
Abstract
Production of vibrationally excited N2 (N2(v)) in atmospheric pressure nonthermal plasma and loss of N2(v) by gas-phase reactions and reactions on catalytic surfaces are analyzed to examine the role of N2(v) in NH3 formation by plasma catalysis. Vibrational state-to-state kinetic models complemented with molecular beam mass spectrometry (MBMS) measurements demonstrate that N2(v> 0) is produced with densities 100× greater than the density of N radicals by a radiofrequency atmospheric pressure plasma jet. The experimentally measured loss of N2(v) corresponds with a state-to-state kinetic model that describes loss of N2(v) by surface-mediated vibrational relaxation without consideration of reactions that convert N2(v) to NH3 over the catalyst surface. Rate constants for vibrational relaxation of N2(v) on catalyst surfaces exceed upper bounds on proposed rate constants for NH3 formation reactions from N2(v) over Fe when v < 9, Ni when v < 18, and Ag when v < 39, which indicates that only higher vibrational levels can possibly contribute to catalytic NH3 formation faster than they undergo vibrational relaxation on the surface. Densities of N2(v> 8), vibrational levels that can possibly react over Fe to form NH3 faster than they undergo vibrational relaxation, are less than or similar to N densities at the inlet of the catalyst bed and measured NH3 formation for the investigated conditions in this work, while densities of N2(v> 17) and N2(v> 38) are orders of magnitude below the N density at the inlet of the catalyst bed and the measured NH3 formation. The loss of N2(v) by vibrational relaxation on the surface limits the ability of N2(v) to contribute to catalytic NH3 formation and explains why N2(v) does not produce NH3 in quantities that are comparable to NH3 formation from N even though N2(v > 0) is more abundantly produced by the plasma.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Nonequilibrium plasma has the potential to enhance catalytic conversion of molecules with strong bonds through activation of these molecules in the plasma phase before reacting on the catalytic surface [1, 2]. In the context of NH3 formation from N2 and H2 by plasma catalysis, high-energy electrons can activate N2 and H2 by vibrational excitation, electronic excitation, bond dissociation, or ionization [3]. These activated species may increase rates of reaction on catalytic surfaces by lowering or removing activation barriers for surface reactions [4]. Of the N2-derived plasma-activated species, vibrationally excited N2 (N2(v)) has the lowest energy of formation, ∼0.3 eV per vibrational quantum, compared to a minimum of 6 eV for electronic excitation, 10 eV for dissociation, and 15.6 eV for ionization [5, 6]. With proper choice of plasma source and operating conditions, the majority of the plasma dissipated energy can be deposited into vibrational excitation [3]. As a result, the abundant production of N2(v) by nonequilibrium plasma followed by effective utilization of these species in catalytic surface reactions that form NH3 would result in the lowest energy cost for NH3 formation by plasma catalysis [7].
Mehta et al [8] reported that N2(v) can enhance rates of NH3 formation by orders of magnitude compared to N2 alone, based on microkinetic models that probe catalytic reactions for NH3 formation from N2(v) and H2, with the underlying assumption that N2(v) participates in dissociative adsorption reactions with a lowered activation barrier dependent on its vibrational energy. Lower barriers for NH3 formation in the presence of plasma have been experimentally observed by Rouwenhorst et al [9], where they measure NH3 formation at multiple gas temperatures in a dielectric barrier discharge (DBD) packed with promoted and unpromoted Ru/γ-Al2O3. They attribute the decreased activation energy to N2(v) having lower barriers for dissociative adsorption, although no direct measurements of the N2(v) flux or its correlation with the observed NH3 flux were reported. Plasma-derived species other than N2(v) also have the potential to enhance NH3 production from plasma catalysis. Engelmann et al [10] show through microkinetic models which consider N2(v), N, H, NH, and NH2 species densities predicted from a kinetic model of a packed bed DBD that catalytic reactions involving radical species produce NH3 at rates faster than N2(v) and H2 alone. Radical species may drive catalytic NH3 formation in DBDs because N2(v) production may be too small to contribute to NH3 formation. DBDs operate with reduced electric fields on the order of 100 Td [11], which results in a smaller fraction of energy deposition into vibrational excitation than plasma sources that operate with lower reduced electric fields, like microwave plasmas [12], non-self-sustained plasmas [13], and atmospheric pressure radiofrequency (RF) discharges [14]. Our previous experimental study of NH3 formation from plasma-derived species generated by an atmospheric pressure RF Ar/N2/H2 plasma jet over Fe, Ni, and Ag wools demonstrated that surface reactions involving N radicals are responsible for NH3 formation, even though estimates of N2(v) densities suggest that ∼30% of N2 is vibrationally excited and the total N2(v> 0) density exceeds the N density by two orders of magnitude [15]. A detailed analysis of N2(v) formation and loss pathways for different gas mixtures, temperatures, and catalytic surfaces is needed to determine the necessary conditions for N2(v) to contribute to NH3 formation by plasma catalysis.
The vibrational distribution function (VDF) of N2 is determined by rates of vibrational excitation of N2 by electron impact in the plasma, vibrational–vibrational (VV) energy transfer reactions in the gas phase, and processes that transfer vibrational energy away from N2(v) in the gas phase (VT) and on the surface. Richards et al [14] and Jiang et al [16] show through coherent anti-stokes Raman spectroscopy (CARS) measurements, molecular beam mass spectrometry (MBMS) measurements, and state-to-state kinetic modeling that the N2 VDF in the afterglow of RF-driven Ar/N2 plasma cannot be accurately described by a Treanor distribution, an analytical distribution often used to describe populations of N2(v) in plasma catalysis [8, 17]. These observations highlight the necessity of experimentally validated state-to-state kinetic modeling to determine the VDF of N2 beyond assumptions of Treanor-like distributions in order to accurately investigate the formation of N2(v) in plasmas and loss of N2(v) from gas-phase and surface processes.
In addition to dissociative adsorption reactions that are proposed to be responsible for NH3 formation from N2(v), N2(v) can quench in gas-phase collisions and in collisions with surfaces that do not contribute to NH3 formation. Through ab initio molecular dynamics, Kedalo et al [18] demonstrate that N2(v) adsorbed on Ru undergoes vibrational relaxation on sub-ns timescales, and as a result, only higher vibrational levels of N2(v) undergo dissociative adsorption faster than vibrational relaxation. Vibrational relaxation of N2(v) on surfaces has also been experimentally measured by Black et al [19] using Raman scattering and by Marinov et al [20] using quantum cascade laser absorption spectroscopy, where they show that N2(v= 1) has a relaxation probability of ∼10−3 for every collision with a surface, irrespective of whether the surface is SiO2, Al2O3, TiO2, stainless steel, Al, Cu, or Teflon. Vibrational relaxation also occurs in the gas phase through vibrational–translational (VT) collisions between N2(v) and other gas molecules. Richards et al [14] and Jiang et al [16] show that vibrational temperatures of N2(v), an indication of the relative populations of N2(v= 1) compared to N2(v= 0) [21], decrease in the afterglow of RF-driven Ar/N2 plasma over millisecond timescales due to VT relaxation. Rates of VT relaxation in the gas phase are dependent on the composition of the gas. Kinetic models of N2/H2 plasmas demonstrate that depopulation of higher levels of N2(v) becomes more pronounced as the H2 content in the plasma increases due to faster rates of VT reactions with H and H2 [22, 23]. These observations demonstrate that VT relaxation in the gas phase and vibrational relaxation on the surface may occur to an extent that precludes N2(v) from reacting on catalytic surfaces to form NH3. These reactions, along with surface reactions that contribute to NH3 formation, need to be considered to quantitatively and accurately assess the ability of N2(v) to react in NH3 formation reactions.
In this work, we investigate the production of N2(v) in an atmospheric pressure RF Ar/N2/H2 plasma jet and loss pathways of N2(v) over catalytic surfaces using a combination of MBMS measurements and state-to-state kinetic modeling to answer the following questions:
- 1.What is the N2 VDF, and what parameters determine the shape of the VDF?
- 2.How does N2(v) react in the gas phase and on catalytic surfaces?
- 3.Why does N2(v) not measurably contribute to NH3 formation for the investigated experimental operating conditions?
- 4.What conditions (dis)favor NH3 formation from N2(v) by plasma catalysis?
We show that the total N2(v> 0) density exceeds the N density at the inlet of the catalyst bed by 100×. The total N2(v) density is higher when the inlet concentration of N2 in the jet is increased, and increasing the inlet concentration of H2 in the jet depopulates densities of N2(v> 8) due to VT relaxation. Vibrational relaxation of N2(v) on the surface is the dominant loss process of N2(v) in the packed bed for the investigated experimental conditions, and no measurable NH3 formation from N2(v) is detected in packed beds of Fe, Ni, and Ag. A comparison between rates for vibrational relaxation of N2(v) on the surface with upper bounds on DFT-based rates for dissociative adsorption of N2(v) [8] shows that only N2(v> 8) (which makes up less than 2% of N2(v> 0)) can possibly undergo dissociative adsorption faster than vibrational relaxation, which limits the ability of N2(v) to contribute to NH3 formation by plasma catalysis.
2. Methods
2.1. Plasma source and reactor setup
An RF-driven atmospheric pressure plasma jet (inner diameter = 2 mm, outer diameter = 3 mm) coupled with a packed bed of metal wools downstream of the plasma was used in this study. The plasma jet was driven at 12.8 MHz with two duty cycle modulations, one at 20 kHz with a 50% duty cycle and one at 50 Hz with a 50% duty cycle to allow for species densities background subtraction in the MBMS. The volumetric gas flow rate was kept at 16.67 cm3 s−1, referenced to standard temperature and pressure (1 bar, 298 K) throughout this study. The Ar/N2/H2 flow was passed through an O2/H2O trap (Agilent, OT3-4) to remove impurities before entering the plasma jet. More details about the setup and operation of the jet are described in prior work [15].
The reactor setup is displayed schematically in figure 1. The distance from the end of the cylindrical ground electrode to the nozzle (end of the quartz tube) was varied from 5 to 15 mm. The sampling orifice of the MBMS was placed adjacent (<0.2 mm) to the nozzle to sample the gas composition at the effluent of the reactor. When catalyst was used, the distance between the end of the ground electrode and the catalyst bed was kept constant at 5 mm, such that MBMS measurements made 5 mm from the ground to the nozzle are representative of species densities at the inlet of the catalyst bed. The length of the catalyst bed was varied from 2–10 mm with 0.0024 g mm−1 Fe, 0.0036 g mm−1 Ni, or 0.0033 g mm−1 Ag wool loaded as catalyst.

Figure 1. Experimental setups with and without catalyst present. The ground electrode extends from 0 to 3 cm. Catalyst, when present, was always placed at 3.5 cm (0.5 cm from the ground electrode). The position of the end of the quartz tube was varied from 3.5 cm to 4.5 cm (0.5–1.5 cm after the ground electrode).
Download figure:
Standard image High-resolution image2.2. Materials
Fe (99.5% purity by EDS), Ni (99.6% purity by EDS), and Ag (97.1% purity by EDS) metal wools (EA consumables) are used as catalysts. Scanning electron microscopy was used to determine that the diameter of the metal wool fibers was ∼70 μm (as displayed in our prior work) [15]. Surface areas of the fibers were calculated based on the assumption that fibers were cylinders with infinite length.
2.3. MBMS methods
The experimental MBMS setup and methods for detection, background subtraction, ionizer electron energy calibration, and determination of response factors for N, H, and NH3 are described in detail in previous work [15]. MBMS signal was quantified when the signal reached steady-state within the 50 Hz modulation cycle, and densities of species were not observed to change over longer (seconds-minutes) timescales with or without the 50 Hz modulation. Error bars associated with reported species densities or MBMS signals represent 95% confidence intervals that arise from the acquisition of MBMS signal.
Densities of N2(v) predicted by kinetic models are validated by the procedure outlined by Jiang et al [16] and are summarized below. MS signal is measured for m/z = 28 (N2) for MS ionizer energies between 12.1 and 15.6 eV, values at or below the threshold energy for ground-state N2 ionization. Background-subtracted signals  were normalized to the signal measured at 15.6 eV for each operating condition as described in equation (1) to yield the normalized signal at the given electron energy (
were normalized to the signal measured at 15.6 eV for each operating condition as described in equation (1) to yield the normalized signal at the given electron energy ( ):
):
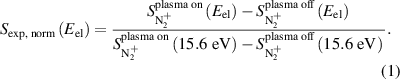
Background subtraction was enabled by modulating the plasma at a frequency of 50 Hz, which allowed for the measurement of the plasma-on signal and the background signal with a multichannel scaler in a single measurement. Experimentally measured signal is compared to densities of individual vibrational levels predicted by the state-to-state kinetic model, based on the proportionality of mass spectrometer signal to the summation of ionization cross sections of individual vibrational levels at the given electron energies ( ) multiplied by the density of the corresponding individual vibrational level
) multiplied by the density of the corresponding individual vibrational level  as shown in equation (2):
as shown in equation (2):
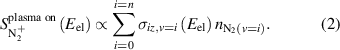
Ionization cross sections are obtained from NIST [24]. Ionization cross sections for N2(v) are assumed to be the same as ionization cross sections for ground state N2 with the electron energy shifted by the vibrational energy as described in previous work [15]. Ionization cross sections were corrected to account for the finite electron energy width of the ionizer as described previously [15]. It has been shown that electronically excited N2 does not contribute to the plasma-on signal for similar operating conditions as those investigated in this work [16]. For the case of the plasma-off signal, only ground state nitrogen contributes to the signal, as shown in equation (3):
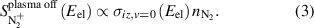
Equations (2) and (3) can be combined and normalized to relate normalized, background subtracted signal with the densities of N2(v) in the sampled gas, as shown in equation (4):
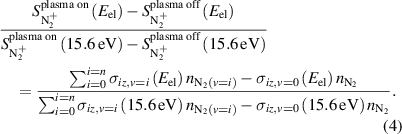
Normalized, background subtracted mass spectrometer signal can be predicted at a given ionizer electron energy using the obtained densities of N2(v) from the state-to-state kinetic model as shown in equation (5) to obtain a kinetic-model predicted MS signal ( ):
):
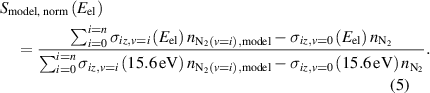
The experimental signal is normalized per equation (1) and compared to the signal predicted by the state-to-state kinetic model in equation (5) to assess the accuracy of the VDF predicted by the kinetic model.
2.4. Gas-phase kinetic modeling
The N2 VDF for different experimental conditions is determined with a quasi-zero-dimensional model that simultaneously solves the electron energy equation, the heavy species (e.g. Ar, N2, H2, N, H, etc) energy equation, and the equations for individual species balances using the ANSYS Chemkin-Pro software. The model incorporates electron impact reactions [5, 6], including vibrational excitation, electronic excitation, dissociation, and ionization; state-specific vibration–vibration (VV) energy transfer for N2–N2 [25] and vibration–translation (VT) relaxation for N2–Ar [26, 27], N2–N2 [26, 27], N2–N [28, 29], N2–H2 [30], and N2–H [30]; chemical reactions among radical species [30–32]; energy transfer involving excited electronic states of Ar and N2 [30, 32–39]; and ion recombination reactions [30, 32, 37, 40] to describe the evolution of species densities and the VDF. Rates for electron impact reactions at different electron temperatures were determined by solving the Boltzmann equation using Bolsig for the inlet composition of the plasma jet [41]. Functional forms of the rate constants used in the state-to-state kinetic model are described in the supporting information in section S1.
Collisional processes between N2(v) and NHx species were not considered in this model, since these species densities are orders of magnitude lower than the species that most contribute to VT relaxation, and their VT rate coefficients are not expected to be larger than the VT rate coefficients of other species by orders of magnitude (see section 3.1). Vibrational excitation of H2 was not considered in the kinetic model because its inclusion is not expected to significantly alter N2(v) and H densities. H2(v) densities are expected to be low, since rates of VT relaxation of H2(v) by H are >1000× faster than VV energy transfer from N2 to H2 due to the differences in their single vibrational quantum energies [30, 42]. Because of the fast VT relaxation of H2(v), vibrationally enhanced H2 dissociation is estimated to be negligible in comparison to dissociation of H2 by electron impact or dissociation of H2 from quenching of excited states of N2 or Ar [36, 43, 44]. Rate constants for VV energy transfer from N2 to H2 are lower than VV energy transfer from H2 to N2 because H2 has a higher single vibrational quantum energy than N2 [22]. As a result, VV energy transfer tends to depopulate H2(v) and populate N2(v), so it is not expected that VV energy transfer from N2 to H2 will depopulate N2(v) more than VT relaxation.
Inputs into the model include the inlet gas composition, inlet gas and electron temperature, flow rate, reactor geometry, radial heat transfer coefficient, and power deposition profile (displayed in section S2 of the supporting information). The plasma volume is considered to be the volume between the needle electrode and the ground electrode plus the region of emission beyond the electrodes. The total power deposited into the plasma is equal to the experimentally measured power. The volumetric power density input from the beginning of the plasma to the end of the ground electrode is constant, and then drops linearly from the end of the ground electrode to the end of the visible emission of the plasma jet (figure S1(a)) [16]. The power deposition profile in the model neglects the 20 kHz modulation cycle used in experiments but operates with the same average power as in experiments, since models of RF atmospheric pressure plasma jets demonstrate that the 20 kHz modulation cycle does not significantly alter species densities measured experimentally or predicted by the model but does significantly increase the computation time [45].
The output of the model includes species densities, gas temperature, and electron temperature as a function of the gas residence time or axial position. These outputs are displayed in the supporting information (section S2). A small but finite power deposition in the afterglow is necessary to enable Chemkin to solve the electron energy equation in the afterglow. Simulations that compare species densities and temperature outputs with increasing power deposition in the afterglow show that these outputs do not significantly change as the power deposition in the afterglow varies, indicating that the power deposition does not significantly impact the results and conclusions of this study (figure S3). The heat transfer coefficient is estimated to match the gas temperature predicted by the model with the experimentally measured gas temperature at the 3.5 cm axial position, although simulations with different heat transfer coefficients show that the variation of this parameter does not significantly alter species densities predicted by the model (figure S4).
2.5. Modeling of loss of N2(v) by surface reactions
Loss pathways for N2(v) in the catalyst bed were analyzed through inclusion of surface-mediated vibrational relaxation into a subset of the previously described gas-phase reaction set that considers N2 VV and VT processes. Inlet N2(v) densities were taken from the plasma kinetic model at the axial position of the start of the catalyst bed and densities of other species were taken from MBMS measurements. The gas temperature was determined from thermocouple measurements with continuous operation of the plasma, and the surface temperature was estimated to be the average gas temperature when a 50 Hz modulation cycle is included, as described in previous work [15]. Decay of N and H in the catalyst bed was fitted to an exponential function that passes through the densities of N and H at the inlet and the detection limits of these species (1.5 × 1013 cm−3 and 5 × 1014 cm−3, respectively) at 2 mm of catalyst bed, although this assumption does not significantly influence the N2 VDF because neither of these species are the dominant quenchers of N2(v) at the investigated operating conditions (see section 3.1).
Vibrational relaxation on the surface is modeled according to the approach of Marinov et al [20], which uses collision theory to describe collisions between N2(v) and the surface and loss coefficients ( ) to express the probability of vibrational relaxation when N2(v) collides with the surface. Loss coefficients are assumed to scale linearly with vibrational level (v). The rationale behind this assumption is discussed by Marinov et al [20] We use a value of
) to express the probability of vibrational relaxation when N2(v) collides with the surface. Loss coefficients are assumed to scale linearly with vibrational level (v). The rationale behind this assumption is discussed by Marinov et al [20] We use a value of  ≈ 0.001–0.0026 as measured by Black et al [19] over stainless steel and Cu. Equation (6) displays the rate constant (
≈ 0.001–0.0026 as measured by Black et al [19] over stainless steel and Cu. Equation (6) displays the rate constant ( ) for vibrational relaxation of N2(v) on a surface, which is the thermal flux to the surface (
) for vibrational relaxation of N2(v) on a surface, which is the thermal flux to the surface ( ) multiplied by the loss probability:
) multiplied by the loss probability:
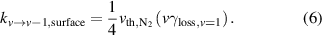
The rate of N2(v) loss by surface-mediated vibrational relaxation ( ) is written as shown in equation (7):
) is written as shown in equation (7):
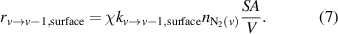
 represents the surface area of the fibers per volume of reactor, and
represents the surface area of the fibers per volume of reactor, and  is the number density of vibrationally excited N2 of level v. The
is the number density of vibrationally excited N2 of level v. The  term in equation (7) captures the effect that external mass transfer has on rates of quenching [46].
term in equation (7) captures the effect that external mass transfer has on rates of quenching [46].  is defined in equation (8), with the mass transfer coefficient for N2 (
is defined in equation (8), with the mass transfer coefficient for N2 ( ) calculated by methods described in previous work [15]:
) calculated by methods described in previous work [15]:

NH3 is proposed to be formed from N2(v) by dissociative adsorption of N2(v) over transition metal surfaces to form N adatoms, which then undergo hydrogenation and desorption to form NH3 [8]. Rate constants for dissociative adsorption of N2(v) are calculated according to the approach used by Mehta et al [8] The rate constant for dissociative adsorption consists of the pre-exponential factor (A) multiplied by an exponential factor that accounts for an activation barrier modified by the vibrational energy of N2(v), which is shown in equation (9):
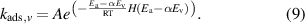
The exponential factor changes as a function of vibrational level through subtraction of the activation energy for ground-state N2 dissociative adsorption (Ea) by the vibrational energy (Ev) of the corresponding vibrational level multiplied by a Fridman–Macheret parameter (α). DFT-derived values for N2(v) dissociative adsorption activation energies are computed using the scaling relations reported by Mehta et al [8] using dissociative adsorption energies for N2 on terrace sites reported in the CatApp database [47], since terrace sites should be the most abundant sites on the metal wools with 70 μm diameter used in experiments. The Fridman–Macheret parameter, α, describes how effectively vibrational energy can be used to overcome activation barriers.  is defined as the activation barrier for the forward reaction and
is defined as the activation barrier for the forward reaction and  is the activation barrier for the reverse reaction, which together define α in equation (10):
is the activation barrier for the reverse reaction, which together define α in equation (10):

When  exceeds
exceeds  , the exponent is set to zero through use of a Heaviside function (H). Equation (11) describes the rate of N2(v) loss to dissociative adsorption (
, the exponent is set to zero through use of a Heaviside function (H). Equation (11) describes the rate of N2(v) loss to dissociative adsorption ( ), with
), with  taking the same form as in equation (8) with
taking the same form as in equation (8) with  replaced by
replaced by  :
:
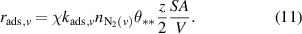
In equation (11), the  term accounts for coordination effects associated with N2 adsorption on a surface. It is assumed that z = 6, which corresponds to dimer adsorption on the (111) plane of a face-centered cubic lattice. Assumptions about the values for the pre-exponential factor (A) and
term accounts for coordination effects associated with N2 adsorption on a surface. It is assumed that z = 6, which corresponds to dimer adsorption on the (111) plane of a face-centered cubic lattice. Assumptions about the values for the pre-exponential factor (A) and  (fractional coverage of sites available for dimer adsorption) will be described in more detail in section 3.3. Values of
(fractional coverage of sites available for dimer adsorption) will be described in more detail in section 3.3. Values of  ,
,  ,
,  , and
, and  used to determine reaction rates for the different metals are reported in table S.7.
used to determine reaction rates for the different metals are reported in table S.7.
3. Results and discussion
3.1. Production of N2(v) by the plasma jet
N2 VDFs predicted by the state-to-state gas-phase kinetic model for different inlet compositions at the 3.5 cm axial position, which corresponds to the location of the front of the catalyst bed when a catalyst bed is included in experiments, are displayed in figure 2(a). Over 40% of N2 is vibrationally excited for all of the investigated operating conditions. The VDFs differ from Treanor or Boltzmann distributions (figure S5), which highlights the necessity of kinetic modeling to accurately determine the VDF and investigate N2(v)-enabled reactions. A comparison between the normalized signal measured by MBMS with the kinetic model-predicted mass spectrometer signal at different electron energies is displayed in figure 2(b) for the different process conditions. The mass spectrometer signal predicted by the state-to-state kinetic model (equation (5)) displays quantitative agreement with the experimentally measured signal (equation (1)), showing that the VDFs predicted by the kinetic model are representative of the distribution of N2(v) available to react in the catalyst bed.

Figure 2. (a) N2(v) densities predicted by the state-to-state kinetic model at the 3.5 cm axial position. (b) Correspondence between experimentally measured MBMS signal with kinetic model-predicted MS signal. (c) Experimentally measured N and H densities at the 3.5 cm axial position. 0.5% N2 and 0.5% H2 in Ar, 5% N2 and 0.5% H2 in Ar, or 0.5% N2 and 2.5% H2 in Ar. 16.67 cm3 s−1, 2.1 W average power, 12.8 MHz, 20 kHz modulation with 50% duty cycle, 50 Hz modulation with 50% duty cycle.
Download figure:
Standard image High-resolution imageFigures 2(c) and (d) display experimentally measured N and H densities at the same axial position as the N2(v) measurements for comparison. Figure S2 compares experimentally measured values for N, H, and NH3 densities with those predicted by the state-to-state kinetic model. The kinetic model predicts H densities accurately within ∼2×, overpredicts N densities by 2.4–6.4×, and underpredicts NH3 densities by around an order of magnitude. Possible explanations for these discrepancies are discussed in the supporting information in section S2, and the implications that these discrepancies have on N2(v) densities predicted by the kinetic model are discussed below.
A comparison between N2(v), N, and H densities in figure 2 shows that the combined N2(v> 0) densities are at least 10× greater than H densities and 100× greater than N densities, with densities of N2(v) becoming comparable to N densities around v = 9, 12, and 8 for 0.5% N2 and 0.5% H2 in Ar; 5% N2 and 0.5% H2 in Ar; and 0.5% N2 and 2.5% H2 in Ar conditions, respectively. As N2 concentration is increased from 0.5% to 5% with a constant 0.5% H2 concentration, the corresponding densities of N2(v) increase by around a factor of ten, while the N density only increases by a factor of two. As H2 concentration is increased from 0.5% to 2.5% with a constant 0.5% N2 concentration, the densities of N2(v= 0−5) vary by less than 10% but densities of N2(v> 8) are at least an order of magnitude lower because the higher H and H2 densities result in increased rates of VT relaxation for the higher vibrational levels [30]. The H density increases by a factor of 3.5 as the H2 concentration is increased from 0.5% to 2.5% at constant 0.5% N2 because more H2 is available to react in gas-phase dissociation processes. N2(A) densities predicted by the kinetic model at the inlet of the catalyst bed are ∼1011 cm−3, which is orders of magnitude lower than N2(v> 0) densities and the N densities at the inlet of the catalyst bed, and is orders of magnitude lower than the observed NH3 formation in the catalyst bed, even though the densities of N2(A) reported by the model in the afterglow may be overestimated because of afterglow power deposition required by Chemkin.
To determine how inaccuracies in N, H, and NH3 densities predicted by the state-to-state kinetic model may influence the VDF due to changes in VT rates, a sensitivity analysis was performed in which this model was run with VT rate constants for N, H, or H2 changed to be the same as VT rate constants for Ar (or N2, as it is assumed in the model that  =
=  [14]), since Ar is the species with the lowest VT rate constants. These results are displayed in figure 3 and show that the inclusion of N2(v) VT relaxation by H2 with the VT rate constants for N and H the same as Ar produces a VDF that is closest to the VDF produced with the inclusion of the species-specific VT rate constants for all species. VT relaxation of N2(v) by N or H contributes comparatively less than VT relaxation of N2(v) by H2 to the VDF. The fact that H2 has the strongest influence on the VDF explains why the kinetic model can accurately predict the VDF even if there are discrepancies between N, H, and NH3 densities that are experimentally measured and predicted by the model. It also affirms the observation that increasing the H2 density in the discharge results in a loss of N2(v) densities in the tail of the distribution. This is detrimental to NH3 formation from N2(v), since high levels of N2(v) are proposed to be the most reactive for dissociative adsorption, and thus NH3 formation, over catalytic surfaces [8].
[14]), since Ar is the species with the lowest VT rate constants. These results are displayed in figure 3 and show that the inclusion of N2(v) VT relaxation by H2 with the VT rate constants for N and H the same as Ar produces a VDF that is closest to the VDF produced with the inclusion of the species-specific VT rate constants for all species. VT relaxation of N2(v) by N or H contributes comparatively less than VT relaxation of N2(v) by H2 to the VDF. The fact that H2 has the strongest influence on the VDF explains why the kinetic model can accurately predict the VDF even if there are discrepancies between N, H, and NH3 densities that are experimentally measured and predicted by the model. It also affirms the observation that increasing the H2 density in the discharge results in a loss of N2(v) densities in the tail of the distribution. This is detrimental to NH3 formation from N2(v), since high levels of N2(v) are proposed to be the most reactive for dissociative adsorption, and thus NH3 formation, over catalytic surfaces [8].

Figure 3. (a) VDFs produced by the kinetic model at 3.5 cm when rate constants for VT reactions involving N, H, and H2 are altered. (b) Comparison of experimentally measured MBMS signal with kinetic model-predicted MS signal with altered rate constants. 0.5% N2 and 0.5% H2 in Ar, 16.67 cm3 s−1, 2.1 W average power, 12.8 MHz, 20 kHz modulation with 50% duty cycle, 50 Hz modulation with 50% duty cycle.
Download figure:
Standard image High-resolution image3.2. Experimentally observed loss of N2(v) in packed beds of catalyst and comparison with NH3 formation
MBMS measurements and state-to-state kinetic modeling simulations were performed to investigate consumption pathways of N2(v) in the packed bed. Figure 4(a) displays densities of N2(v) determined by the kinetic model at 35 and 45 mm (locations of inlet and outlet of catalyst bed when present) with an empty tube (only gas-phase reactions considered). Small changes in N2(v) densities from 3.5 to 4.5 cm are predicted by the model due to VT relaxation in the gas phase, which is quantitatively consistent with MBMS measurements that show small changes in the N2(v) signal at 14.6 and 15.1 eV in figure 4(b).

Download figure:
Standard image High-resolution image
Figure 4. N2(v) densities at the inlet of a packed bed (3.5 cm axial position), 2 mm of packed bed, and 10 mm of packed bed with (a) no catalyst, (c) 0.0024 g Fe/mm, (e) 0.0036 g Ni/mm, or (g) 0.0033 g Ag/mm predicted by the state-to-state kinetic model. Comparison between experimentally measured MBMS signal with kinetic model-predicted MS signal at the inlet of a packed bed (5 mm from ground), 2 mm of packed bed, and 10 mm of packed bed with (b) no catalyst, (d) 0.0024 g Fe/mm, (f) 0.0036 g Ni/mm, or (h) 0.0033 g Ag/mm. 0.5% N2 and 0.5% H2 in Ar, 16.67 cm3 s−1, 2.1 W average power, 12.8 MHz, 20 kHz modulation with 50% duty cycle, 50 Hz modulation with 50% duty cycle. Error bars in figures 4(c), (e), and (g) and the shaded regions in figures (d), (f), and (h) represent uncertainty in the value of  = 0.0018 ± .0008 reported in literature [19] that are used in the kinetic model.
= 0.0018 ± .0008 reported in literature [19] that are used in the kinetic model.
Download figure:
Standard image High-resolution imageVibrational relaxation of N2(v) on the catalyst surface is included in the reaction set of the state-to-state kinetic model with  = 0.0018 ± 0.0008, as described in section 2.5, to model loss of N2(v) in the packed bed of catalyst. Figures 4(c), (e) and (g) display densities of N2(v) determined by the kinetic model after 2 and 10 mm of the Fe, Ni, or Ag catalyst bed with the inclusion of vibrational relaxation on the surface in the reaction set. Kinetic model-predicted MS signals are in quantitative agreement with loss of MBMS signal as displayed in figures 4(d), (f) and (h). These results show that quenching of N2(v) on the surface can account for the observed decrease in N2(v) MBMS signal when catalyst is present without consideration of reactions that consume N2(v) to form NH3.
= 0.0018 ± 0.0008, as described in section 2.5, to model loss of N2(v) in the packed bed of catalyst. Figures 4(c), (e) and (g) display densities of N2(v) determined by the kinetic model after 2 and 10 mm of the Fe, Ni, or Ag catalyst bed with the inclusion of vibrational relaxation on the surface in the reaction set. Kinetic model-predicted MS signals are in quantitative agreement with loss of MBMS signal as displayed in figures 4(d), (f) and (h). These results show that quenching of N2(v) on the surface can account for the observed decrease in N2(v) MBMS signal when catalyst is present without consideration of reactions that consume N2(v) to form NH3.
3.3. Possible NH3 formation from N2(v)
NH3 is proposed to be formed from N2(v) through dissociative adsorption on the catalyst surface. The rate of this reaction is described by equations (9)–(11). N2(v) could contribute to NH3 formation when it can undergo dissociative adsorption faster than vibrational relaxation on the surface, which occurs when  . Ignoring mass transfer effects, this criterion is expressed by equation (12):
. Ignoring mass transfer effects, this criterion is expressed by equation (12):
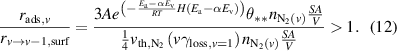
To determine which vibrational levels have  , values for A and
, values for A and  must be known. A can be determined using transition state theory with knowledge of the transition state entropy of the adsorption process, and the determination of
must be known. A can be determined using transition state theory with knowledge of the transition state entropy of the adsorption process, and the determination of  requires microkinetic modeling to determine steady-state coverages of adsorbates. An upper bound on
requires microkinetic modeling to determine steady-state coverages of adsorbates. An upper bound on  for vibrational level v,
for vibrational level v,  , can be defined by setting
, can be defined by setting  , which is an upper bound on the pre-exponential factor and assumes that the adsorbate retains two degrees of translational freedom on the catalytic surface, and by setting
, which is an upper bound on the pre-exponential factor and assumes that the adsorbate retains two degrees of translational freedom on the catalytic surface, and by setting  , which assumes that all surface sites are unoccupied. After cancellation of identical terms in the numerator and denominator,
, which assumes that all surface sites are unoccupied. After cancellation of identical terms in the numerator and denominator,  takes the form shown in equation (13):
takes the form shown in equation (13):
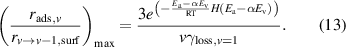
Vibrational levels for which  represent an upper bound on vibrational levels that can participate in NH3-forming surface reactions faster than they lose their energy to vibrational relaxation on the surface. Figure 5 plots
represent an upper bound on vibrational levels that can participate in NH3-forming surface reactions faster than they lose their energy to vibrational relaxation on the surface. Figure 5 plots  as a function of vibrational level at 400 K over Fe, Ni, and Ag terrace sites, and table 1 tabulates values of v, the percentage of N2(v), and percentage of vibrational energy at the inlet of the catalyst bed for which
as a function of vibrational level at 400 K over Fe, Ni, and Ag terrace sites, and table 1 tabulates values of v, the percentage of N2(v), and percentage of vibrational energy at the inlet of the catalyst bed for which  .
.

Figure 5. Upper bounds on the ratio of rates of dissociative adsorption to the rates of vibrational relaxation of N2(v) over Fe, Ni, and Ag metal wool catalysts as a function of vibrational level at the experimental surface temperature (400 K).
Download figure:
Standard image High-resolution imageTable 1. Vibrational levels, % of N2(v), and % of vibrational energy at the inlet of the catalyst bed for which  >
>  for the process conditions described in figure 2.
for the process conditions described in figure 2.
| Metal | v | % of N2(v) | % of vibrational energy |
|---|---|---|---|
| Fe | >8 | <2 | <7 |
| Ni | >17 | <10−7 | <10−6 |
| Ag | >38 | ∼0 | ∼0 |
Figure 6 compares the density of N2(v) at the inlet of the catalyst bed for which  , the N density at the same location, and the experimentally measured NH3 formation (NH3 density at 4.5 cm with catalyst minus NH3 density at 3.5 cm) in the packed bed of Fe, Ni, or Ag. Measurements of NH3 density at 3.5 cm and 4.5 cm without catalyst show that the change in NH3 density from 3.5 to 4.5 cm is below the limit of uncertainty (1.0 × 1014 cm−3) for each process condition (figure S2), indicating that any changes in the NH3 density with catalyst present can be attributed to reactions on the metal wools. Figure 5 and table 1 show that for Ni and Ag,
, the N density at the same location, and the experimentally measured NH3 formation (NH3 density at 4.5 cm with catalyst minus NH3 density at 3.5 cm) in the packed bed of Fe, Ni, or Ag. Measurements of NH3 density at 3.5 cm and 4.5 cm without catalyst show that the change in NH3 density from 3.5 to 4.5 cm is below the limit of uncertainty (1.0 × 1014 cm−3) for each process condition (figure S2), indicating that any changes in the NH3 density with catalyst present can be attributed to reactions on the metal wools. Figure 5 and table 1 show that for Ni and Ag,  when v > 17 and v > 38, respectively. The density of N2(v> 17) is at least 6 orders of magnitude lower than the N density and the observed NH3 formation in the Ni catalyst bed (figure 6(b)). Similarly, figure 6(c) shows that the density of N2(v > 38) is orders of magnitude lower than the N density and the observed NH3 formation in the Ag catalyst bed. Thus, N2(v) does not measurably contribute to NH3 formation over Ni and Ag even though the densities of N2(v> 0) exceed N densities at the inlet of the catalyst bed by 100× because most of the N2(v) loses its energy to the catalyst faster than it can dissociate to form NH3.
when v > 17 and v > 38, respectively. The density of N2(v> 17) is at least 6 orders of magnitude lower than the N density and the observed NH3 formation in the Ni catalyst bed (figure 6(b)). Similarly, figure 6(c) shows that the density of N2(v > 38) is orders of magnitude lower than the N density and the observed NH3 formation in the Ag catalyst bed. Thus, N2(v) does not measurably contribute to NH3 formation over Ni and Ag even though the densities of N2(v> 0) exceed N densities at the inlet of the catalyst bed by 100× because most of the N2(v) loses its energy to the catalyst faster than it can dissociate to form NH3.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. Comparison of N densities and the densities of N2(v) for v where  at the inlet of the catalyst bed with the experimentally measured NH3 formation in an (a) Fe, (b) Ni, or (c) Ag catalyst bed. 10 mm of packed bed beginning at 3.5 cm axial position with 0.0024 g Fe/mm, 0.0036 g Ni/mm, or 0.0033 g Ag/mm. 0.5% N2 and 0.5% H2 in Ar, 5% N2 and 0.5% H2 in Ar, or 0.5% N2 and 2.5% H2 in Ar. 16.67 cm3 s−1, 2.1 W average power, 12.8 MHz, 20 kHz modulation with 50% duty cycle, 50 Hz modulation with 50% duty cycle. N2(v) densities predicted by the kinetic model are below 1 cm−3 when represented by an error bar.
at the inlet of the catalyst bed with the experimentally measured NH3 formation in an (a) Fe, (b) Ni, or (c) Ag catalyst bed. 10 mm of packed bed beginning at 3.5 cm axial position with 0.0024 g Fe/mm, 0.0036 g Ni/mm, or 0.0033 g Ag/mm. 0.5% N2 and 0.5% H2 in Ar, 5% N2 and 0.5% H2 in Ar, or 0.5% N2 and 2.5% H2 in Ar. 16.67 cm3 s−1, 2.1 W average power, 12.8 MHz, 20 kHz modulation with 50% duty cycle, 50 Hz modulation with 50% duty cycle. N2(v) densities predicted by the kinetic model are below 1 cm−3 when represented by an error bar.
Download figure:
Standard image High-resolution image{kind=link}
Figure 5 and table 1 show that for Fe, the transition metal that binds nitrogen the most strongly and has the lowest activation barrier for dissociative adsorption of N2 of the three investigated metals,  when v > 8. Figure 6(c) shows that the density of N2(v> 8) is on the order of 1013–1015 cm−3 for the investigated operating conditions. This accounts for <2% of N2(v> 0) and <7% of energy deposited into N2(v) (table 1). N2(v> 8) densities depend sensitively on the process conditions and can be either less than or greater than the N densities at the inlet of the catalyst bed, depending on the operating conditions (figure 6(a)). Qualitatively, this suggests that Fe could potentially dissociate N2(v) in quantities that would contribute to NH3 formation similar to the measured amounts. However, the data in figure 6(a) suggest that NH3 does not form from N2(v), since NH3 formation does not correlate with N2(v> 8) densities varying from 1013–1015 cm−3. NH3 formation instead correlates with N consumption for the investigated operating conditions, as discussed in previous work [15].
when v > 8. Figure 6(c) shows that the density of N2(v> 8) is on the order of 1013–1015 cm−3 for the investigated operating conditions. This accounts for <2% of N2(v> 0) and <7% of energy deposited into N2(v) (table 1). N2(v> 8) densities depend sensitively on the process conditions and can be either less than or greater than the N densities at the inlet of the catalyst bed, depending on the operating conditions (figure 6(a)). Qualitatively, this suggests that Fe could potentially dissociate N2(v) in quantities that would contribute to NH3 formation similar to the measured amounts. However, the data in figure 6(a) suggest that NH3 does not form from N2(v), since NH3 formation does not correlate with N2(v> 8) densities varying from 1013–1015 cm−3. NH3 formation instead correlates with N consumption for the investigated operating conditions, as discussed in previous work [15].
A lack of NH3 formation from N2(v> 8) can be attributed to inaccuracies in the upper bound estimates on N2(v) reactivity described in equation (13). DFT-based microkinetic modeling that not only considers kinetics for dissociative adsorption but also kinetics for other reactions that can happen on the transition metal surfaces suggests that vibrational excitation does not enhance rates of NH3 formation over Fe [8, 10], likely because the dissociative adsorption step is not the rate limiting step in NH3 formation over Fe. The overestimation of the pre-exponential factor for N2(v) dissociative adsorption can also contribute to the overestimation of vibrational levels that can contribute to NH3 formation reactions. Ultra-high vacuum studies of N2 chemisorption on Fe(111) report that the pre-exponential factor for adsorption should be 0.03–0.003× lower than the upper-bound estimate used in equation (13) [48]. Reducing  by a factor of 0.03× increases the minimum v for which
by a factor of 0.03× increases the minimum v for which  from 9 to 12 over Fe, reducing the density of N2(v) that can undergo dissociative adsorption faster than vibrational relaxation by an order of magnitude or more for the investigated operating conditions.
from 9 to 12 over Fe, reducing the density of N2(v) that can undergo dissociative adsorption faster than vibrational relaxation by an order of magnitude or more for the investigated operating conditions.
3.4. Discussion
The use of equation (12) to assess the ability of N2(v) to contribute to NH3 formation can be applied to systems beyond this work with appropriate considerations. In packed bed DBDs, metal particles supported on a metal oxide are often used as catalysts. Metal particles with sizes below ∼10 nm have fractions of step sites >0.1 [49, 50]. These step sites have lower activation barriers for N2 dissociative adsorption than terrace sites [51, 52]. As a result, a larger range of vibrational levels would be able to undergo dissociative adsorption faster than vibrational relaxation (figure S6). However, these metal nanoparticles are dispersed on a support material with a surface area much larger (i.e. ∼100×) than the surface area of the metal particles [53, 54]. N2(v) undergoes vibrational relaxation on metal oxide supports with similar loss probabilities to metals [19, 20] but cannot participate in NH3 formation reactions on the metal oxide supports, resulting in a larger fraction of N2(v) directed away from possible NH3-forming reactions.
Elevated surface temperature can also increase  , since the rate of dissociative adsorption has an exponential dependence on surface temperature. Figure S7 shows
, since the rate of dissociative adsorption has an exponential dependence on surface temperature. Figure S7 shows  as a function of vibrational level at a surface temperature of 700 K. Even at 700 K, the lowest vibrational level where
as a function of vibrational level at a surface temperature of 700 K. Even at 700 K, the lowest vibrational level where  for Ni is v = 17, which is only one vibrational level less than the lowest level at 400 K. For Fe, the lowest vibrational level where
for Ni is v = 17, which is only one vibrational level less than the lowest level at 400 K. For Fe, the lowest vibrational level where  is v = 5. N2(v> 4) still only represents ∼10% of N2(v>0) for the VDFs in figure 2(a), although the shapes of the VDF will change with temperature, since rates of VV and VT processes are temperature dependent, and rates of EV processes will change due to changes in electron temperature and density with temperature [55]. At temperatures near and above 700 K, NH3 decomposition is catalyzed over transition metal surfaces [56] at rates which will limit the net NH3 formation from N2(v). Hence, while increasing the gas temperature would likely enhance dissociative adsorption of N2(v), the beneficial effect for NH3 formation will likely be limited in addition to requiring increased energy deposition.
is v = 5. N2(v> 4) still only represents ∼10% of N2(v>0) for the VDFs in figure 2(a), although the shapes of the VDF will change with temperature, since rates of VV and VT processes are temperature dependent, and rates of EV processes will change due to changes in electron temperature and density with temperature [55]. At temperatures near and above 700 K, NH3 decomposition is catalyzed over transition metal surfaces [56] at rates which will limit the net NH3 formation from N2(v). Hence, while increasing the gas temperature would likely enhance dissociative adsorption of N2(v), the beneficial effect for NH3 formation will likely be limited in addition to requiring increased energy deposition.
Another method to maximize NH3 formation from N2(v) is to increase the densities of the vibrational levels of N2(v) that can dissociate faster than relax. Figure 2(a) shows that by increasing N2 from 0.5% to 5% with constant 0.5% H2 produces a ∼10× increase in the N2(v) density at each vibrational level. Increasing the fraction of N2 further (for example, to 50%) would require the use of an alternative plasma source. DBDs can operate without a noble gas carrier but operate with E/N ∼100 Td, which favors energy deposition into electronic excitation or dissociation rather than vibrational excitation [11]. This results in relatively lower N2(v) formation than N formation in the plasma compared to the investigated operating conditions using a RF source and consequently a lower relative contribution of N2(v) to NH3 formation compared to N. Microwave discharges or gliding arc discharges operate in regimes that favor vibrational excitation and can be sustained with molecular gases [12]; however, they also operate with gas temperatures >1000 K [57, 58], which can prevent net NH3 formation due to the prominence of NH3 decomposition above 700 K over catalytic surfaces [56]. For these discharges, the gas would have to be cooled to an appropriate temperature before reaching the surface over a timescale that does not result in significant loss of N2(v) in the gas phase due to VT relaxation. Figure 2(a) also shows that increasing H2 from 0.5% to 2.5% at constant 0.5% N2 reduces N2(v> 7) densities by at least an order of magnitude. Operating with lower % H2 would increase densities of N2(v) that are more reactive for dissociative adsorption; however, relatively lower H2 densities could adversely affect rates of hydrogenation steps on the catalyst surface, and as a result reduce NH3 formation rates and yields. Proximity of the plasma to the catalyst also dictates the densities of N2(v) that can react to form NH3. Figures S2(g)–(i) show that loss of N2(v) from the end of the plasma to the beginning of the catalyst bed (∼300–650 μs) is most pronounced for the operating condition with the highest fraction of H2, which exhibits a ⩾2× decrease in N2(v) densities for v> 9 due to VT relaxation in the gas phase. Loss of N2(v) in the gas phase would become more significant as the fraction of H2 increases (for example, at or beyond 50%) and the transport timescale increases.
The choice of catalyst, plasma source, reactor design, gas/surface temperature, and gas composition dictate the production of N2(v) in the plasma, loss of N2(v) by relaxation in the gas phase and on surfaces, and utilization of N2(v) to form NH3 over the catalyst. While conditions that enhance the role of vibrational excitation in plasma-catalyzed NH3 synthesis more than in this study might be found, the results in this work and literature precedent does not suggest that most, or even a significant fraction, of the energy directed towards vibrational excitation can be directed towards pathways that contribute to NH3 formation, which limits the ability of N2(v) to produce NH3 by plasma catalysis with high energy efficiency.
4. Conclusion
Production of N2(v) by nonthermal plasma and loss pathways for N2(v) in the gas-phase and over catalytic surfaces are investigated by MBMS measurements and state-to-state kinetic modeling. State-to-state kinetic models that are validated by threshold ionization MBMS measurements demonstrate that N2(v) is produced by the plasma in quantities that exceed the N density by two orders of magnitude. Total N2(v) density is governed by the N2 density in the jet at otherwise identical operating conditions. Increasing the density of H2 in the discharge depopulates densities of N2(v> 8), vibrational levels which are proposed to be more reactive for catalytic NH3 forming reactions [8].
N2(v) is consumed more quickly when a catalyst is placed in the afterglow, which is attributed to vibrational relaxation of N2(v) on the catalyst surface, based on state-to-state kinetic modeling and MBMS measurements. A comparison between rates of vibrational relaxation of N2(v) on the catalyst surface and upper bounds on proposed rates of dissociative adsorption of N2(v) on the catalyst surface shows that at most only N2(v> 8) (<2% of N2(v> 0)), N2(v> 17), and N2(v> 38), can undergo dissociative adsorption faster than vibrational relaxation over Fe, Ni, and Ag, respectively, for the investigated operating conditions. Densities of N2(v> 17) and N2(v> 38) are orders of magnitude lower than N densities at the inlet of the catalyst bed, and densities of N2(v> 8) are similar to or less than the N densities at the inlet of the catalyst bed, which can explain why N2(v) does not measurably contribute to NH3 formation for the investigated operating conditions. Excluding particular scenarios in which very high N2(v) densities can be acquired largely in absence of N radicals and in which the catalyst can react the N2(v) to form NH3 faster than relaxation of N2(v) in the gas phase or on solid surfaces of catalysts and catalytic supports, it is unlikely that availability and reactivity of N2(v) is relevant for plasma-catalyzed NH3 synthesis.
Acknowledgments
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Fusion Energy Sciences General Plasma Science program under Award Number DE-SC0020232. The work heavily relied on equipment and methods developed within project DE-SC0001939. The authors thank Dr Jingkai Jiang and Professor Yolanda Aranda Gonzalvo (University of Minnesota Twin Cities) for assistance with the MBMS system and thank Professor Bryan Goldsmith (University of Michigan Ann Arbor) for insightful comments on the manuscript.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Supplementary data (0.5 MB PDF)