

Abstract
Two-dimensional transition metals borides TixBx have excellent magnetic and electronic properties and great potential in metal-ion batteries and energy storage. The thermal management is important for the safety and stability in these applications. We investigated the lattice dynamical and thermal transport properties of bulk-TiB2 and its two-dimensional (2D) counterparts based on density functional theory combined with solving phonon Boltzmann transport equation. The Poisson's ratio of bulk-TiB2 is positive while it changes to negative for monolayer TiB2. We found that dimension reduction can cause the room-temperature in-plane lattice thermal conductivity decrease, which is opposite the trend of MoS2, MoSe2, WSe2 and SnSe. Additionally, the room temperature thermal conductivity of mono-TiB2 is only one sixth of that for bulk-TiB2. It is attributed to the higher Debye temperature and stronger bonding stiffness in bulk-TiB2. The bulk-TiB2 has higher phonon group velocity and weaker anharmonic effect comparing with its 2D counterparts. On the other hand, the room temperature lattice thermal conductivity of mono-Ti2B2 is two times higher than that of mono-TiB2, which is due to three-phonon selection rule caused by the horizontal mirror symmetry.
Export citation and abstract BibTeX RIS
1. Introduction
Bulk metal borides, such as MgB2, TaB2, Nb3B4 and AlB6, are special functional materials, which process broad application prospects in the fields of superconductivity, semiconductors, super-hard materials, catalysis, etc [1–11]. Based on the rich AlB2–type bulk structures (ScB2, TiB2, VB2, YB2, CrB2, MoB2, TaB2, HfB2, etc), two-dimensional (2D) transition metal (TM) borides were suggested. Due to the unpaired d-orbital electrons of transition metal atoms, 2D TM borides have the excellent magnetic properties for spintronic devices. 2D MnB shows the robust metallic ferromagnetism with high Curie temperature of 345 K and it can be enhanced by −F and −OH groups functionalizations [8]. 2D Ti2B exhibits the room-temperature ferromagnetism with the Curie temperature of 39.06 K [9]. So, the two-dimensional metal borides have also stimulated the research interests of many researchers [10, 11].
Compared with 3D metallic bulk, 2D TM borides presented novel electronic properties and have potential application as an anode material for batteries. Two-dimensional Tix Bx is a special 2D transition metal boride and shows great significance in theory and applications [9,12–19]. Recently, two-dimensional TiB has been successfully synthesized in experiments by dealloying of the parent Ti2InB2 at high temperature [16]. The band structure of M-TiB2 was found to be characterized with anisotropic Dirac cones with ultrahigh electron mobility [20]. TiB2 has a complex nodal-net structure and shows several topological novel states. The linearly and quadratically dispersed surface Dirac points were also found [21]. On the other hand, 2D Tix Bx can be used for Li-ion, Na-ion and Mg-ion batteries. The calculated specific capacities of the Ti2B2 monolayer are 456 mAhg−1 and 342 mAhg−1 for Li and Na, respectively [19]. For TiB monolayer, the theoretical specific capacity for Li or Na is 480 mAhg−1. The Ti2B monolayer has ultra-high specific capacity of 3018 mAhg−1 for Mg ion [22]. The results are larger than that for carbon MXenes such as Ti3C2, Ti2C and Mo2C [23, 24]. In addition, TiB4 monolayer is an excellent energy storage material [12]. TiB4–2CH4 obtained the effective energy storage of 10.14 wt% under ambient conditions. The heat transport is important for the applications in nanoelectronic device, battery, energy storage and energy harvesting [25–32]. But the investigation of thermal transport property for 2D TMs borides has not been investigated up to now.
On the other hand, dimensional changes can effectively tune the physics and chemical properties of materials. It is well known that the phonon thermal conductivity of graphite is smaller than that of graphene. For MoS2 [33], MoSe2 and WSe2 [34], SnSe [35], it is also found that dimensional reduction makes the lattice thermal conductivity increase. However, the lattice thermal conductivity of monolayer ZrS3 is obviously smaller than that for bulk ZrS3 along both the x and y directions [36]. Then, how does the phonon thermal transport change from bulk TiB2 to monolayer structures?
In this work, based on density functional theory (DFT) combined with solving phonon Boltzmann transport equation (PBTE), we compared the lattice thermal transport properties of bulk-TiB2 and its 2D counterparts. We found that the room-temperature in-plane thermal conductivity of bulk-TiB2 is 83.3% lower than that of mono-Ti2B2 and thermal conductivity of mono-Ti2B2 is about three times of that in mono-TiB2. The big difference in thermal transport property was explained.
2. Theoretical method
The lattice thermal conductivity is calculated by using an ab initio method based on the DFT calculations combined with PBTE. This microscopic transport description explicitly considers mode-dependent phonon-scattering processes and their entangled distribution functions as the Boltzmann equation are solved self-consistently. In particular, a small applied temperature gradient ∇T perturbs the phonon distributions from equilibrium, resulting in a drifting phonon flux which is balanced by phonon scatterings. For cases where ∇T does not drive the phonon populations far from equilibrium, the single-mode relaxation time approximation can give a reasonably accurate solution to the BTE. Explicitly, the lattice thermal conductivity tensor can be expressed as:
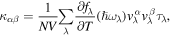
where λ comprises both a phonon branch index j and a wave vector q.  T, N,
T, N,  V and
V and  mean the Planck constant, absolute temperature, the number of uniformly spaced wave vector q points in the Brillouin zone (BZ), the phonon distribution function, the volume of the unit cell and the relaxation time, respectively.
mean the Planck constant, absolute temperature, the number of uniformly spaced wave vector q points in the Brillouin zone (BZ), the phonon distribution function, the volume of the unit cell and the relaxation time, respectively.  is the frequency of phonons with mode j and wave vector q in the BZ, and
is the frequency of phonons with mode j and wave vector q in the BZ, and  is the phonon group velocity in the α/β direction. Under relaxation time approximation, phonon relaxation time can be described as [37]:
is the phonon group velocity in the α/β direction. Under relaxation time approximation, phonon relaxation time can be described as [37]:
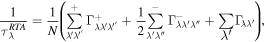
where  and
and  represent the scattering rates due to absorbing and emitting three-phonon intrinsic processes, respectively.
represent the scattering rates due to absorbing and emitting three-phonon intrinsic processes, respectively.
All ab initio calculations are performed by using the Vienna ab initio simulation package [38–40]. The Perdew–Burke–Ernzerhof functional of generalized gradient approximation is chosen as the exchange–correlation functional [41]. The energy convergence criterion is chosen to be 10–8 eV and the cutoff energy of the plane-wave is 600 eV. A Monkhorst-Pack [42] k-mesh of 25 × 25 × 25 and 25 × 25 × 1 is used to sample the Brillouin Zone (BZ) of bulk and monolayer structure respectively. The vacuum space of at least 20 Å is kept along the z direction, which is enough to avoid the interactions between periodical images. All geometries are fully optimized until the maximal Hellmann–Feynman force is not larger than 10–4 eV Å−1.
In the calculation of phonon dispersion, the harmonic interatomic force constants (IFCs) are obtained from the finite displacement method as implemented in the PHONOPY package [43]. A 4 × 4 × 4 and 5 × 5 × 1 supercell is constructed to ensure the convergence of bulk and monolayer structure respectively. For sampling the BZ of bulk and monolayer structures, the 5 × 5 × 5 and 5 × 5 × 1 k-mesh are used respectively. For the calculation of thermal conductivity, anharmonic third order IFCs are also necessary, besides the harmonic second order IFCs obtained above. The same supercell and the k-mesh are used to obtain the anharmonic third order IFCs, and the interatomic interactions are considered with the cutoff distance up to 0.5 nm. At last, the thermal conductivity is calculated by solving the PBTE using the ShengBTE code [37]. To ensure the conductivity convergence, we choose a 21 × 21 × 21 and 101 × 101 × 1 q-mesh to describe the phonon–phonon interactions in bulk and monolayer structure, respectively. In order to calculate the volume and cross-section area of 2D materials, we define the effective thickness as the summation of the buckling distance and the vdW radius of the out most atom.
3. Results and discussion
3.1. Structure stability and mechanical property
For Ti-B layers alternate stacking bulk-TiB2, different combination of layer types can result in diversity of its 2D structures. According to stoichiometric ratio, there are three types of 2D structures: mono-Ti2B2, mono-TiB2 and mono-TiB4. The structures of mono-TiB2 and mono-Ti2B2 were also presented in earlier researches to study the electronic properties [13, 19]. The crystal structures of bulk-TiB2 and its 2D counterparts are shown in figure 1. The relaxed lattice parameters obtained from structural optimization are summarized in table 1. We find that the lattice constants of them have minor changes and the interlayer distance of mono-TiB2 is remarkable smaller than the others. The space group symmetry has also some differences. The space group symmetry of bulk-TiB2, mono-Ti2B2 and mono-TiB4 structure is P6/mmm while that of mono-TiB2 is P6mm. The broken horizontal reflection symmetry caused the lower space group symmetry for mono-TiB2.

Figure 1. The crystal structures of bulk-TiB2 and its 2D counterparts. The B and Ti atoms are represented in green and blue, respectively.
Download figure:
Standard image High-resolution imageTable 1. The space group and structural parameters of the bulk-TiB2 and 2D counterparts. a is relaxed lattice constant and h is interlayer distance.
| Space group | a (Å) | h (Å) | |
|---|---|---|---|
| Bulk-TiB2 | P6/mmm | 2.997 | 3.167 |
| Mono-Ti2B2 | P6/mmm | 2.954 | 3.07 |
| Mono-TiB2 | P6mm | 3.099 | 1.187 |
| Mono-TiB4 | P6/mmm | 3.030 | 3.077 |
To study the lattice stability, we plot phonon dispersion along high-symmetry lines of these four structures in figure 2. No imaginary frequency mode in phonon dispersion for bulk- TiB2, mono-Ti2B2 and mono-TiB2, which indicates that these structures are dynamically stable. As shown in figure 2(d), however, there is a softened 'U' shape feature near the K point in the mono-TiB4 phonon dispersion. It shows that the mono-TiB4 is unstable. Additionally, the stability of the three structures was also confirmed from the Born–Huang mechanical stability criteria. Table 2 presents the elastic constants (Cij) of bulk-TiB2, mono-Ti2B2 and mono-TiB2. It is found that the calculated results satisfy the Born–Huang mechanical stability criteria of hexagonal crystal [44], namely,  and
and 
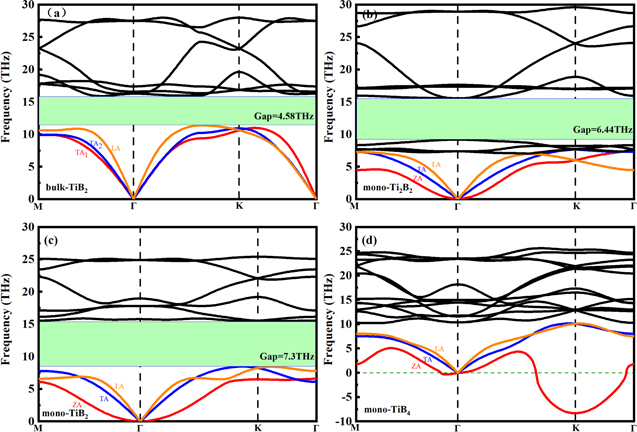
Figure 2. Phonon dispersion of bulk-Ti2B2 and its 2D counterparts.
Download figure:
Standard image High-resolution imageTable 2. The calculated elastic constants (Cij), Young's modulus (Y), in-plane Young's modulus (Y2D) and Poisson's ratio (γ) of bulk- TiB2, mono-Ti2B2 and mono-TiB2.
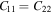 (GPa) (GPa) |
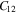 (GPa) (GPa) |
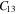 (GPa) (GPa) |
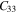 (GPa) (GPa) |
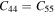 (GPa) (GPa) |
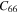 (GPa) (GPa) | Y(GPa) | Y2D (N m−1) | γ | |
|---|---|---|---|---|---|---|---|---|---|
| Bulk-TiB2 | 654.9 | 71.9 | 116.4 | 460.0 | 261.2 | 291.5 | 336.4 | — | 0.292 |
| Mono-Ti2B2 | 110.7 | 16.1 | — | — | — | 47.3 | 318.2 | 216.7 | 0.145 |
| Mono-TiB2 | 77.3 | −2.2 | — | — | — | 39.7 | 341.3 | 154.5 | −0.028 |
Table 2 compares the mechanical properties of bulk-TiB2, mono-Ti2B2 and mono-TiB2 from the Young's modulus (Y), in-plane Young's modulus (Y2D) and Poisson's ratio (γ). Poisson's ratio of mono-Ti2B2 is calculated to be 0.145, which is about half of that for bulk-TiB2. In addition, the Poisson's ratio is changed from positive value to negative value if the bulk structure of TiB2 changes to monolayer TiB2. It also indicates that the mono-TiB2 can be used as auxetic materials. On the other hand, the calculated Young's modulus Y of bulk-TiB2 is comparable with that of mono-Ti2B2 and mono-TiB2. However, the calculated Y2D of mono-Ti2B2 is about 40.3% larger than that of mono-TiB2, indicating mono-Ti2B2 have better mechanical properties. Comparing with other 2D materials, the Y2D of mono-Ti2B2 is much larger than those of silicene [45] (61.87 N m−1), germanene [46] (41.4 N m−1), monolayer SiC [47] (163.5 N m−1) and borophene [48] (armchair: 187.8 N m−1; zigzag: 180.8 N m−1).
To explore the bonding nature in bulk-Ti2B2, mono-Ti2B2 and mono-TiB2, the electron localization function (ELF) is presented, as shown in figure 3. The ELF, defined to have the convenient range of values between 0 and 1, is known to be an important tool to distinguish different bonding interactions in molecules or solids. We can find that the B–B atoms are bonded covalently and the covalent nature in mono-TiB2 is stronger than the other structures. The Ti–B bond exhibits ionic nature derived from the electron transfer from Ti atoms to the B atoms.

Figure 3. The calculated electron localization functions of bulk-Ti2B2, mono-Ti2B2 and mono-TiB2 (a)–(c) with the section of Ti-plane, (d)–(f) with the section of B-plane, (g)–(i) with the section of diagonal-plane.
Download figure:
Standard image High-resolution image3.2. Comparative study of thermal transport property
We firstly present the phonon dispersion of bulk-TiB2 and its 2D counterparts, as shown in figure 2. We can see that there are some obvious differences in phonon dispersion between them. Due to the large atomic weight difference between Ti and B, a large phonon bandgap exists. The phonon bandgap of bulk-TiB2 is 4.58 THz, which is smaller than that for mono-Ti2B2 (6.44 THz) and mono-TiB2 (7.3 THz). The larger phonon bandgap can make forbid the scattering between the acoustic and optical phonon modes. On the other hand, the acoustic branches in bulk-TiB2 and mono-TiB2 do not entangle with other optical branches, which is diverse from that in mono-Ti2B2. Since there are four atoms in each unit cell for mono-Ti2B2, there are 3 acoustic and 9 optical phonon branches. Compared with bulk-TiB2 and mono-TiB2, the extra 3 optical phonon branches of mono-Ti2B2 are located near the max of acoustic branches, due to its larger proportion (50%) of Ti atoms. In addition, compared with the bulk structure, the maximum frequency of the acoustic phonons of two-dimensional structure is lower, especially the ZA branch, which is due to the smaller restoring force of the atomic out-of-plane vibration in the two-dimensional material.
Based on the 2nd and 3rd interatomic force constants, we calculated the temperature-dependent phonon thermal conductivity of bulk-TiB2 and its 2D counterparts, as shown in figure 4. The thermal conductivity is decreased with increasing the temperature due to the enhancement of phonon scattering at higher temperature. We find that in-plane and out-of-plane lattice thermal conductivity of bulk-TiB2 at the room temperature is 144 (W mK−1) and 119 (W mK−1), which is slightly higher than the experimentally reported 110 W mK−1 [49]. The room-temperature in-plane thermal conductivity of mono-Ti2B2, and mono-TiB2 is 75 (W mK−1 and 24 (W mK−1), respectively. With the dimensional reduction, the in-plane thermal conductivity of bulk-TiB2 is remarkably reduced, which is opposite of the trend of MoS2 [33], MoSe2 and WSe2 [34], SnSe [35], and in line with that of ZrS3 [36]. The room temperature thermal conductivity of mono-TiB2 is calculated to be 83.3% lower than that of bulk-TiB2. The results can be explained from the phonon dispersion spectrum in figure 2. The maximum frequency of the acoustic branch of bulk-TiB2 is higher than of its 2D counterparts, which leads to the higher Debye temperature and stronger bonding stiffness in bulk-TiB2. Additionally, one of the TA branches in bulk-TiB2 convert into ZA mode due to the dimension reduction. The lowest flexural phonon branch exhibits a quadratic dispersion ω ∼ q2, making the group velocity vanish as q → 0.
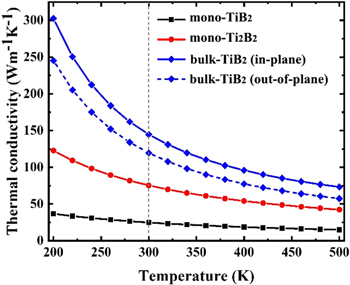
Figure 4. Lattice thermal conductivities as a function of temperature for bulk-Ti2B2, mono-Ti2B2 and mono-TiB2.
Download figure:
Standard image High-resolution imageLattice thermal conductivity is always determined by both phonon group velocity and phonon scattering rate. The phonon group velocity and relaxation time of bulk-TiB2 and its 2D counterparts are compared in figure 5. We can find that the group velocity of bulk-TiB2 is higher than that of its 2D counterparts. The mode-dependent relaxation time (τ) of bulk-TiB2 in the low frequency region is also remarkably larger, indicating the weaker anharmonic effect on phonons. The results lead to the higher thermal conductivity of bulk-TiB2 comparing with that of its 2D counterparts.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. (a) Phonon group velocity as a function of frequency for bulk-Ti2B2, mono-Ti2B2 and mono-TiB2. (b) The comparison of phonon relaxation time as a function of frequency at 300 K.
Download figure:
Standard image High-resolution image{kind=link}
On the other hand, for the 2D allotrope of mono-Ti2B2 and mono-TiB2, the thermal conductivity of mono-Ti2B2 is three times of that in mono-TiB2. It is mainly attributed to three-phonon selection rules [50]. The selection rule on phonon–phonon scattering in two-dimensional materials is revealed by examining 3th order interatomic force constant, which is given by [51]:
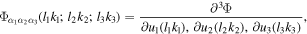
where l1 k1 l2 k2 l3 k3 are refer to any B atom.
Because of the mirror symmetry, the force constants  are found to be zero in mono-Ti2B2. This means that there is no anharmonicity induced by the relative motion among the three B atoms along z direction. However, there is no horizontal mirror symmetry in mono-TiB2 crystals and the third-order anharmonic force constants
are found to be zero in mono-Ti2B2. This means that there is no anharmonicity induced by the relative motion among the three B atoms along z direction. However, there is no horizontal mirror symmetry in mono-TiB2 crystals and the third-order anharmonic force constants  become non-zero. It leads to stronger scattering of ZA phonons and smaller thermal conductivity in mono-TiB2 comparing with mono-Ti2B2. Additionally, figure 5(b) shows that phonon relaxation time of mono-TiB2 in the low frequency region is obviously smaller than that of mono-Ti2B2, which results in lower thermal conductivity.
become non-zero. It leads to stronger scattering of ZA phonons and smaller thermal conductivity in mono-TiB2 comparing with mono-Ti2B2. Additionally, figure 5(b) shows that phonon relaxation time of mono-TiB2 in the low frequency region is obviously smaller than that of mono-Ti2B2, which results in lower thermal conductivity.
4. Conclusions
In summary, we have compared the intrinsic lattice thermal conductivity of bulk-TiB2 and its 2D counterparts from first-principles calculations combined with solving PBTE. The 2D counterparts of mono-Ti2B2 and mono-TiB2 are stable and the mono-TiB4 is unstable. The Poisson's ratio of bulk-TiB2 is positive while it is negative for monolayer TiB2. The in-plane lattice thermal conductivities at room temperature are 144 W mk−1, 75 W mk−1 and 24 W mK−1 for bulk-TiB2, mono-Ti2B2 and mono-TiB2, respectively. It shows that the dimensional reduction makes the thermal conductivity obvious reduction for TiB2. This can be derived from the changing of phonon dispersion and phonon anharmonic effect. In addition, the difference of thermal conductivity between mono-Ti2B2 and mono-TiB2 is originated from horizontal mirror symmetry in mono-Ti2B2 crystals, which leads to longer phonon lifetime caused by the three-phonon selection rule. These results show that the dimensional changes can lead to huge difference of thermal transport for 2D TM borides and it provides an effective method to tune the thermal management for micro-/nano-devices based on metal borides.
Acknowledgments
We gratefully acknowledge funding supporting from Scientific and Technological Research of Chongqing Municipal Education Commission (KJZD-K202100602).
Data availability statement
The data that support the findings of this study are available upon reasonable request from the authors.
Conflicts of interest
There are no conflicts of interest to declare.