

Abstract
The sites of interaction between a cell and its surrounding microenvironment serve as dynamic signaling hubs that regulate cellular adaptations during developmental processes, immune functions, wound healing, cell migration, cancer invasion and metastasis, as well as in many other disease states. For most cell types, these interactions are established by integrin receptors binding directly to extracellular matrix proteins, such as the numerous collagens or fibronectin. For the cell, these points of contact provide vital cues by sampling environmental conditions, both chemical and physical. The overall regulation of this dynamic interaction involves both extracellular and intracellular components and can be highly variable. In this review, we highlight recent advances and hypotheses about the mechanisms and regulation of cell–ECM interactions, from the molecular to the tissue level, with a particular focus on cell migration. We then explore how cancer cell invasion and metastasis are deeply rooted in altered regulation of this vital interaction.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
For higher-order multicellular organisms, physiological adaptation to our surroundings is a crucial part of everyday living. From an evolutionary standpoint, this ability to adapt to changes in the environment is conserved from the whole organism level to organ, tissue, and cellular levels. Hence, sampling and receiving information about the surrounding environment is key to adaptation. At the cellular level, numerous organelles exist to sample environmental conditions, which are generally classified as cell–cell or cell–extracellular matrix (ECM) interactions.
The organelles mediating cell–ECM interactions can be separated into two main categories, focal adhesions and hemidesmosomes, which associate with actin and intermediate filaments, respectively. Hemidesmosomes are found mostly in epithelial tissues associated with a basement membrane. In contrast, focal adhesions can be found in most adhesive cells and represent the major sites of cell–ECM interactions. Once thought to be static structures, focal adhesions are highly dynamic organelles composed of an ever-growing list of structural and signaling components, ranging from transmembrane integrins that directly bind matrix molecules to actin regulatory proteins, such as α-actinin [1]. At these points of cell–ECM contact, cytoskeletal force generated through actomyosin contraction is transmitted out to the surrounding environment. Conversely, focal adhesions can respond to external differences in environmental properties, such as stiffness, tension, and the recently established properties of viscoelasticity and stress relaxation [2]. Focal adhesions also play a major role in controlling the rate of cell migration, with their formation and disassembly being two of the four key steps involved in the migration cycle: (1) protrusion, (2) adhesion assembly, (3) cell body translocation and (4) rear adhesion disassembly [3].
It this review, we focus on the dynamic interplay between cells and the ECM, with emphasis on focal adhesion dynamics and how changes to the microenvironment can alter the cellular response. In addition, we highlight recent mechanisms by which cell–ECM interactions mediate cell migration. We then shift our focus to recent studies in cancer biology exploring how cancer cell interactions with basement membrane and stromal ECMs are associated with tumor invasion. For brevity, we include a brief introduction of the major concepts and players involved in this interplay of cells with the microenvironment but will focus on the most important recent advances in the field.
2. Focal adhesions are a mechanical signaling hub
Cell–ECM interactions are crucial for fibroblast/mesenchymal cell migration. The seminal work of Weiss, Dunn, Abercrombie, Harris, Izzard, and Chen led the way toward understanding the mechanisms of cell migration and the interactions of cells with underlying substrates. Chen first demonstrated the dynamic changes in fibroblast area and 'focal contacts' during cell tail retraction [4]. It is well established that these 'focal contacts' (now known as focal adhesions) form through the direct binding of integrins to underlying ECM proteins such as fibronectin, laminin, or the numerous collagens; they comprise one end of the mechanotransduction machinery/pathway (figure 1(A)). Focal adhesions are membrane-associated organelles that consist of several hundred different proteins that can be classified into four basic categories: (1) ECM-binding integrins, (2) signaling proteins such as paxillin and focal adhesion kinase (FAK), (3) force transduction proteins including talin and vinculin, and (4) actin regulatory proteins, such as VASP and Zyxin. Recent evidence suggests that on flat 2D cell culture surfaces, these protein groups are stratified or laminated and organized according to their functions (figures 1(A)–(C)) [5, 6].

Figure 1. Focal adhesions are mechanical signal hubs. (A) Schematic side view of integrins (orange) in the plasma membrane binding ECM and clustering together with intracellular focal adhesion proteins. The mechanosensitive proteins vinculin (blue) and talin (green) are shown interacting with actin monomers (light blue) that have assembled into stress fibers and are crosslinked with myosin II (red). Inset: focal adhesion proteins are organized in layers. (B) TIRF images of EGFP-paxillin in a fibroblast showing numerous nascent (NA: yellow arrowheads) and focal (FA: blue arrowheads) adhesions that have formed on a 2D ECM. (C) Paxillin (green), α5 integrin (red) and fibronectin (blue)-containing fibrillar adhesions (FX: white arrowheads). Fibroblasts were plated on matrigel and medium was supplemented with labeled soluble fibronectin to observe the process of fibronectin fibrillogenesis. (D) Fibroblast expressing EGFP-VASP (green) and mCherry-actin (magenta) on a fibronectin-coated 2D surface. Blue arrowheads indicate numerous focal adhesions associated with lamellipodia (LA). The kymograph at the right from the white-dashed region shows LA expansion followed by FA formation FAs, establishing the new leading edge. After FA elongation and maturation, a new LA can form. Time is in seconds. Scale bars: 10 μm.
Download figure:
Standard image High-resolution image2.1. Integrins: activation and outside-in/inside-out signaling
Integrins comprise a transmembrane receptor family consisting of 18α and 8β heterodimeric protein subunits that specify the ECM ligand and binding site, respectively, forming 24 different pairs [7, 8]. For example, β1 integrin binds preferentially to the arginine–glycine–aspartic acid (R–G–D) residues of ECM molecules, α1 and α2 subunits promote binding to collagens, and α5 specifies fibronectin binding. Blocking the ligand-binding site (via RGD blocking peptides or antibodies) inhibits ECM binding in a concentration-dependent manner and blocks cell adhesion and migration. The integrin extracellular domain binds ECM ligands (figure 2(A)). The β cytoplasmic tail region interacts only indirectly with actin. Several proteins within the focal adhesion, namely the talins, the tensins, and the kindlins, bind to the cytoplasmic tail of the β subunit and provide a physical link to other proteins, including actin. While there are numerous features of integrin functions, two important and possibly interrelated concepts are key to their role in mechanobiology: integrin activation and integrins acting as catch bonds. Because other proteins of the mechanotransduction machinery are also known catch bonds, we will discuss this concept later in this review.
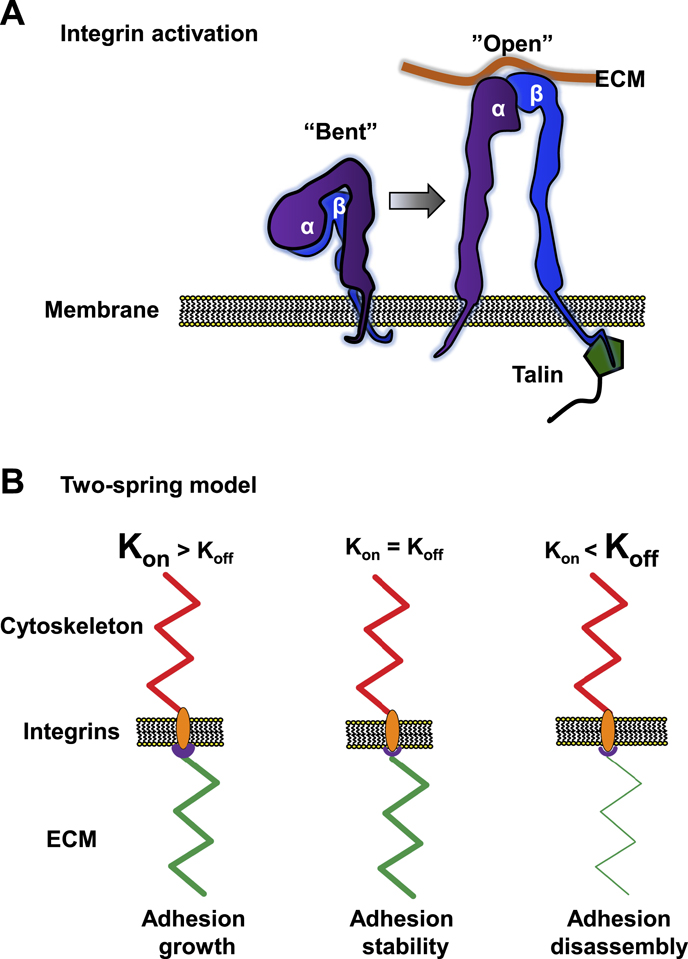
Figure 2. Integrin activation and the two-spring model. (A) Schematic representation of an α and β integrin heterodimer in the 'bent' and 'open' configuration. In the 'bent' configuration, the integrin pair has weak ECM-binding affinity. In the 'open' or activated configuration, both subunits extend away from the membrane and the transmembrane domains and cytoplasmic tails separate. Talin is shown binding to the β cytoplasmic tail, which may assist in the activation process. (B) Schematic representation of three scenarios in the two spring-model of cell adhesion, where the 'springs' represent the ECM (green) and the cytoskeleton (red) with integrin/focal adhesion in between. In the 1st condition, Kon (associated with high ligand binding affinity: purple region) is larger than Koff (associated with contractility). ECM stiffness matches the cytoskeletal 'spring,' leading to adhesion growth. In the 2nd scenario, both 'springs' (Kon and Koff rates) are equal, resulting in adhesion stability. The 3rd situation shows high Koff (contractility higher than the binding of integrins can withstand) together with a softer ECM 'spring,' which leads to focal adhesion disassembly.
Download figure:
Standard image High-resolution imageIntegrin activation is associated with changes in the conformation state of both the extracellular and intracellular domains of the integrin heterodimer [9]. Activated integrins have an open or extended extracellular domain conformation for binding ligand, accompanied by separation of the cytoplasmic tails of the α and β subunits, which is thought to promote integrin clustering, focal adhesion assembly, and signal transduction (figure 2) [8]. These conformational changes increase ligand-binding affinity and the force-bearing capability of an individual monomer, which together can alter integrin off rates and adhesion lifetime. While activation most often refers to extension of the β subunit, α5 integrin has an activation state associated with the binding of the synergy site to fibronectin that contributes to α5β1 load capacity [10].
Activation can also be induced by the intracellular binding of either kindlin or talin, known as inside-out activation. Their binding to the cytoplasmic domains is not solely responsible for activation but is thought to accelerate the process [8, 9]. Force may also be involved: once integrins extend/open and bind to ligand, the resulting talin/kindlin binding and subsequent actin association for force production stabilizes the ligand association. Because of this increased ECM binding affinity, integrin activation is likely a key regulatory point for mechanosensing.
2.2. Adhesion formation and maturation
Focal adhesions act as mechanosensory organs, sensing and adapting to changes in tension, e.g., intracellular contractility or stiffness of the ECM. The three main types of adhesion are (1) nascent adhesions, (2) focal adhesions, and (3) fibrillar adhesions, with the latter being involved in fibronectin fibrillogenesis (discussed elsewhere in this review: table 1 and figure 1). Current consensus is that focal adhesions initially form as force-independent nascent adhesions consisting of integrins, paxillin, vinculin, talin, FAK, Src, GIT, βPix and others (table 1) [11]. Nascent adhesions have short lifespans of seconds to 2 min. To mature to focal adhesions, they require the application of force. Such maturation involves increased numbers of proteins involved in actin polymerization, and its hallmark is growth of the adhesion to a size greater than 1 μm2. Adhesion growth also involves a local increase in the density of integrin cell–ECM receptors to reinforce the added forces from the cytoskeleton. Talin is integral to this maturation process, where actin binding helps to unfold this lengthy structural protein, exposing up to 4 and 11 binding sites for actin and vinculin, respectively [12]. This force-dependent unfolding is thought to structurally reinforce the integrin-talin complex [8]. Recent evidence suggests that two lesser-known adhesion components, KANK and CDK1, may bind directly to talin and regulate its ability to activate integrins and its mechanosensitivity, respectively [13, 14]. Without full-length talin, force transmission is weak, so talin is likely the key mechanosensitive protein required for focal adhesion growth and maturation [15].
Table 1. Characteristics of nascent, focal, and fibrillar adhesions.
| Adhesion type | Nascent adhesions | Focal adhesions | Fibrillar adhesions |
|---|---|---|---|
| Size and shape: | Submicron size; | ⩾1 μm; elongation | >1 μm size; elongation |
| elongation or growth; | with maturation; | over time; | |
| no axial ratio < 1.5:1 | no axial ratio < 1.5:1 | axial ratio > 7:1 | |
| Lifetime: | 30–120 s | >2 min to hours | >2 min to hours |
| Key proteins: | integrins, paxillin, | integrins, paxillin, | α5 integrin, |
| vinculin, talin, FAK, | vinculin, talin, FAK, | paxillin,vinculin, | |
| Src, GIT, βPix | Src, zyxin, VASP | talin, FAK, | |
| α-actinin, Rac, VASP | α-actinin | tensin, zyxin | |
| Cellular location: | Lamellipodia | Lamellipodial/lamellar border | Lamella to beneath |
| the cell | |||
| Key characteristic: | Rac dependent; | Highly dynamic; | Formed |
| No growth | maturation | dynamically | |
| Force dependence: | Force-independent | Force-dependent growth, | Force-dependent |
| formation | maturation, and disassembly | growth and maturation |
While force is integral for adhesion growth and stabilization—treatment of cells with contractility inhibitors such as Rho kinase inhibitor Y-27632 and the myosin II ATPase inhibitor blebbistatin inhibit growth, and only nascent adhesions are formed [16]—force is also responsible for focal adhesion disassembly; likely important is the rate of force loading to individual integrins [17, 18]. Currently, the two-spring model has gained traction as the primary model associated with mechanisms of force-dependent cell–ECM interactions; it has recently been strengthened both theoretically and experimentally [15, 17, 19, 20].
The two-spring model describes two Hookean springs in series, one representing the ECM, the other the cytoskeleton, with dynamic cell adhesion complexes (integrins) between them (figure 2(B)) [17, 20]. The cellular components of this model display variable on rate (Kon) and off rates (Koff), where Kon and Koff are highly dependent on integrin avidity and contractility, respectively, and regulate adhesion assembly and disassembly mechanisms. In essence, the model predicts that the softer of the two springs defines the threshold at which an adhesion reaches steady state (Kon and Koff are equal). For the adhesion to reach steady state, both springs must exert equal tension within the system: too little cytoskeletal force results in adhesions that do not stabilize on a rigid surface, and too much cytoskeletal force leads to adhesion disassembly and rapid protein dynamics on a soft surface. Because the soft spring sets the steady-state threshold for stability, it can vary—hence the system is meant to be dynamic and responsive to both internal and external changes. In other words, it is 'mechanoresponsive'. While initially modeled for 2D systems, there is evidence of a similar mechanism in 3D environments [19].
2.3. The molecular clutch: converting actin flow into extracellular force transmission
The molecular clutch hypothesis originally hypothesized by Mitchison and Kirschner [21] is involved in traction force propagation—the transmission of actomyosin contractility to the underlying/surrounding matrix through cellular adhesions—that is integral to migration. Application of traction force is posited to slow actin flow at the focal adhesion sites formed at the lamellipodia-lamella border, so that actin polymerization can proceed anteriorly to exceed the retrograde flow and thereby push against the cell membrane and extend the leading edge (figures 1(D) and 3) [1]. Hence, the general purpose of this 'clutch' is to promote migration. The hierarchical transmission of actin flow exists between the different core structural and signaling proteins within focal adhesions. Translocation of the actin-binding proteins talin, vinculin, and α-actinin correlate closely with actin flow, moving rapidly, whereas other signaling proteins (paxillin, FAK) and integrins move relatively slowly with respect to actin dynamics [22]. Chan and Odde [23] demonstrated that this 'motor-clutch' varies depending on substrate compliance and is associated with cellular traction forces through which cytoskeletal forces are transmitted to the substrate: stiff substrates lead to frictional slippage with few 'clutches' being engaged at any time, leading to high actin flow rates and low traction. Softer substrates show a load-fail regime with slower actin flow rates, higher tractions and slower speeds as cells grip-and-release repetitively, retracting newly formed adhesions behind the leading edge and reducing migration. These mechanisms are likely due to the system's rate constants, where the rate of tension development leads to an inverse relationship between actin flow and traction force (i.e., rapid tension development leads to lower traction force). Hence, clutch engagement is higher on softer surfaces. While this model does not explain how stiffer substrates promote higher traction force, others have demonstrated that the load-and-fail model does likely occur with oscillations of traction force in fibroblasts on 2D surfaces [24] and the pulling and retraction behavior of adhesions in soft collagen gels [19]. As for the effect on overall cell migration and cell persistence, it is difficult to assess events that vary greatly in time (seconds versus hours) and physical scale (micro versus macro). However, rapidly moving cells such as fish keratocytes, immune cells and Dictyostelium discoideum show lower traction and have relatively low actin flow rates [25].
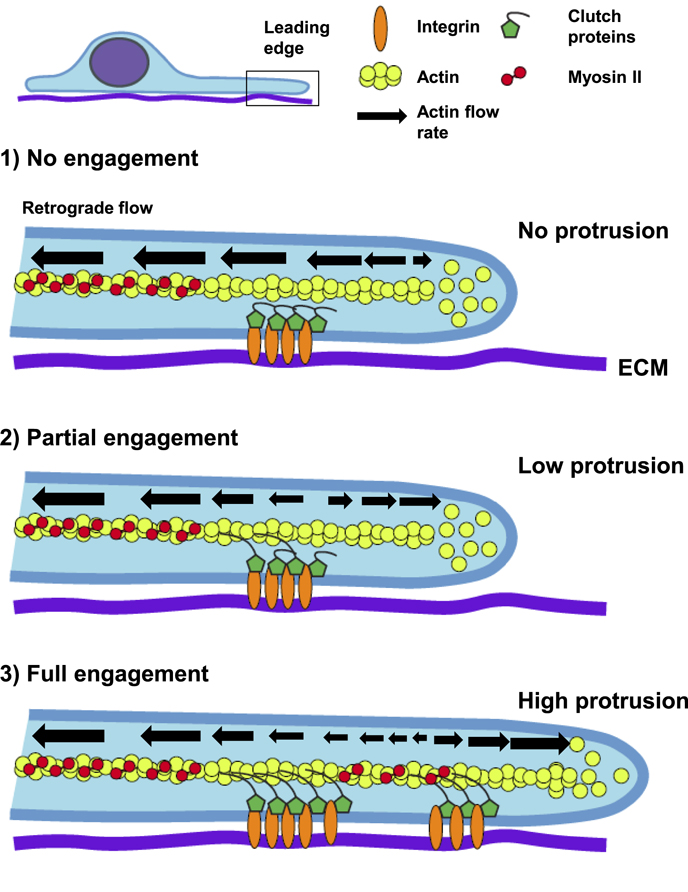
Figure 3. The molecular clutch. A side view of a cell on a 2D ECM (purple) showing the leading edge and crucial components of the molecular clutch. Three scenarios are shown. (1) In the absence of integrin-linked clutch proteins (green) engaging actin (yellow), the retrograde flow of F-actin associated with myosin-II contractility is high, greatly exceeding the rate of G-actin polymerization and no protrusion occurs (actin cannot push against the membrane). (2) With partial clutch engagement, the binding of clutch proteins to F-actin helps to slow retrograde flow at adhesion sites and allow the local actin polymerization rate to equal and then surpass rearward flow to weakly promote protrusion. (3) A fully engaged molecular clutch causes a significant reduction in local actin retrograde flow, promoting high leading-edge protrusion, advancing the lamella and forming a nascent adhesion in the lamellipodia.
Download figure:
Standard image High-resolution imageA key step in the regulation of this clutch machinery is the unfolding of the mechano-sensitive protein talin [15]. Without talin, little to no traction force is generated. Increased traction force likely occurs when additional vinculin and actin binding sites are exposed after talin unfolding. In addition, the force relationship is biphasic, such that a threshold level of force must be reached—otherwise integrins release, and adhesions do not mature and strengthen. This system can be influenced or regulated by not only substrate stiffness but by the relative amount of ECM ligand, which is also known to alter cell migration rates [26]. The type of ECM also affects the transmission of cytoskeletal force depending on the affinity of its bound integrin [27]. This affinity affects how much the molecular clutch will 'slip' with altered dynamics as shown after changing the ECM [27] or after changing the integrin that associates with a particular ECM [18].
2.4. Catch bonds: force-strengthening molecular bonds
Cell adhesion dynamics and the molecular clutch depend on catch bonds. Although most non-covalent protein–protein interactions are considered to be slip bonds, where increased pulling force decreases bond lifetime, integrins and several other proteins of the mechanotransduction machinery can form catch bonds. With catch bonds, increasing tension between two proteins increases bond lifetime. A simple example of a catch bond mechanism is the Chinese finger trap puzzle, where stronger pulling in opposite directions increases the tension within the trap. Numerous catch bonds exist in biology [28]. Recently, a simple tweezer-like design has simulated catch bond kinetics for a number of proteins including integrins and actin [29]. However, increasing tension beyond the loading capacity of a catch bond results in catastrophic rupture, e.g., bond failure of individual integrins [17]. Force versus bond-lifetime curves are biphasic, suggesting an optimal range for their biological function. There are likely at least four catch bonds involved in cell–ECM interactions: integrins, vinculin (now hypothesized to be a 'one-way' catch bond), actin, and myosin II [30, 31]. Interestingly, many processes, including focal adhesion formation and many aspects of cell migration, are biphasic and are likely due to the presence of these force-sensitive bonds.
Single-molecule experiments have shown that β1 integrin activation will affect catch bond strength or affinity as well as the absolute force the bond can withstand, indicating an interrelationship between these important functions. Integrin-ECM bond dynamics change as force increases in three regimes: (1) an initial low load rate (slow building of force) leads to spontaneous bond detachment before a substantial load occurs. (2) As the load rate increases, single bond forces increase and reduce the Koff rate below the Kon rate, leading to reinforcement by the addition of other integrins to increase their local density (integrin clustering). (3) If the loading continues to increase, Koff can elevate above Kon, resulting in the release of individual bonds before others can form, thereby decreasing force transmission [18]. Hence, integrins functioning as catch bonds in the molecular clutch can explain the growth, stabilization, and disassembly of focal adhesions.
3. ECM properties: a key modulator of focal adhesion regulation
As noted previously, the type of ECM can affect the integrins that bind and will likely change the dynamic interplay between focal adhesions and the environment. ECM ligand density also plays an important role in regulating migration speed: low levels of ECM reduce adhesion formation, affecting leading edge protrusion at the front, while high densities reduce cell retraction at the rear [32]. In both cases, migration rate is reduced with an optimum in between. This effect is due to adhesion dynamics and can be modified by altering the relative level of intracellular contractility [26].
The type of ECM can govern the type of cell-matrix adhesion. Fibrillar adhesions can arise from focal adhesions in association with the force-dependent process of fibrillogenesis of fibronectin [33]. Fibrillar adhesions contain the α5β1 integrin and tensin, often observed to extend toward the cell center with a highly elongated shape and likely lower force-transmission capabilities. Recent evidence suggests that the mechanism of fibril formation is highly dependent on the type of 2D ECM. Basement membrane components such as collagen IV and laminin promote robust fibronectin fibrillogenesis three-fold more than vitronectin. They induce fibrillar adhesions elongating behind focal adhesions that dynamically slide centripetally toward the cell center, leaving fibronectin fibers behind (figures 4(A) and (B)) [34]. This mechanism is associated with the switching of integrins from α2β1/α3β1 to α5β1 and uses a myosin IIA-based 'contractile winch' where stress fibers shorten and translocate focal adhesions and deposit fibronectin matrix towards the cell center, suggesting that the type of ECM, integrins, and myosin II contraction are intimately involved in fiber formation and ECM organization.
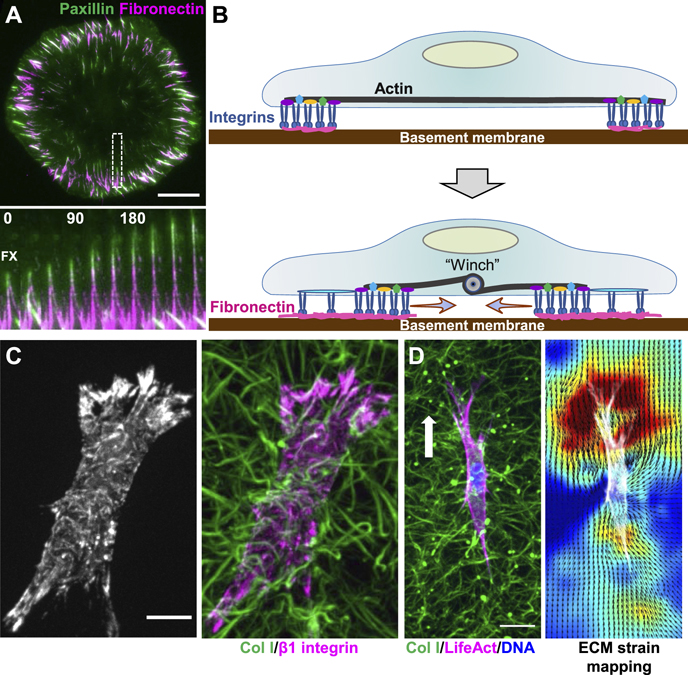
Figure 4. Fibrillar adhesion dynamics and 3D cell migration. (A) A fibroblast expressing EGFP-paxillin (green) pulls soluble fibronectin (magenta) into fibrils shortly after plating on Matrigel. Inset kymograph of the white-dashed box shows the dynamic movement of paxillin upward toward the cell center, polymerizing fibronectin fibrils behind it. Time is in min. (B) Schematic representation of how fibrillar adhesions can slide inwards using a contractile 'winch'. (C) A fibroblast stained for activated β1 integrin (left image; magenta in right) crawling through a 3D collagen hydrogel (green). (D) A fibroblast expressing TagGFP2-LifeAct crawling through a collagen gel. Right image shows the ECM strain map, where warmer colors depict higher strain at the leading edge. Arrow indicates migration direction. Scale bars: (A) and (C), 10 μm; (D) 20 μm.
Download figure:
Standard image High-resolution imageIn addition to ECM composition, its rheological properties can play a profound role in regulating adhesion dynamics and hence the associated clutch mechanism. Lo and Wang [35] established that fibroblasts can detect graded variations in substrate stiffness in a process termed durotaxis, preferring to migrate from soft to stiff substrata. Other cells, including numerous cancer cells, mesenchymal stem cells, and epithelial cell sheets, can undergo durotaxis. Cells within an epithelial sheet can work together to sense changes in ECM stiffness over long distances by utilizing the molecular clutch [36]. Numerous human cancer cells undergo durotaxis preferentially in very soft 2–7 kPa regions of a gradient 2D ECM in a mechanism requiring Arp 2/3 but not contractility [37]. In addition, cells can sense ECM rigidity using oscillatory forces at individual focal adhesion sites to 'test' the local stiffness, and in doing so guide cell migration [24].
Another intriguing property of the ECM is viscoelasticity. Currently, there is limited direct evidence concerning how ECM viscoelasticity alters the dynamics of the molecular clutch within focal adhesions. However, recent studies suggest that the rate of stress relaxation—a key aspect of viscoelastic materials—can affect adhesion formation in 3D hydrogels [38] and on 2D surfaces [2], 2D and 3D cell spreading [38, 39], traction stress [2], migration rate and persistence [2]. Indeed, ECMs that are dynamic and undergo fast stress relaxation, where the ECM/material rapidly responds to applied stress, promotes filopodia-based migration and 3D adhesion formation [39]. Viscoelasticity is likely present in many biological tissues with multiple complex effects on cells. For more in-depth discussions, please see reviews by Chaudhuri et al [40] and Elosegui-Artola [41].
3.1. ECM properties: 3D environments
While most studies use two-dimensional linear elastic or Hookean hydrogels coated with an adsorbed or conjugated ECM, the majority of biological in vivo 3D ECMs are nonlinear elastic in nature and can undergo strain-stiffening or strain-softening, where the gels either stiffen or soften with added strain. A recent intriguing concept applicable to both 2D and 3D ECMs is that stress relaxation of an ECM can alter adhesion dynamics. As a steady-state level of strain is applied to a material, the relative amount of stress decreases over time, which can be a fast or slow mechanism. Focal adhesions can respond to either fast or slow stress relaxation of hydrogels [2]. For example, higher traction forces can be sustained by fast gels and are associated with filopodia and not lamellipodia, resulting in altered cell migration. While the dynamics of cell–ECM adhesions under stress relaxation remains to be characterized, it is clear that the rheological properties of ECM can dictate the dynamics of cell–ECM adhesions.
Planar 2D ECMs have been a mainstay for biophysical analyses of cell–ECM interactions for many decades due to their ease of generation and repeatability. However, 3D matrices, such as collagen type I, laminin, and fibrin hydrogels, and cell-derived matrices (3D CDM) have emerged as the next relevant in vitro step toward directly studying cell–ECM dynamics in vivo. Other synthetic gels and biomaterials including electrospun fiber made of dextran methacrylate or polycaprolactone have also been used [42, 43], but for brevity we will not discuss these here. Biological hydrogels differ from 3D CDMs in several ways. Hydrogels are: (1) polymerizable, (2) sensitive to gelation conditions, (3) often composed of a single matrix protein, and (4) nonlinear elastic. In contrast, 3D CDMs are dependent on the cells that generate them (e.g., fibroblasts) and are composed of a developmental matrix with fibronectin, collagen I, and proteoglycans, and they are primarily linear elastic. In 3D laminin gels, some cancer cells alter their intracellular mechanics to mimic the stiffness of the surrounding ECM [44], suggesting that cells respond a 3D ECM similarly to a 2D pliable substrate.
Although thousands of publications have used 3D matrices (synthetic or biological) to characterize cell migration and adhesion components, relatively few have studied the dynamics of adhesions in these highly pliable environments, mainly due to the difficulties of imaging 3D volumes at depths of ⩾50 μm. Adhesion lifetimes are substantially longer in 3D CDMs than 2D focal adhesions [16, 19]. Adhesions in 3D collagen gels show a variable lifetime and zyxin turnover rate that is highly dependent on the local stiffness of ECM fibrils—stiffer fibrils promote longer lifetimes, while softer fibrils lead to adhesion disassembly through retraction [19]. In fibrin gels, cell adhesions undergo retrograde movement away from the leading edge with the rate of movement dependent on the protein, e.g., α-actinin moves faster than paxillin [45]. Together, these findings suggest that a similar molecular clutch mechanism is involved in 3D adhesion formation, stability, and disassembly.
Cells in 3D environments apply traction forces and strains to the ECM. Initially, forces were quantified using well-defined linear-elastic polyethylene glycol hydrogels containing both adhesive RGD and degradable proteolytic sites [46]. More recently, nonlinear elastic collagen and fibrin gels have demonstrated cell–ECM strain dynamics that are often cell type-dependent. MDA-MB-231 cells, a commonly used metastatic human breast cancer cell line, produce low ECM strains in 3D collagen. Differing reports describe (1) nearly equal-and-opposite strains [47], (2) high posterior strains [48], or high anterior strains [49]. These discrepancies may be due to different phases of the migration cycle or migratory persistence of individual cells. Overall, however, these cells cannot sustain constant ECM strain for extended periods. In contrast, mesenchymal fibroblasts generate anisotropic ECM strains nearly four-fold higher. These strains are two-fold higher anterior than posterior and are maintained during directional migration. Matrix prestrain in front of migratory cells is also exhibited by mesenchymal fibrosarcoma and melanoma cells [49, 50]. Formation and maintenance of this prestrain depends on levels of activated integrins and myosin IIA—both key regulators of adhesion dynamics.
The intracellular site of contractility in mesenchymal cells, based on 3D imaging of adhesion and cytoskeletal components, is at the anterior of the cell between leading edge and cell body, producing an adjacent pinch-like contraction of the matrix [45, 49]. We recently demonstrated that this anterior contraction is associated temporally with leading edge protrusion to establish a 3D migration cycle. We hypothesize that increasing contractility behind the leading edge stabilizes 3D adhesions and enhances molecular clutch engagement, permitting actin polymerization at the leading edge to exceed the slowing retrograde flow of actin to promote protrusion. Thus, concepts associated with 2D cell–ECM interactions also contribute to mechanosensing in more complex 3D microenvironments.
4. Cancer
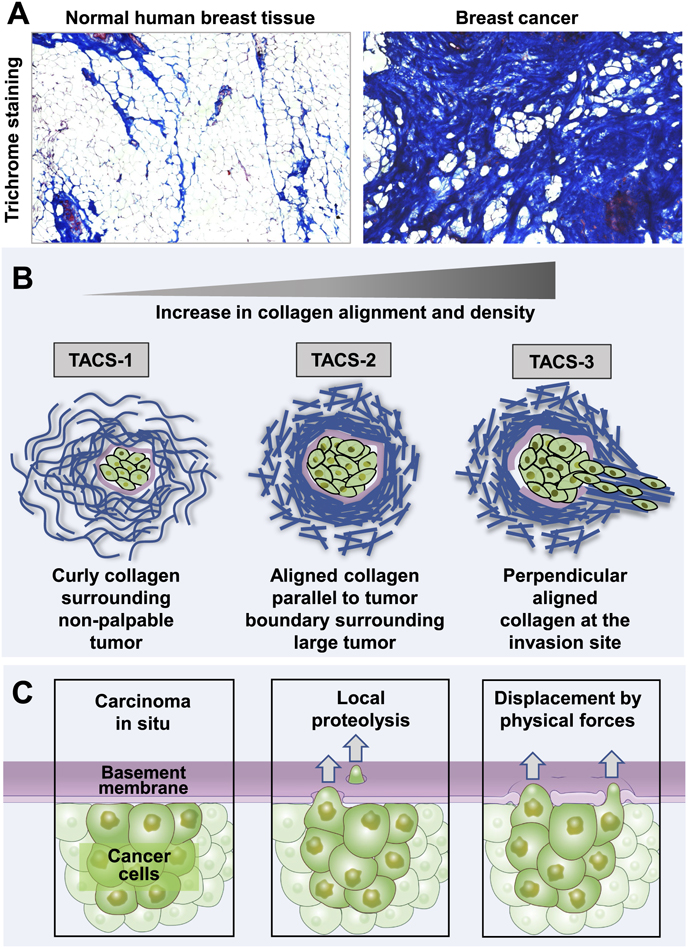
The type of cell can greatly influence the dynamic interplay at cell–ECM contact sites. A prime example is how cancer cells interact with and remodel their microenvironment. In the tumor microenvironment, early cancer cells remain confined by a sheetlike basement membrane and are often surrounded by a stromal ECM, which includes matrix proteins, cancer-associated fibroblasts (CAFs) and immune cells [51]. Cancer cells can interact with stromal cells by stimulating matrix component deposition or by locally crosslinking the ECM. Both result in an increase in matrix density and stiffness, a common attribute of the tumor microenvironment [52, 53]. Crosslinking is accomplished through lysyl oxidase (LOX) and transglutaminase 2. LOX is typically overexpressed in many cancers, which is often correlated with poor a prognosis. CAFs and carcinoma cells also deposit increasing amounts of glycoproteins, such as collagen I, fibronectin, laminin, osteopontin, and tenacin C in the tumor microenvironment, resulting in a dense ECM termed desmoplasia (figure 5(A)) [53, 54] Together, the increase in ECM stiffness and density alters cancer and stromal cell migration [37, 52, 53].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Matrix remodeling during tumor progression and mechanisms of cancer cell invasion. (A) Breast cancer progression correlates with higher collagen density demonstrated by trichrome blue staining to measure collagen density in normal compared to tumor tissue from the same patient. (B) Matrix remodeling closely correlates with tumor progression. Normal curly collagen fibrils surround a non-palpable tumor with localized collagen density around the periphery of the tumor mass (TACS-1). As the tumor enlarges, collagen is remodeled and appears more linear, dense, and aligned parallel to the tumor boundary (TACS-2). During cancer cell invasion, collagen becomes perpendicular to the tumor boundary at the invasion site (TACS-3). (C) During cancer invasion, cancer cells can breach the basement membrane barrier by chemically degrading the matrix using proteolysis or physically displacing the matrix by pushing through the basement membrane using invasive protrusions.
Download figure:
Standard image High-resolution image{kind=link}
The organization of the local matrix changes during tumor progression, especially in breast cancer. The normal architecture of collagen in breast tissue consists of wavy, flexible bundles, which permits tissue deformation, involution, expansion during ductal growth, and resistance to tension that can damage the tissue [55]. Keely and Provenzano identified a sequence of dynamic alterations in collagen architecture around mouse and human tumors (figure 5(B)). First is an increase in dense, wavy collagen in thin fiber bundles initially around tumors, termed tumor associated signature 1 (TACS-1) [55]. With tumor progression, the collagen fibers become stretched, elongated, and oriented parallel to the outer boundary of the tumor (TACS-2). While the wavy fibers serve to support the gland, the straightened collagen fibers may act to transmit mechanical signals across the basement membrane. Further cancer progression leads to TACS-3 as cancer cells locally invade out of the tumor with collagen fibers becoming oriented perpendicular to the tumor boundary. High TACS-3 signatures correlate with poor cancer survival [56].The cells and forces causing this alignment of collagen fibers perpendicular to cancer margins remain to be established definitively, but they could involve active ECM spatial remodeling by cancer associated fibroblasts (CAFs). Cancer cells may in turn use these collagen fibers for invasion [57]. In vitro experiments using aligned 3D-collagen matrix demonstrated that collagen alignment enhances the overall efficiency of migrating cells by keeping the cells oriented in a single direction [58]. In contrast, cell migration was impeded by matrix stiffness after increasing collagen density, perhaps due to the increase in friction caused by the additional ECM binding sites. Increased stiffness associated with collagen alignment, however, enhanced migration efficiency by promoting the directional persistence of cell migration [58].
The local density of collagen can also alter cellular responses. Experimentally increasing collagen density in vitro results in focal adhesion clustering at the cell–ECM interface [59]. FAK, vinculin, and paxillin are increasingly co-localized in high-density collagen gels, which can lead to rho activation, enhanced myosin light-chain mediated contractility, and further focal adhesion clustering and maturation [59]. When cultured in high collagen-density 3D gels, normal MCF10A mammary cells begin to express known epithelial-mesenchymal transition markers seen in cancer with more invasive cell morphologies [60]. Thus, ECM tissue density and collagen architecture may be important contributors to tumor progression.
4.1. ECM barriers to invasion
The majority of cancer-related deaths (∼90%) are caused by metastatic disease rather than the primary tumor [61]. For cancer cells to metastasize, they must first break through a basement membrane barrier and then migrate through the stromal ECM, both having pores narrower than the cells. Thus, to transmigrate and invade, cancer cells must navigate through both the basal/basement membrane and stromal matrices, through either localized proteolysis and/or physical forces (figure 5(C)).
The basement membrane is a thin, sheet-like network of proteins, composed of laminin, collagen IV, perlecan, nidogen, and proteoglycans. Laminin directly binds to cell surface receptors such as β1 integrin and dystroglycan, and it self-assembles into a dense sheet. Collagen IV then polymerizes to form a second covalently crosslinked network [62, 63]. Basement membrane is a nanoporous structure that restricts the diffusion of large molecules while being permeable to small molecules. The sizes of basement membrane pores vary depending on the tissue type, with average pore sizes ∼10–100 nm in diameter in different tissues [64, 65]. During the transition from carcinoma in situ (local cancer) to invasive carcinoma, cancer cells must enlarge basement membrane pores and migrate in the fibrillar ECM toward the circulatory or the lymphatic system. With the nucleus being the largest organelle of the cell (∼10 microns), successful invasion requires cells to widen substantially the nanometer-sized pores of the basement membrane to break through this barrier.
Once cancer cells have breached the basement membrane, they need to traverse tissues that often contain a fibrillar collagen matrix with pores that are roughly 2–10 μm in diameter in vivo [66]. CAFs can remodel ECM independent of proteolysis, through widening of pre-existing breaks or proteolytic perforations in the basement membrane [67]. Invasion of cancer cells does not depend on the stiffness of the matrix but instead on cellular contractility [67]. Interestingly, CAFs are also shown to act as leader cells, leaving 'microtracks' behind them for cancer cells to follow during invasion [53]. In contrast, another type of stromal cell in the tumor microenvironment, tumor-associated myoepithelial cells, can act as an additional barrier between the BM and cancer cells. In a 3D organoid model, myoepithelial cells act as a passive physical barrier to epithelial cells and can recapture escaping cells [68]. In addition to these barriers, a cell's geometric shape [69, 70] and the geometry of the surrounding ECM [71] can also affect the orientation of cell's leading edge and thus the direction of its migration, as demonstrated using micropatterning techniques [69–72].
4.2. Invasion by chemical degradation
Classically, cancer cell invasion has been thought to require the proteolytic degradation of both the basement membrane and stromal ECM. Both membrane-bound and secreted proteases have been implicated in basement membrane degradation and tumor cell invasion with matrix metalloproteinases (MMPs) being essential for each process [64, 73]. MMPs are zinc-dependent endopeptidases categorized into groups according to their substrate specificity; some are secreted, and others are membrane-bound (MT-MMPs) [74]. The two main post-transcriptional regulations of MMP activity are activation of the precursor form of the MMP and inhibition of the active MMP by tissue inhibitors of metalloproteinases (TIMPs) [75]. MMPs are synthesized as an inactive pro-enzyme through the interaction of a cystine-bound motif at the pro-peptide domain with a zinc ion at the catalytic site. MMPs are activated extracellularly by the removal of the pro-peptide domain [75, 76]. MMP expression is correlated with increased cancer invasion, metastasis, advanced tumor stage, and higher mortality. The expression of three membrane bound MMPs, MT1-MMP (MMP-14), MMP15 and MMP16, is especially important in tumor cell invasion of basement membrane matrix [77].
4.2.1. Invadopodia
Cancer cells can use specialized protrusions termed invadopodia to deliver localized proteases for ECM remodeling and degradation. Invadopodia are often long and slender, 2 μm long in vitro and 20 μm in some 3D environments [78]. Invadopodia differ from focal adhesions in their composition and organization, most notably by using matrix-degrading MMPs for local ECM degradation, including by MMP2, MMP9, and MMP14 (MT1-MMP) [76]. Besides MMPs, invadopodia typically express cortactin, Tks4, and Tks5, while being induced by oncogenes such as Src, as well as by highly dense fibrillar collagen in the tumor microenvironment [79, 80]. The MT1-MMP protease known to be present in invadopodia that chemically degrades the matrix can also bind to collagen fibers and initiate a signaling cascade leading to Tks5 recruitment and actin polymerization at the protrusion independent of its proteolytic capabilities, ultimately leading to pushing forces that can contribute to invasion [81].
4.3. Invasion using physical forces
Although initial invasion of cancer cells through the basement membrane barrier was thought to depend on proteolytic degradation through MMPs, emerging evidence has implicated other mechanisms in breaching of the basement membrane for cell invasion. Multiple clinical trials using MMP inhibitors have failed to reduce mortality [62, 64, 82]. This failure of anti-MMP clinical trials might have been due to insufficient or inadequately targeted drug concentrations due to side effects. Nevertheless, these findings raised the possibility that cells may be able to breach the basement membrane barrier through mechanisms independent of proteases during cancer progression. Additionally, although tumor or other cells can proteolytically remodel the stromal ECM, they may also require physical force to mechanically reorganize this matrix [64].
During cancer cell invasion, stromal cells in the tumor microenvironment—CAFs, macrophages, and myoepithelial cells—have been documented to exert force and remodel the basement membrane, opening pre-existing pores in the basement membrane, or leaving micro-tracks for cells to follow for cancer cell invasion [67, 83]. What remains unclear is whether force-driven breaching by the invading tumor cells themselves is equally important in cancer cell invasion and transmigration. In a Caenorhabditis elegans model of invasion, anchor cells use force generated by an actin network via the arp2/3 complex to deform and displace the basement membrane, resulting in accumulation of laterally displaced basement membrane in a ring around perforations in the absence of proteolysis [84]. Interestingly, invadopodia in C. elegans cells devoid of MMPs increase in size five-fold compared to normal invadopodia and are enriched in Arp2/3, ATP, and mitochondria [84]. Moreover, during salivary gland morphogenesis, epithelial cells require myosin II contractility in addition to proteases to perforate and remodel the basement membrane [85]. Although a recent paper demonstrated very similar MMP-induced perforations in the basement membrane required for early mouse embryonic development, the role of myosin II contractility remains to be explored [86]. These findings in normal developing embryos underscore the importance of ECM remodeling for both normal and malignant cells.
5. Conclusions and future directions
Recent advances in understanding the mechanisms and functions of cell–ECM dynamics have opened many opportunities for future research. Studies using 2D cell culture have revealed how focal adhesions link cells with their microenvironment through bidirectional mechanical communication, e.g., through integrin activation and the 'molecular clutch.' However, initial studies in 3D microenvironments identify differences, such as different modes of migration [87] and the importance of local cellular deformation of the ECM [49]. Unresolved questions include whether the molecular clutch is important for cell migration in all 3D microenvironments and whether specific roles and activation states of various integrin receptors and cell–ECM dynamics are altered in different cancers.
Although we now understand some mechanisms of mechanotransduction linked to ECM properties, signaling, and cellular contractility, there are probably many new modes of cell–ECM communication to be identified that will depend on both specific cell type and the physical/chemical nature of the local 3D microenvironment. Besides ECM density and stiffness, effects of viscoelasticity need further exploration at the focal adhesion level. Current computational models need much greater sophistication, not only by adding more newly identified molecular components, but also by incorporating their dynamics in terms of individual rate constants, feedback loops, and upstream genetic/epigenetic regulators. A particular challenge will be to generate accurate 3D models of dynamic cell–ECM interactions.
Rich opportunities for understanding disease processes in depth will appear as researchers unravel complex interactions underlying cell–ECM dynamics—not only in cancer, but in many genetic diseases, disorders and aging. Novel approaches to clinical therapy will arise with better understanding of mechanisms of pathogenesis. For example, the few small molecule and antibody reagents that target specific integrin-ECM binding interactions should expand as many new molecular targets are identified. The goal will be to develop potential drugs with greater specificity for practical, rather than just experimental, applications. Moreover, tissue engineering and regenerative medicine will depend on more deeply understanding cell–ECM dynamics, e.g., for establishing and maintaining robust stem cell environments and subsequent tissue homeostasis.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, NIDCR, Cell Biology Section (Z01 DE0005241, Z01 DE000719 and Z01 DE000718). We thank Jiaoyang Lu and Shaohe Wang for critical comments and the NIDCR Imaging Core (ZIC DE0007502) for support. The authors declare no conflicting interests.
Data availability statement
No new data were created or analysed in this study.