26. Carbon for N2 reduction
Zengxi Wei1, Yuezhan Feng2 and Jianmin Ma1
1 School of Physics and Electronics, Hunan University
2 Key Laboratory of Materials Processing and Mold (Zhengzhou University), Ministry of Education, Zhengzhou University
Status
NH3 production is one of the most popular topics that is widely used in industrial and agricultural applications. However, industrial NH3 production still uses the Haber–Bosch process, which consumes more than ∼1.5% of global energy and releases 300 million metric tons of carbon dioxide annually. Designing and developing clean and free methods to replace the Haber–Bosch process is the most challenging issue at this time. The electrochemical NRR uses the ingredients of water and N2 with an applied potential at room temperature, and is known to be the most promising sustainable alternative for NH3 production [313–315]. Unfortunately, the low selectivity and FE have impeded the development of electrochemical NRR. Thus, it is vital to screen the ideal catalysts to enhance the catalytic activity and inhibit the competing HER. Nowadays, the noble metals, such as Pt, Ru, Au, Rh and etc, are known to have high conductivity, high selectivity and activity as efficient electrocatalysts for the NRR. However, the high cost and scarcity of noble metals limit their industrial applications, which motivated the effort to search for low-cost electrocatalysts for the NRR.
Recently, it was demonstrated that carbon-based materials were able to be used as effective electrocatalysts for NRR, which is ascribed to their lowcost, easy synthesis, high conductivity, high selectivity, etc [316]. Liu et al reported an N-doped porous carbon (NPC) as a cost-effective electrocatalyst for the NRR with a high NRR production rate of 1.4 mmol g−1 h−1 at −0.9 V vs RHE (figure 39(a)) [317]. Pyridinic and pyrrolic nitrogen have been proposed as the active sites and the porous structure of NPC provided many active sites for N2-fixation (figure 39(b)). Lv et al found that a metal-free polymeric carbon nitride catalyst with nitrogen vacancies (PCN-NV) was able to effectively adsorb N2 molecules on the adjacent carbon atoms of PCN-NV in an end-on manner, leading to the activation of a N≡N triple bond [318]. Li et al reported that carbon cloth could be a catalyst for NRR following the introduction of rich defects [319]. The thermally treated carbon cloth exhibited high electrocatalytic activation with an NH3 production rate of 15.85 μg h−1 cm−2 in a 0.1 M Na2SO4 + 0.02 M H2SO4 mixture. However, carbon-based materials still need an improvement in their catalytic activity for industrial application.

Figure 39. (a) Ammonia production rate of NPC-750 at −0.5 V to −1.1 V and (b) ten consecutive cycles at −0.9 V. (c) Schematic illustration of NPC preparation.
Download figure:
Standard image High-resolution imageCurrent and future challenges
Although carbon-based materials have been proposed as electrocatalysts for the NRR, their low FE and production rate impeded their industrial use. Thus, it is key to design effective carbon-based materials with high activity and selectivity.
Carbon-based-material-supported single atom catalysts (SACs) have been demonstrated as promising catalysts for the NRR. Geng et al reported that Ru single atoms on nitrogen-doped carbon (Ru SAs/N–C) were able to enhance the electrocatalytic NRR by an atomic mass of 0.18 wt% [320]. The high-efficiency catalytic activity of Ru SAs/N–C exhibited a production rate of 120.9 μgNH3mg−1 h−1 at −0.2 V. Wang et al reported a Fe–N/C–CNT catalyst with built-in Fe–N3 active sites [321]. This catalyst exhibited high catalytic activity with an NH3 production rate of 34.83 μg h−1 mgcat −1 and an FE of 9.28% at −0.2 V vs RHE. DFT calculations demonstrated that the Fe–N3 species had a larger spin moment of 3.16 μB, compared to that of Fe–N4 species of 2 μB, suggesting that the Fe–N3 species has high reaction activity for N2 adsorption. Choi et al performed DFT calculations demonstrating that SACs anchored on defective graphene were able to enhance the NRR selectivity, compared to that of the existing bulk metal surface due to strong suppression of the HER [243]. The Ti@N4 and V@N4 exhibited low free energies of 0.69 eV and 0.87 eV, respectively, which was ascribed to strong back-bonding between the hybridized d-orbital metal atom (Ti or V) and the π* orbital of the adsorbed N2.
Apart from metal doping, the metal-free element doping also exhibited considerable catalytic activity. Since Légaré et al found that the B atom was able to enhance N2 adsorption and activation through its empty p orbital, numerous studies have drawn attention to nonmetal doping on carbon-based materials. Yu et al reported that boron-doped graphene was able to efficiently enhance the activation of N2 [322]. DFT calculations revealed that the BC3 structure enabled the lowest energy barrier for NRR. The catalyst was able to achieve an NH3 production rate of 9.8 μg hr−1 cm−2 and an FE of 10.8% at −0.5 V vs RHE at an atomic doping rate of 6.2%. Ji et al reported that a boron interstitial (Bint)-doped C2N layer was able to act as an efficient electrocatalyst for the NRR using DFT calculations [323]. The calculations demonstrated that the Bint-doped C2N was able to efficiently activate the N2 molecule through the 'acceptance-donation' process with an ultra-low limiting potential of −0.15 V (figures 40(a) and (d)). Other non-metal atomic dopants have also been demonstrated to act as electrocatalysts for the NRR, such as S, P, N, F, etc.

Figure 40. Free energy diagrams of boron-doped C2N in the (a) distal, (b) alternating, and (c) enzymatic mechanisms. (d) The schematic diagram of boron-doped C2N.
Download figure:
Standard image High-resolution imageAlthough functionalized carbon-based electrocatalysts for the NRR have been widely improved through atomic doping, there are many issues and challenges to be overcome before industrial production can be achieved. Firstly, the deficient evaluation criterion for the accurate quantification of the NH3 production rate, which causes huge errors in real experimental results. Second, the mechanism of the electrocatalytic NRR is still ambiguous, impeding the development of an ideal electrocatalyst for the NRR. Third, it is a challenge to identify catalytic activity sites using current characterization techniques. Furthermore, the atomic mass rate of SACs and stability are still the key issues for designing efficient electrocatalysts for the NRR.
Concluding remarks
As discussed in this chapter, functionalized carbon-based electrocatalysts could be promising candidates for the NRR. However, it is crucial to improve the test criterion and enhance the catalytic activation and cycling stability of catalysts.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant Nos. 51302079, 51702138), the Natural Science Foundation of Hunan Province (Grant No. 2017JJ1008), and the Key Research and Development Program of Hunan Province of China (No. 2018GK2031).
27. C3N4 for N2 reduction
Yaping Liu and Ke Chu
School of Materials Science and Engineering, Lanzhou Jiaotong University
Status
The electrocatalytic nitrogen reduction reaction (ENRR) powered by renewable electricity under ambient conditions represents a sustainable route for artificial NH3 production, in contrast to the Haber–Bosch process which is energy-intensive and increases greenhouse gas emissions [324]. However, the efficiency of the NRR process is largely restricted by the unsatisfactory NH3 yield rate and low FE, as a result of poor N2 activation on the catalyst's surface and severe competition from the HER. The development of low-cost, robust and active electrocatalysts to effectively activate the N2 molecule and suppress the HER is urgently desired, but remains challenging. Recent advances have provided a wide range of potential NRR catalysts, including precious metals, TM-based compounds, and non-metallic materials [11, 302, 325, 326].
Graphitic carbon nitride (C3N4) and related materials have been considered as attractive electrocatalysts because of their unique 2D nanostructure, enriched N species, tailorable electronic structure, high durability, facile synthesis, and environmental benignity (figure 41(a)). Although C3N4 is, by itself, catalytically inert in the NRR, defect/vacancy engineering and construction of heterostructures offer promising approaches for endowing C3N4 with enhanced NRR activity. A pioneering work by Yu et al demonstrated that engineering defects by creating nitrogen vacancies (NVs) on C3N4 was able to create an active metal-free NRR catalyst. The C3N4 with rich NVs delivered an NH3 yield of 8.09 μg h−h mg−g and an FE of 11.59% at −0.2 V vs. RHE [318]; the NH3 yield was increased by an order of magnitude in comparison with pristine C3N4. Theoretical studies based on DFT calculations revealed that the NVs enabled the construction of a dinuclear end-on bound structure for spatial electron transfer into the antibonding orbital of the N2, which was conducive to capturing the N2 molecule and rupturing the N≡N covalent triple bonds (figure 41(b)). Nevertheless, under the N2-supply conditions of the NRR process, the active NVs tend to be poisoned by N occupation, engendering deactivated NVs and a dramatic decrease in NRR activity/stability for the NV-containing C3N4. To circumvent such a problem, we proposed the approach of filling NVs with S dopants (figure 41(c)), and the resultant S–NV–C3N4 with a 5.2 at.% S concentration showed an NH3 yield of 32.7 μg h−h mg−g and an FE of 14.1% at −0.4 V vs. RHE [327], significantly exceeding those of pristine C3N4 and NV-containing C3N4. S–NV–C3N4 also possessed long-term durability with negligible activity decay following 20 h of continuous electrolysis. Mechanistic investigations revealed that filled S-dopants can break the *N2H–*NH2 scaling relation to balance the binding energy for optimal adsorption of reaction intermediates, consequently leading to a significantly reduced energetic barrier and boosted intrinsic NRR activity. On the other hand, owing to the unique 2D structure composed of abundant and uniform heptazine heterorings, C3N4 can provide abundant anchoring/coordination sites for the construction of heterostructures which exhibit the synergistic functions that promote electrocatalytic activity. In this regard, we developed a 2D/2D MoS2/C3N4 heterostructure which exhibited an NH3 yield of 18.5 μg h−h mg−g and an FE of 17.8% at −0.3 V vs. RHE [328], which are far superior to those of the individual MoS2 or C3N4. DFT calculations disclosed that the interfacial MoS2–C3N4 electronic coupling not only provides enhanced *N2H binding on the Mo edge to decrease the reaction energy barrier but also prevents the active Mo edge from competing with the HER (figure 41(d)).
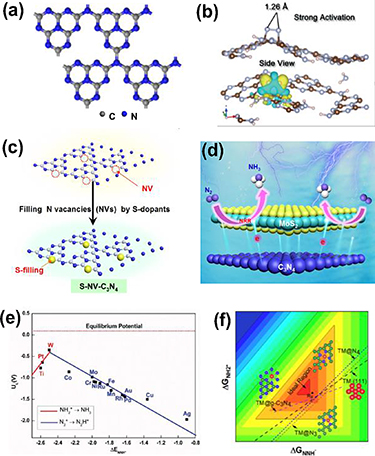
Figure 41. (a) Scheme of tri-s-triazine-based atomic configuration of C3N4. (b) The charge density difference of the N2-adsorbed C3N4 with NVs. Adapted from [318] with permission. Copyright 2018, Wiley–VCH. (c) Scheme of NVs filled with S-dopants in C3N4. Adapted from [327] with permission. Copyright 2020, Elsevier Science Publishers. (d) Scheme of 2D/2D interface engineering of a MoS2/C3N4 heterostructure for boosting the NRR. Adapted from [328] with permission. Copyright 2020, American Chemical Society. (e) The computed NRR limiting potential (UL) as a function of the adsorption energy of NNH* species (ΔENNH*) for various SACs supported on C3N4. Adapted from [329] with permission. Copyright 2019, Wiley–VCH. (f) Color-filled contour plots of the limiting potential as a function of the Gibbs free energy of *NNH (ΔGNNH*) and *NH2 (ΔGNH2*) for various TM-SACs for an improved NRR. Adapted from [6] with permission. Copyright 2019, American Chemical Society.
Download figure:
Standard image High-resolution imageRecently, SACs with isolated metal centers have attracted increasing attention, thanks to their unique electronic properties of a single active site and maximum atomic efficiency. C3N4, which possesses plentiful nitrogen coordinators with rich electron lone pairs, can be used as an ideal platform to stably anchor SACs for more favorable catalytic properties. Zhao et al performed theoretical screening of a series of SACs supported on C3N4 for the NRR [329]. According to several screening benchmarks such as SAC anchoring stability, N2 adsorption energy, *H adsorption free energy, and potential-determining step (PDS) energy barrier (figure 41(e)), they demonstrated that W@C3N4 is quite a promising SAC for the NRR, with high stability, favorable N2 adsorption, a low PDS energy barrier and suppression of the HER. The outstanding NRR activity of W@C3N4 originates from its unique electronic structures, including the positive charge and large spin moment of the W atom, high electrical conductivity and moderate binding strength for NRR intermediates. Lately, using DFT calculations, Qiao's group built up a full picture of TM-SACs on nitrogen-doped carbons and C3N4 for the NRR by considering the three key factors of activity trends, electronic origins, and design strategies (figure 41(f)) [8]. They showed that the intrinsic activity of TM-SACs correlates well with their nitrogen adatom adsorption energies (ΔEN*), and that the variation of ΔEN* stems from the change in the bonding/antibonding orbital populations of the metal centers. Based on these findings, they demonstrated that Ru@C3N4 is the most promising NRR candidate, and that TM-SACs on C3N4 can be further optimized by tailoring the adsorption strengths of the key intermediates. Although SACs supported on C3N4 are theoretically attractive for NRR, there is still a lack of experimental validation.
Current and future challenges
Despite some progress, as outlined above, in the exploration of C3N4 for the NRR, from both experimental and theoretical perspectives, this research field is still in its infancy and remains largely unexplored. There are lots of challenges to overcome, and we propose that future research can be strengthened in the following areas:
First, the reported NH3 yield and FE of C3N4-based catalysts are still far from satisfactory and also uncompetitive with state-of-the-art NRR catalysts because of several unsolved issues, such as the limited density of active sites, poor N2 activation, a high reaction energy barrier and unsatisfactory HER suppression. From the viewpoint of catalyst design, maximizing the number of active sites and enhancing the intrinsic activity per active site represent the two general strategies, which can be achieved by a series of well-established approaches, such as surface atomic regulation, heterostructure coupling, defect/vacancy engineering, heteroatom doping, pore fabrication, component modification, phase manipulation, strain engineering, etc. The current regulation methods for C3N4-based catalysts are limited to defect/vacancy engineering and heterostructure coupling only. Other efficient approaches, especially heteroatom doping and surface atomic regulation are recommended for fine tuning of the electronic/surface configurations of C3N4 to acquire the desired NRR activity. In addition to catalyst design, the NRR performance, in particular the FE, can be further optimized from the perspective of regulating the electrode–electrolyte interface by controlling the factors including N2 solubility near the catalyst, the pH/cation type/concentration of the electrolyte, the environmental temperature/pressure, and the local availability of protons. Furthermore, catalytic stability is equally important, whereas the current C3N4-based catalysts are generally only assessed for 2–20 h, which is not sufficient for practical applications. Extending the test duration to several days or weeks is highly necessary for the rational evaluation of the real stability performance of C3N4-based catalysts.
Second, fully unveiling the reaction mechanisms will help to guide the rational design of efficient catalysts. At present, DFT calculations are normally employed to provide theoretical insights into the NRR mechanisms of C3N4-based catalysts. Nonetheless, there is generally a large gap between theoretical modeling based on idealized assumptions and the real electrochemical conditions of the catalysts. In this context, some advanced in-situ characterization methods, such as in-situ Fourier transform infrared (FTIR) spectroscopy, in-situ Raman spectroscopy, in-situ scanning transmission electron microscopy (STEM), etc, may provide solid experimental evidence by monitoring in real-timechanges in reaction kinetics and energetics, as well as in the chemical configurations of the NRR intermediates. This may help to bridge the gap between theoretical simulations and real electrocatalysis and further provide a comprehensive understanding of the NRR mechanisms of C3N4-based catalysts. More recently, the combined use of in-situ XRD/in-situ Raman spectroscopy and computational simulations has been applied to understand the NRR process catalyzed by boron-rich covalent organic frameworks through electrochemical excitation [330]. These types of in-situ technique are thus highly recommended to complement theoretical calculations, thereby uncovering the origins of catalysis in C3N4-based catalysts when used in the NRR.
Last but not least, since C3N4 itself, and its synthesis procedure involve nitrogen-containing chemicals, more rigorous experiments should be performed with a combination of Ar control and 15N isotope tracer experiments to validate experimental accuracy and avoid the risk of any nitrogen contaminants. In this regard, it is vital to apply at least two detection methods from among the indophenol blue test, nuclear magnetic resonance, and ion chromatography, to determine the concentration of NH3 produced.
Concluding remarks
According to our summary, although the significant challenges still exist, there is no doubt that C3N4-based catalysts are very promising NRR electrocatalysts. Using rationally engineered strategies, C3N4-based catalysts with novel physicochemical properties and potentially high NRR performance can be fabricated. The combination of in-situ techniques and theoretical simulations would contribute to a fundamental understanding of the NRR kinetics and energetics, which may further set a direction for the more precise design and exploration of C3N4-based catalysts for efficient electrochemical N2 fixation with superior activity, selectivity and durability.
28. Single-atom catalysts for N2 reduction
Yuan Qiu and Xijun Liu
Tianjin Key Lab of Advanced Functional Porous Materials, School of Materials and Engineering, Tianjin University of Technology
Status
The TNRR can be used to synthesize NH3 directly from renewables (N2 and H2O) under ambient conditions. While this process is a promising alternative to the energy-intensive Haber–Bosch process, it is largely impeded by the lack of efficient NRR catalysts. SACs, in which isolated single atoms are dispersed on support materials, were first proposed in 2011 [117]. They show appealing properties such as maximum atomic utilization efficiency, unique electronic structures, and coordinately unsaturated metal sites [273, 331–333]. Recently, with the development of techniques such as aberration-corrected electron microscopy and XAS, SACs have been widely investigated and have shown impressive performance in various catalytic fields. The use of SACs for the NRR has rapidly expanded since 2018 (figure 42(a)).

Figure 42. (a) Publication numbers reporting the use of SACs and NRR over the past eight years in the Web of Science. (b) Performance map of the reported NRR catalysts in terms of the NH3 yield and FE. Some values were estimated from the reported values throughusingthroughby using simple unit conversion to provide a better comparison. A solid or hollow square indicates experimental results that were or were not verified using control experiments, respectively. Current as atof 29 February 2020.
Download figure:
Standard image High-resolution imageMost of the reported SACs that have been used as NRR catalysts (figure 42(b)) include noble-metal-based (Au, Ru, etc), transition-metal-based (Fe, Mo, Ni, etc), or even rare-earth-based (Y, Sc) materials. Most are M–Nx –C type electrocatalysts, in which the metal centers are coordinated with nitrogen atoms, usually at graphitic carbon defects. These materials are typically prepared via the high-temperature annealing of carbon, nitrogen, and metal precursors under an inert gas atmosphere. In early publications, the metal loading was usually less than 0.1 wt%. As synthetic strategies have matured, the introduction of high-surface-area carbon materials has allowed the density of M–Nx –C sites to be greatly increased by more than an order of magnitude, and loadings of 5–9 wt% can now be achieved. Among the noble-metal-based materials, Ru SACs, such as Ru atoms on NPC, are the most efficient materials, both experimentally and theoretically. On the other hand, inspired by MoFe active centers in nitrogenase, Mo- and Fe-based SACs have been the most thoroughly investigated non-noble-metal NRR catalysts. Our group first synthesized Mo and Fe SACs anchored to NPC as cost-effective catalysts for NRR, which achieved high NH3 yield rates of 34.0 ± 3.6 μgNH3 h−1 mgcat. (FE, 14.6 ± 1.6%) and 62.9 ± 2.7 μgNH3 h−1 mgcat. (FE, 18.6 ± 0.8%), respectively [334, 335].
The structural simplicity of active sites in SACs has inspired DFT calculation studies. The volcano plot, which relates the theoretical limiting potential to the adsorption energies of N-containing intermediates, is an effective tool for predicting the trends of NRR catalysts [293]. By comparing their theoretical limiting overpotentials and competition from the HER, SACs with different metal centers on conductive substrates (graphitic carbon, g–C3N4, MXene, 2D sulfide, etc) have shown potential to serve as NRR electrocatalysts [8, 331]. Computational studies have generally shown good consistency with experimental results. SACs have been experimentally proven to be efficient catalysts for NRR, such as the above-mentioned Ru, Mo, and Fe SACs, which are typically located near the top of the volcano plot obtained from DFT calculations. Furthermore, theoretical calculations are expected to predict experimental results. In 2014, Tian and co-workers first evaluated the catalytic profile of a MoSAC for nitrogen fixation [336], while the first experimental report of a MoSAC was published in 2019 by our group.
The electronic structure of SACs, such as the d-orbitals of the metal centers, can be altered due to the unique coordination environments of the supports. This makes it possible to regulate the linear scaling relations of adsorption energies among nitrogen-containing intermediates, which indirectly affects their activity. As shown in figure 2(b), the coordination of supports in TM-SACs shifts the scaling relations for the adsorption of intermediates [8]. Therefore, the NRR pathways can be optimized by precisely modulating and engineering the atomic structure of SACs, which can be realized by advances in both experiments and theoretical computations. On the other hand, in situ/in operando characterizations such as x-ray diffraction, infrared spectroscopy, Raman spectroscopy, XPS, and XAS can offer detailed structural and chemical information about the reactions that occur under working conditions. These results can provide mechanistic insight into the NRR and guide the rational design of electrocatalysts.
The supply of N2, electrons, and protons is important to obtain a highly efficient NRR. The selective transfer of N2 by nanostructured catalysts or synergistic catalysts can most likely optimize a selective NRR by combining efficient active sites with kinetically preferred material transfer processes. For example, a three-phase (electrocatalyst–electrolyte–gas) interface can be constructed to provide higher N2 accessibility using electrocatalysts. Furthermore, kinetic modeling of the competing HER and the mass transport of reactants in electrolytes and catalyst surfaces can also improve the understanding and optimization of the NRR process.
Recently, benchmarking protocols, including a standardized set of control experiments and quantitative isotope measurements with 15N2 gas, have been developed to definitively quantify the electroreduction of N2 to NH3 [337]. This has resulted in significant improvements in the data reproducibility and accuracy of NRR test experiments. Further screening of SACs based on this consensus will ensure that reliable data are reported.
Current and future challenges
Despite the rapid progress in developing the NRR, the performance of the NRR catalysts investigated still lags behind the requirements of practical applications. For example, the target of the US Department of Energy's ARPA-E REFUEL program is an NH3 yield of 10−6 mol s−1 cm−2 and an FE of 90%. Both of these values are much higher than the current best performances of the NRR obtained using SACs [338]. Thus, more materials should be developed and significant breakthroughs are needed for NRR to replace the Haber–Bosch process.
First, the NRR generally exhibits a high overpotential. In a typical NRR process, proton/electron pair transfer leads to the consecutive protonation of nitrogen species via dissociative and associative pathways. The PDS is determined by the most endothermic step, which is generally related to key intermediates, such as *NNH and *NH2. However, the extremely stable N≡N triple bond (with a bond energy of 941 kJ mol−1) and the linear scaling relations of adsorption energies among nitrogen-containing intermediates inevitably lead to high NRR overpotentials [293].
Furthermore, the competing HER accompanying the NRR will reduce the FE of ammonia production. As illustrated in the partial Pourbaix diagram for the N2–H2O system in figure 43(a), the potential window between the NRR and HER is quite narrow, making it difficult to simultaneously achieve both high selectivity and catalytic activity [339]. N2 also has extremely low solubility in water (0.66 mmol l−1 under ambient conditions), which significantly retards the supply of N2 and consequently lowers the NRR selectivity. As shown in figure 43(b), no SACs have both high Faradaic efficiencies and NH3 yield rates, and most of the reported Faradaic efficiencies are below 30%.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
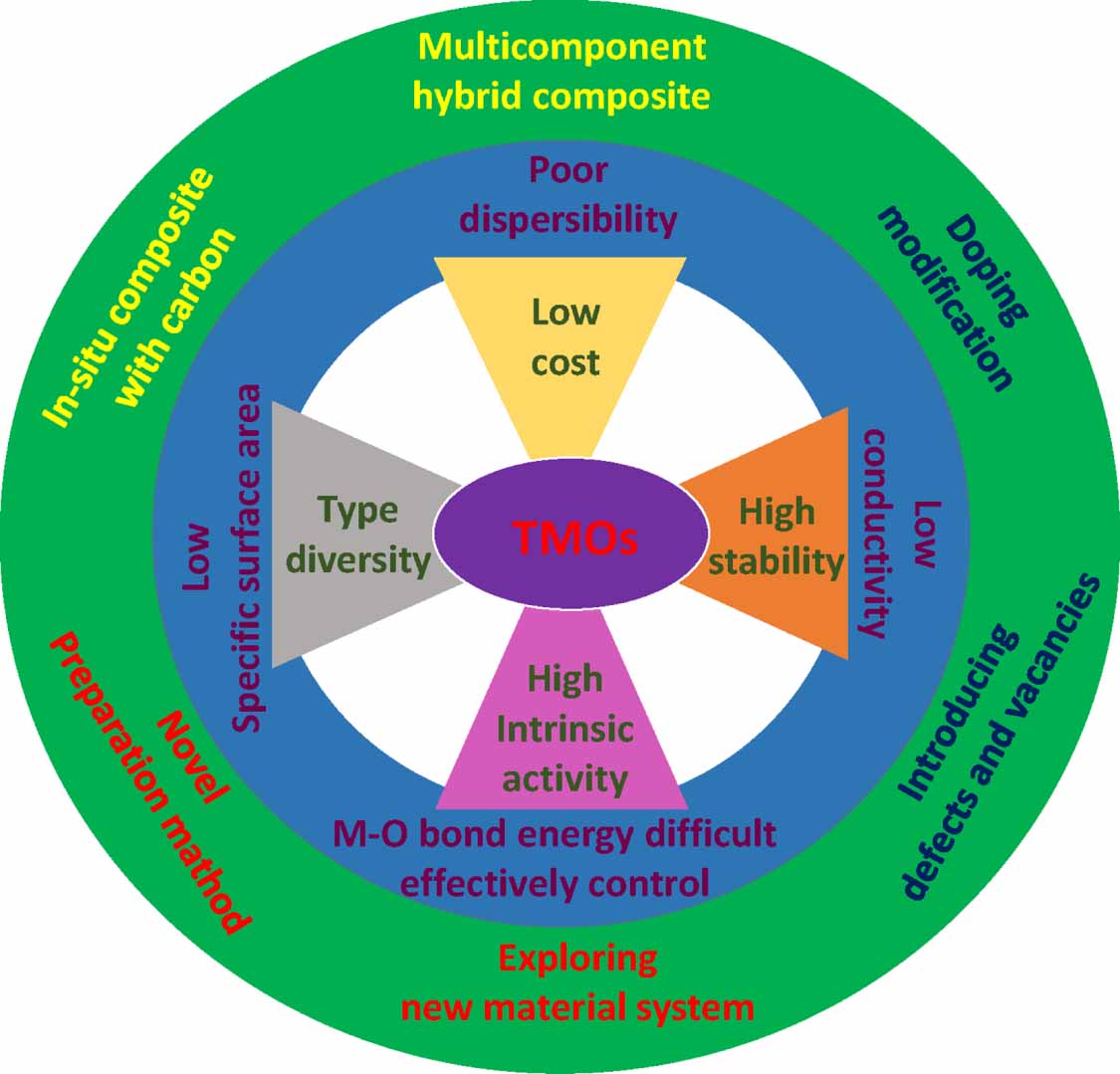{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
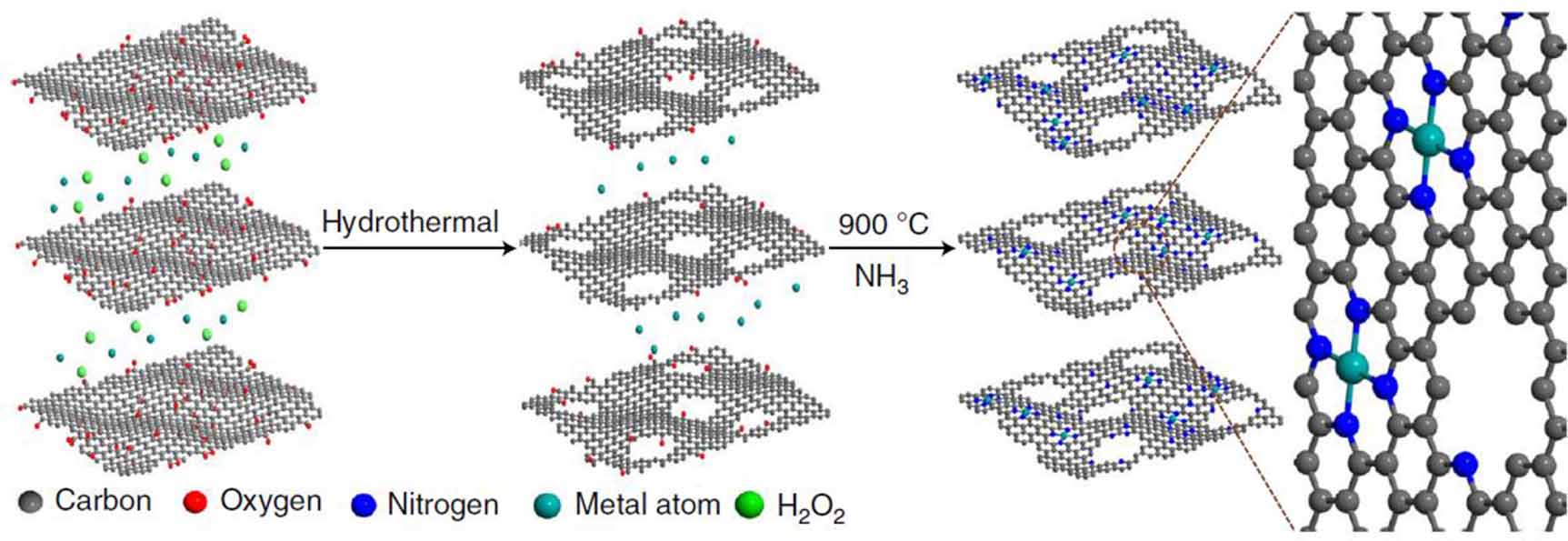{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 43. (a) Partial Pourbaix diagram for the N2–H2O system. Copyright 2018, American Association for the Advancement of Science. Figure reprinted from [338] with permission. (b) Comprehensive comparisons of the limiting potential of SACs with different metal centers and supports versus a pure metal (111) surface included as a reference. Copyright©2019 American Chemical Society. Figure reprinted from [8] with permission. with
Download figure:
Standard image High-resolution image{kind=link}
This scenario is further aggravated by ammonia contamination during test procedures (such as in the air, human respiration, latex gloves, raw chemicals, and vessels), which makes the NRR experiments more difficult. In particular, in the case of M–Nx –C materials, the possibility of background x–NH3 produced by unstable N-species should be accounted for. Furthermore, because of diverse operating procedures and different control experiments, it is difficult to systematically compare the catalytic performances of SACs reported in different works, and the repeatability and accuracy of the results cannot be guaranteed [337].
Concluding remarks
In recent years, there has been an ever-increasing number of both experimental and theoretical studies of the fabrication of SACs for the NRR under ambient conditions. Although the performances (NH3 yield and FE) of SACs are still far from those required by practical applications, with continuous research efforts, more advanced NRR catalysts are expected to be developed in the future. These new materials and insights can provide breakthroughs for conducting the NRR under ambient conditions, and will help to replace the Haber–Bosch cycle.
Acknowledgment
This work was financially supported by the National Natural Science Foundation of China (21601136).