
Abstract
Purpose. To overcome the insufficiency of conventional photodynamic therapy (PDT) for treating metastatic melanoma, the combination of smart nanoparticles and PDT with immunotherapy was used to achieve a higher efficiency by accumulating more photosensitizers in tumor areas and triggering stronger immune responses against tumors after PDT. Methods. In this study, we designed a nanoliposome co-encapsulation of chlorin E6 (Ce6) and SB-3CT to realize significant antitumoral proliferation and metastasis efficacy after laser irradiation in A375 cells. The morphology, size distribution, and loading efficiency of Ce6–SB3CT@Liposome (Lip-SC) were characterized. The reactive oxygen species (ROS) generation and cytotoxicity were evaluated in A375 cells, and the mechanisms of natural killer (NK) cell-mediated killing were assessed. Results. Lip-SC showed good stability and was well-dispersed with a diameter of approximately 140 nm in phosphate-buffered saline. The nanoliposomes could accumulate in tumor areas and induce apoptosis in cancer cells upon 660 nm light irradiation, which could trigger an immune response and induce the expression of NK group 2 member D (NKG2D) ligands. The subsequently released SB-3CT could further activate NK cells effectively and strengthen the immune system by inhibiting the shedding of soluble NKG2D ligands. Discussion. Taken together, the synergistic effects of SB-3CT on nanoliposomes for Ce6-mediated PDT were analyzed in detail to provide a new platform for future anti-melanoma treatment.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Abbreviations
| PDT | photodynamic therapy |
| NKG2D | activating receptor natural killer group 2, member D |
| Ce6 | chlorin E6 |
| DPPC | 1,2-Dipalmityl-SN-glycerol-3-phosphocholine |
| DOTAP | (2,3-Dioleoyloxy-propyl)-trimethylammonium-chloride |
| DSPE-PEG20001 | 2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethyleneglycol)] |
| Cryo-TEM | Cryo-transmission electron microscopy |
| DMEM | Dulbecco's modified Eagle's medium |
| MMP | matrix metalloproteinase |
1. Introduction
Recently, the incidence of melanoma has increased year after year, and it has become one of the cancers that threaten human life and health due to its high metastasis [1, 2]. A single treatment method, especially traditional methods, such as chemoradiotherapy, is often ineffective to restrain melanoma and causes serious side effects and tumor resistance. PDT has become a new therapeutic modality for melanoma that has received much attention recently due to its effectiveness, safety, synergy, and relatively low treatment cost [3–5]. However, conventional PDT is insufficient to overcome tumors, especially those with strong metastasis, such as melanoma. Smart nanoparticles combined with other therapeutic modalities were required to activate the immune system to achieve stronger immune responses against tumors and significantly increase the efficiency of PDT [6–9]. P PDT agents can directly induce tumor cell necrosis and apoptosis through the reactive oxygen species (ROS) generated under the light [10, 11]. In this process, local inflammation occurs, and several tumor cell antigens are exposed, which could induce local immune responses and provide an excellent microenvironment for immunotherapy. Therefore, the enhancement of immune activity by additional immunomodulatory agents may be promising to improve the antitumor effects of PDT.
NKG2D is involved in both innate and adaptive immunity and functions as a 'master switch' in determining the activation status of natural killer (NK) cells [12, 13]. The NKG2D–NKG2D ligand (NKG2DL) pathway has been considered a promising target for immunotherapy due to the selective expression of 'stress-induced ligands' on tumor cells and the strong activating potency of NKG2D [14, 15]. Treatments that target the NKG2D–NKG2DL pathway have been used in clinical practice [16]. Chimeric antigen receptor (CAR)–NK cells transduced with NKG2D fused to the cytotoxic ζ-chain of T-cell receptors could be used to target suppressive myeloid cells and improve the infiltration and function of subsequent infusions of tumor-specific CAR-T cells [17]. Moreover, studies have shown that PDT can enhance NKG2DL expression in tumors, and the combined application of PDT and NKG2D-related immunotherapy can improve the antitumor immune response induced by PDT [18]. A new nano-platform composed of innate immune activator Astragaloside III (As) and the PDT reagent Ce6 (As + Ce6)@MSNs-PEG nanoparticles induce a strong adaptive immune response and activate NK cells by elevating the expression of NKG2D and interferon-gamma [19]. Furthermore, a NK cell membrane-decorated nanoparticle was designed for tumor-targeting cell-membrane immunotherapy by inducing immunogenic PDT-enhanced NK cell-membrane immunity to produce an enhanced antitumor immunity, which not only eradicated primary tumors but also inhibited the growth of pre-existing distal tumors [20].
Several pathways have been identified to participate in the regulation of NKG2DLs [21, 22]. For instance, pharmacological inhibition of MMPs reduced the level of released NKG2DLs, which could increase the tumor cell surface expression of NKG2DLs, reverse their immunosurveillance escape properties, and make them easier to be cleared by immune cells (mainly NK cells) [23–27]. Treatment of lung adenocarcinoma (ADC)-Coco cells with the MMP-2/MMP-9 inhibitor IV (MMPI-IV) potentiates NK cell-dependent cytotoxicity, which is mainly mediated by NKG2D [28].
Herein, we designed and synthesized a PS-MMP inhibitor nanoliposome (i.e. Ce6-SB3CT@Liposome [Lip-SC]), which combines the PS, Ce6, and a MMP inhibitor (i.e. SB-3CT). The synthesis and mechanism of Lip-SC are shown in figure 1. The liposomes can increase the stability of Ce6, avoiding its prior release. The nanoliposomes efficiently internalized into cancer cells, delivered the PS to the tumors, and generated ROS under 660 nm laser irradiation, which results in robust cytotoxicity. SB-3CT was designed to induce the expression of NKG2DLs in tumor cells, which benefits the recognition of NK cells. Additionally, SB-3CT can effectively prevent cell adhesion and invasion, which is a key issue that should be solved during PDT. Therefore, combining the aforementioned mechanisms together, Lip-SC can provide a potential platform for melanoma treatment.

Figure 1. The schematic illustration of co-delivery system-enhanced photodynamic therapy. The chlorin E6 (Ce6)-SB3CT@Liposome (Lip-SC) were taken by tumor cells and entrapped into lysosomes (1). Ce6 and SB-3CT, a matrix metalloproteinase inhibitor, were released into cytosol (2). PDT can induce apoptosis and necrosis (4) and increase the expression of natural killer group 2 member D ligands (NKG2DLs) (5). Besides, SB-3CT could inhibit the shedding of soluble NKG2DLs from tumor cells (3), which enhanced the immunotherapeutic effects of PDT.
Download figure:
Standard image High-resolution image2. Methods and materials
2.1. Materials
DPPC, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy-(polyethyleneglycol)-2000] (DSPE-PEG2000), DOTAP, and cholesterol were purchased from Avanti Polar Lipids (USA), Ce6 was purchased from Luminescence Technology (China), and SB-3CT was obtained from TargetMol, Boston, MA (USA).
2.2. Preparation and characterization of Lip-SC
Lip-SCs were synthesized using a film dispersion and extrusion method according to our previous study [29]. Briefly, a mixture of DPPC, DSPE-PEG2000, DOTAP, and cholesterol in a mass ratio of 15:3:3:4 (total mass, 12.5 mg) was dissolved in chloroform and incubated for 5 min CHCl3 was removed under nitrogen protection to form a thin phospholipid film. Phospholipid films were hydrated with 1 ml phosphate-buffered saline (PBS) at 48 °C for 20 min. For Ce6 and/or SB-3CT encapsulation, 5 mg ml−1 Ce6 or 3 mg ml−1 S−1 B−1 3CT in acetone was mixed with chloroform solution to form thin lipid films, followed by hydration. After hydration, liposomes with uniform particle sizes were obtained by extrusion 11 times through 100 nm polycarbonate membranes using a mini-extruder system (Avanti Polar Lipids, Inc., AL, USA). The morphology of the liposomes was characterized using cryogenic transmission electron microscopy (Cryo-TEM) (Talos F200C, FEI). The size and polydispersity index (PDI) were characterized using dynamic light scattering (DLS) (Mastersizer 3000, Malvern Instruments, UK).
The encapsulation efficiency was measured using fluorescence spectroscopy (F-4500, Hitachi, Japan) for Ce6 and ultraviolet–visible spectrophotometry for SB-3CT.
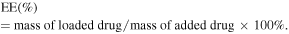
2.3. In vitro PDT-triggered drug release
The PDT-triggered Ce6 release of liposomes was measured via dialysis. Briefly, 1 ml Lip-SC solution was injected into dialysis devices with a cutoff of 100 kDa (Float-A-Lyser, Spectrum Labs) in 2 l PBS at 37 °C for 48 h. PDT irradiation was performed using a 660 nm diode continuous laser (50 mW cm−2, 5 min) 2 h after the beginning of dialysis. The concentration of Ce6 remaining in the dialysis devices at different time points was measured using fluorescence spectroscopy.
2.4. Cell culture
A375 (human skin melanoma cell line) and NK-92MI (NK cells from patients with human malignant non-Hodgkin's lymphoma) cells were purchased from American Type Culture Collection (Virginia, USA). A375 cells were incubated in DMEM (Hyclone, GE Healthcare Bio-Sciences Corp., Massachusetts, USA) containing 10% fetal bovine serum (Gibco, Thermo Fisher Scientific, Massachusetts, USA) and 1% penicillin-streptomycin (10 000 U ml−1) in a 37 °C/5% CO2 humidified chamber. NK92-MI cells were grown in minimum essential medium alpha (Gibco, Thermo Fisher Scientific, Massachusetts, USA) with 0.2 mM Inositol, 0.1 mM β-mercaptoethanol, 0.02 mM folic acid, 12.5% horse serum, and 12.5% FBS.
2.5. In vitro singlet oxygen generation and detection
Singlet oxygen (1O2) production was measured using Singlet Oxygen Sensor Green (SOSG), a fluorescent 1O2 probe (Invitrogen, USA). SOSG (final concentration, 10 μM) was mixed with 1 μg ml−1 Ce6 or Lip-SC (containing 1 μg ml−1 Ce6) and exposed to 660 nm laser irradiation (50 mW cm−2) for 1, 2, and 5 min. The fluorescence intensity was measured using fluorescence spectroscopy (Ex/Em = 504/525 nm).
2.6. Cellular uptake of Lip-SC
Cellular uptake was characterized using fluorescence spectroscopy and fluorescence microscopy. To quantitatively measure the cellular uptake of Ce6-loading liposomes, A375 cells were seeded in 6-well plates with 1 × 105 cells ml−1 and incubated with Lip-SC at 37 °C. Then, the cells were collected at different time points (4 h, 6 h, 8 h, 10 h, and 12 h) for fluorescence spectrum analysis. For fluorescence microscope detection, A375 cells were seeded in confocal dishes and incubated with Lip-SC for 8 h. The nucleus was stained with 4',6-diamidino-2-phenylindole (DAPI, Invitrogen, USA) for 5 min. Then, the cells were washed with PBS to be observed under a fluorescence microscope (ECLIPSE Ti, Nikon Corporation, Japan).
2.7. Intracellular ROS generation for PDT
Dichlorodihydrofluorescein diacetate (DCFH-DA) was used as a fluorescent probe to detect intracellular ROS generation in A375 cells, which was characterized using fluorescence microscopy. The A375 cells were seeded in confocal dishes at a density of 1 × 105 cells ml−1 and incubated with 1 μg ml−1 Ce6, Lip-Ce6, and Lip-SC (containing 1 μg ml−1 Ce6) for 4 h. Untreated cells were considered controls. After incubation, the cells were washed with PBS and then treated with 660 nm laser irradiation (50 mW cm−2) for 5 min. Fluorescence images were captured using a fluorescence microscope under fluorescein isothiocyanate (FITC) channels.
2.8. Antitumor effect in vitro
In vitro cytotoxicity was measured using the Cell Counting Kit-8 (CCK-8) assay. A375 cells were seeded in a 96-well plate and treated with DMEM containing Lip-S, Lip-Ce6, and Lip-SC, with different concentrations, for 8 h. The cells were divided into two groups: one group was irradiated with 660 nm laser for 5 min and further incubated for 24 h. The other group was not irradiated and was incubated for 24 h. For viability tests, CCK-8 was added to each well, which was subsequently incubated for 1 h. The absorbance at 450 nm was measured on a microplate reader (Multiskan FC, Thermo Fisher Scientific, USA). Cell viability was calculated as follows:

where [A]sample refers to OD450 values of the cells under different treatments, [A]con refers to OD450 values of the cells only, and [A]blank refers to OD450 values of the CCK-8 solution.
For live–dead cell staining, A375 cells were seeded in 35 mm diameter confocal dishes and incubated with Lip-S, Lip-Ce6, and Lip-SC for 8 h. Then, the cells were treated in the dark or with irradiation (660 nm, 50 mW cm−2), followed by incubation for another 24 h. After that, co-staining of calcein-acetoxymethyl ester (AM) and propidium iodide (PI) was performed. Fluorescence images were captured using fluorescence microscopy.
The enhanced PDT effects of Lip-SC on A375 cells were estimated using flow cytometry. A375 cells (1 × 106 cells ml−1) were seeded on 24-well plates overnight and treated with blank liposomes, Lip-SB3CT, Lip-Ce6, or Lip-SC for 8 h and exposed to a 660 nm laser (50 mW cm−2, 5 min), followed by incubation for another 12 h. Then, the cells were harvested and stained using the Annexin V-FITC/PI Apoptosis Detection Kit (V13241, Thermo Fisher Scientific, Massachusetts, USA). The treated cells were analyzed using the FACScan system (BD Biosciences, NJ, USA), an automated flow cytometer.
2.9. Erythrocyte hemolysis test
500 μl blood sample was obtained from mouse and collected into a test tube containing heparin sodium. Then centrifugation at 3000 rpm for 10 min to precipitated the erythrocytes and wash with PBS twice. The precipitate of erythrocyte cells was resuspended by 100 μl PBS. Next, 10 μl of erythrocyte cells were treated with 1 ml PBS containing various concentration of Lip-SC (4, 2, 1, 0.5 and 0.25 μg ml−1). After incubation at 37 °C for 4 h, the mixtures in tubes were centrifuged at 3000 rpm for 10 min and then taken photos. The absorbance at 540 nm of the supernatant was measured. The absorbance is based on hemoglobin solubilized from the erythrocytes. Thus, the absorbance corresponds to the number of hemolyzed erythrocytes. Hemolysis induced with ddH2O and PBS was taken 100% and 0%, respectively.
2.10. Western blotting of NKG2DLs
A375 cells were seeded in 6-well plates and treated with Lip-S, Lip-Ce6, and Lip-SC. After 48 h, the cells were washed twice with PBS, and the cells in each group were lysed with the radioimmunoprecipitation (RAPI) assay buffer containing 1 mM phenylmethanesulfonylfluoride (Solarbio, China). Protein samples (20 μg) were electrophoresed on 10% sodium dodecyl sulfate-polyacrylamide gels. Then, the proteins were transferred onto a nitrocellulose filter (NC) membrane. After preincubation in blocking solution (5% milk) at room temperature for 1 h, the NC membrane was incubated with rabbit anti-human monoclonal antibody of MICA, MICB, ULBP1, ULBP2, MMP2, and MMP9 (1:1 000) (Abcam, UK) and rabbit anti-human β-actin (Cell Signaling Technology Inc., USA) at 4 °C overnight. After being washed thrice for 10 min with Tris-buffered saline containing 0.1% Tween-20 (TBST) (Solarbio, China), the membrane was incubated with a secondary antibody linked to horseradish peroxidase (goat anti-rabbit immunoglobulin G; Jackson Immunoresearch Laboratories Inc., USA) for 1 h at room temperature. After washing with TBST thrice for 10 min, the membranes were incubated with the substrate of chemiluminescence kit (Pierce ECL, Thermo Fisher Scientific, Massachusetts, USA). Images were captured using ClinxChemiScope (Clinx Science Instruments Co. Ltd, Shanghai, China). The grayscale band value was measured using Image-Pro Plus (Media Cybernetics, Inc., Maryland, USA).
2.11. Enzyme-linked immunosorbent assay (ELISA) for soluble NKG2DLs
The cell culture supernatant was collected before or after 48 h exposure to the various treatments. Soluble major histocompatibility complex class I-related chain A and B (MICA and MICB) were measured using Human MICA ELISA Kit and Human MICB ELISA Kit (Solarbio, China) according to the manufacturer's instructions. Plates were read at OD450 nm.
2.12. Metastasis inhibition analysis
The inhibition of tumor cell migration was determined using adhesion and invasion assays. A375 cells were incubated with liposomes and treated with 660 nm laser irradiation (50 mW cm−2) for 5 min After being treated for 48 h, the cells were collected and counted.
For adhesion assay, the cells (1 × 105 cells/well) were added to 96-well plates, incubated for 30 min, and washed with PBS to remove nonadherent cells. The remaining cells were stained with 0.1% crystal violet for 5 min and then dissolved using 30% acetic acid. Absorption was obtained at 590 nm. The percentage of inhibition was expressed using 100% being the control.
Cells (5 × 105 cells/well) treated with serum-free medium were seeded into transwell upper chambers (Corning, Inc., NY, USA) with 8 μm pore membranes coated with Matrigel, and 600 μl medium with 20% FBS was added to the bottom chamber. The cells were allowed to invade for 9–12 h at 37 °C. The invaded cells were stained with 0.1% crystal violet and photographed.
2.13. NK cell-mediated cytotoxicity
NK cell-mediated cytotoxicity was measured using lactate dehydrogenase (LDH) release assay. Briefly, the A375 cells were incubated with Lip-SB3CT, Lip-Ce6, and Lip-SC in a 96-well plate and then treated in dark or with 660 nm laser irradiation. NK cells were incubated with treated target cells for 4 h at a 4:1 E–T ratio. After 4 h of incubation, 100 μl supernatant was transferred to a new 96-well plate. LDH released in the supernatant was detected using the LDH Detection Kit according to the manufacturer's instructions.
2.14. In vivo fluorescence imaging
BALB/c nude mice (4−6 weeks old, 17 ± 4 g, male and female in half) were purchased from Xi'an Keaoke Biological Technology Co., Ltd (Xi'an, China) for animal studies. All animal procedures were approved by the Animal Ethics Committee of Xi'an Jiaotong University. A total of 1 × 106 A375 cells in 200 μl of PBS medium were hypodermically injected into the subcutaneous tissue of all mice.
In vivo fluorescence imaging of mice was initiated as the tumor volume increased to approximately 200 mm3. Ce6 and lip-Ce6 (100 μl, 200 μmol l−1) were injected intravenously into tumor-bearing mice. The mice were imaged by an IVIS Spectrum (Perkin Elmer) at different post-injection times (0, 2, 8, 12, 24 h). In addition, 24 h after injection, the mice were dissected, and tumor tissues and main organs (i.e. heart, liver, spleen, lung, and kidney) were imaged ex vivo. During the imaging process, the excitation wavelength was 645 nm and the collected emission wavelength was 680–720 nm.
2.15. Statistical analysis
The results were statistically analyzed using GraphPad Prism 9 (GraphPad Software, Inc., California, USA). The results are expressed as mean ± standard deviation. Comparisons between multiple groups were conducted using one-way analysis of variance. Statistical significance was defined as p < 0.05 (*), p < 0.01 (**), and p < 0.001 (***).
3. Results
3.1. Preparation and characterization of liposomes
The liposomes were prepared using a film dispersion and extrusion method. The chemical structures and molecular weights of lipids, Ce6, and SB-3CT are shown in figure S1 (available online at stacks.iop.org/NANO/32/455102/mmedia). The Cryo-TEM images showed that the liposomes could keep regular morphologies (figure 2(a)). Meanwhile, no aggregates were observed in the Cryo-TEM images. As seen in the DLS results (figure 2(b)), the sizes of the liposomes were approximately 140 nm with PDIs of approximately 0.05 (table 1). Figure S2a implies the successful loading of Ce6 and the synthesis of Lip-SC. The encapsulation of Ce6 by liposome was further investigated by FT-IR spectra (figure S2b). Compared to the spectra of Ce6 and the mixture (blank liposome and Ce6), the characteristic vibrations band of Ce6 at 1600 cm−1 disappeared in that of lip-Ce6 due to the embedding, confirming successful encapsulation of Ce6. Liposomes had high drug-encapsulating efficiency for both SB-3CT and Ce6 (table 1 and table S1). When Ce6 and SB-3CT were mixed with the phospholipid at drug/lipid (D:L) ratios (mass/mass) of 1:20 and 1:10, the drug loading efficiency could reach 87.28% and 66%, respectively. However, when co-encapsulated, the encapsulation rates slightly decreased to 71.3% for Ce6 and 60.01% for SB-3CT.

Figure 2. Characterization of liposomes. (a) The Cryo-SEM images of liposomes with Ce6 and/or SB-3CT (bar = 100 nm). (b) The size distribution of liposomes with Ce6 and/or SB-3CT. (c) The stability of liposomes in PBS at 4 °C. (d) The release curve of Ce6 in Lip-Ce6 at 4 °C. (e) Quantitative analysis of singlet oxygen (1O2) in vitro. (f) Fluorescence images of intracellular reactive oxygen species generated by free Ce6, Lip-Ce6, and Lip-SC under laser irradiation (scale bar = 50 μm). Data are shown as mean ± standard deviation (n = 3).
Download figure:
Standard image High-resolution imageTable 1. Encapsulation rate of SB-3CT and Ce6 in liposome.
| Liposomes | D/L ratio a | Diameter | PDI | Encapsulation efficiency (%) |
|---|---|---|---|---|
| Empty liposomes | — | 140.07 ± 1.16 | 0.054 | — |
| Lip-Ce6 | 1:20 | 150.17 ± 1.37 | 0.066 | 87.29% |
| Lip-SB3CT | 1:10 | 141.97 ± 1.41 | 0.057 | 66% |
| Lip-SC | 1:2:20 | 134.93 ± 0.50 | 0.044 | 71.3% (Ce6) 60.01% (SB-3CT) |
a Data represent as mean ± SD. D/L ratios mean drug and liposome in the mass ratio.
To evaluate the stability of liposomes, we investigated changes in sizes after 2 weeks. After being stored at 4 °C for 14 d, the particle sizes did not change significantly, indicating that the prepared liposomes had good stability (figure 2(c)). Additionally, figure S2c proves the long-term (50 h) release of Ce6 from Lip-Ce6 with or without irradiation. The release of Ce6 was slow without irradiation; approximately 15.2% of Ce6 had been released from liposomes after shaking for 5 h in a dialysis bag and up to 22.3% after 10 h. However, the release efficiency increased sharply from 15.3% to 43.7% after irradiation for 5 min and reached the maximum efficiency of 81.4% at 30 h.
Due to low solubility, the hydrophobic Ce6 was encapsulated into the lipid bilayer of the liposomes, and the presence of Ce6 in the lipid bilayer makes the lipid shell of liposomes susceptible to disruption by laser irradiation, allowing the encapsulated drugs to effectively escape from the liposomes. To test the release of liposomes, they were incubated with PBS at 37 ℃ with different stimuli. Under the 660 nm laser irradiation, a drastic increase in Ce6 was observed in the surrounding medium. The Ce6 released was approximately 86.73% at 12 h after irradiation with 660 nm laser, whereas it was approximately 38.26% without irradiation (figure 2(d)). Additionally, the fluorescence spectra of the released Ce6 from Lip-Ce6 were also examined to prove the release of Ce6 under different conditions (figure S2d).
The generation of SOSG in liposomes under 660 nm irradiation was studied. The fluorescence intensity of Lip-Ce6 and Lip-SC were higher than that of free Ce6, and both exhibited the same ROS generation ability (figure 2(e)). Meanwhile, the fluorescence intensity of Lip-SB3CT remained extremely low after irradiation. Afterward, to confirm the antitumor effect of liposomes, the ROS levels generated by free Ce6, Lip-Ce6, and Lip-SC in A375 cells after laser irradiation were detected using fluorescence microscopy. Lip-Ce6 and Lip-SC generated higher ROS levels in A375 cells than free Ce6 after 660 nm laser irradiation (50 mW cm−2, 5 min), as represented by the fluorescence signal (figure 2(f)).
3.2. Cellular uptake and distribution of liposome nanoparticles
The effective uptake of nanoliposomes by tumor cells is important for tumor treatment. To examine the cellular uptake efficiency of Lip-SC, fluorescence spectroscopy was used to detect the fluorescence intensity of Ce6 contained in the A375 cells after incubation for 4, 6, 8, 10, and 12 h. The fluorescence intensity of Ce6 gradually increased in a time-dependent manner within 4–8 h and reached a maximum intensity at 8 h (figure 3(a)). Then, the fluorescence intensity decreased with the extension of incubation time. These data indicated that Lip-SC is effectively taken up by A375 cells. Simultaneously, the distribution of Lip-SC in A375 cells after 8 h incubation was evaluated using fluorescence microscopy. An obvious fluorescent signal of Ce6 can be observed from the cells (figure 3(b)), and the results were in line with those of fluorescence spectroscopy. Moreover, Ce6 was mainly distributed in the cell membrane and cytoplasm after release.

Figure 3. Uptake of Lip-SC by A375 cells. (a) Fluorescence intensity of Ce6 in cells after Lip-SC incubation for different durations. (b) Fluorescence imaging of A375 cells after Lip-SC incubation for 8 h (bar = 20 μm). *** p < 0.001.
Download figure:
Standard image High-resolution image3.3. The Synergistic cytotoxicity effect of Lip NPs
To assess the toxicity of nanoliposomes to A375 cells, cell viability was measured using the CCK-8 assay. In the absence of laser irradiation, blank liposomes show non-cytotoxicity to A375 cells, with cell viability as high as 92%, indicating that blank liposomes had good biocompatibility and no obvious cytotoxicity to A375 cells. Meanwhile, SB-3CT, as an MMP inhibitor, displayed dose-dependent toxicity (figure 4(a)). Comparatively, lower cytotoxicity effects were observed of Ce6 with a concentration lower than 1 μg ml−1. To examine the effects of PDT, cells with different treatments were exposed to laser irradiation. Blank liposomes were non-cytotoxic to cells (figure 4(b)). Lip-SC-treated cells showed the highest cell death rates compared with those treated with Lip-Ce6 at the same concentration. Lip-Ce6 and Lip-SC showed a dose-dependent cytotoxicity, and the cell survival rates dropped to 42.6% ± 0.7% and 46.4% ± 3.7%, respectively, at a dose of 0.1 μg ml−1. Meanwhile, the rates decreased to 10.3% ± 1.0% and 4.2% ± 0.5% for Lip-Ce6 and Lip-SC, respectively, at a concentration of 0.4 μg ml−1.

Figure 4. (a) Cell viability of A375 cells treated with different concentrations of nanoliposomes without laser irradiation. (b) Cell viability of A375 cells treated with different concentrations of nanoliposomes under 660 nm laser irradiation. (c) Fluorescence images of calcein-AM and propidium iodide co-stained A375 cells treated with blank liposomes, Lip-SB3CT, Lip-Ce6, and Lip-SC (all drug concentrations were 40 μM for SB-3CT and 0.2 μM for Ce6). Scale bar: 40 μm. (d) Flow cytometry analysis of apoptosis of A375 cells with different treatments. * Was compared with group of cells. ** p < 0.01 and *** p < 0.001.
Download figure:
Standard image High-resolution imageSubsequently, the cell-killing effect of liposomes was examined using live–dead cell co-staining. For live–dead cell co-staining, PI stains dead cells and emits red fluorescence, whereas calcein-AM stains live cells and emits green fluorescence. A large percentage of cells treated with blank liposomes were stained with calcein-AM, indicating that blank liposomes did not cause significant cell death (figure 4(c)). Compared with control cells, the number of cells treated with Lip-SB3CT stained with calcein-AM decreased, indicating that SB-3CT inhibited cell proliferation rather than killing effect. Once the 660 nm laser was applied, in the Lip-SC group, most cells were stained with PI, which showed greater significant toxicity than any single treatment (PDT or SB-3CT alone). These results proved that Lip-SC plus irradiation treatment had the most effective cancer-killing efficacy, which were consistent with the CCK-8 results.
Moreover, the anticancer efficacy was also studied using Annexin V-FITC/PI staining using flow cytometry. Blank liposomes induced little apoptosis (6.7%) in A375 cells after irradiation (figure 4(d)). Lip-SB3CT caused apoptosis regardless of the presence (54.4%) or absence (55.2%) of laser irradiation, and most apoptotic cells were in early apoptosis (38.9% and 43.0% with or without irradiation, respectively). However, compared with cells treated with Lip-SC without irradiation, cells that underwent laser irradiation had a sharp decrease in early apoptosis (from 46.8% to 30.3%) and moved to late apoptosis (from 3.63% to 25.2%), indicating significant cell apoptosis.
In addition, we performed the hemolysis test to assess biocompatibility of the Lip-SC nanoliposomes. The percentage of hemolysis induced by Lip-SC nanoliposomes was lower than 2% even at the concentration of 4 μg ml−1 (figure S3). This indicated the low hemolytic and excellent biocompatibility of Lip-SC.
3.4. Cell adhesion and invasion assays
The escape of tumor cells from the primary tumor to the metastatic site is a complex multistep process, with reduced cell adhesion and increased migration and invasion capabilities. To determine the effects of Lip-SC treatment on melanoma metastasis, cell adhesion and invasion assays were performed. Lip-SB3CT can significantly inhibit the cell adhesion (44.27% ± 0.96%) and invasion (75.67% ± 6.49%) abilities of A375 melanoma cells (figures 5 and S4). Before light was applied, Lip-Ce6 had no effect on cell adhesion (91.48% ± 6.11%) and invasion (97.81% ± 5.71%). After exposure to 660 nm laser for 5 min, the cell adhesion (71.39% ± 3.49%) and invasion (64.78% ± 1.65%) abilities of the Lip-Ce6 group were significantly reduced. Additionally, the cell adhesion and invasion abilities of the Lip-SC group after light exposure were also lower than those without light, although no significant difference was observed. Therefore, combining PDT with MMP inhibitor treatment could efficiently inhibit cancer cell adhesion and invasion.

Figure 5. Adhesion and invasion assays of A375 cells. (a) Effects on the adhesion ability of A375 cells after treatment with nanoliposomes. (b) Effects on the invasion ability of A375 cells after treatment with nanoliposomes. (c) Representative images of the invasion ability of A375 cells treated with nanoliposomes (scale bar: 200 μm). * Was compared with group of cells. *p < 0.05, **p < 0.01, and *** p < 0.001.
Download figure:
Standard image High-resolution image3.5. NKG2DL expression and possible molecular mechanism
Several mechanisms have demonstrated that high concentrations of soluble NKG2DLs derived from tumor cells may inhibit tumor immunity and NK cell-mediated target cell lysis by downregulating the expression of NKG2DLs or proteolysis of MICA or MICB, thereby contributing to tumor immune escape. MMP participates in the shedding of soluble NKG2DLs, making it an immunosuppressive molecule that mediates the escape of malignant tumors. SB-3CT can inhibit the production of soluble NKG2DLs by inhibiting the synthesis of MMP-2 (figure S5a). Therefore, to understand the immunomodulatory effects of SB-3CT, the relationship between NKG2DL expression and MMP activity in A375 cells was examined after Lip-SC treatment. The upregulation of MMP activity can induce the downregulation of NKG2DL expression in A375 cells, decreasing sensitivity to NK cells and promoting tumor cells to escape. SB-3CT can significantly inhibit the expression of MMP-2, thereby inducing the expression of NKG2DL (figure S5b), while reducing the production of soluble NKG2DLs (figure S5c); thus, NK cells can better recognize and kill tumor cells.
Lip-SB3CT and Lip-SC can inhibit the expression of MMP-2 (figure 6(a)). Simultaneously, Lip-SB3CT and Lip-SC can restore the expression of MICB and ULBP2. Before light exposure, Lip-Ce6 did not affect the expression of MMP-2 and MMP-9 but can induce the expression of MICB and ULBP2; meanwhile, after 660 nm laser irradiation, Lip-Ce6 can inhibit the expression of MMP-2, and the specific mechanism has been further explored. Additionally, Lip-Ce6 can significantly induce the expression of ULBP2 after laser exposure. Before light exposure, Lip-SB3CT and Lip-SC can inhibit the production of soluble MICA, and the production of MICB changed significantly (figure 6(b)). After exposure to 660 nm laser for 5 min, Lip-Ce6 can also inhibit the production of soluble MICA (figure 6(c)), which is consistent with the suppression effects of MMP-2 expression on Western blotting. Similarly, the generation of soluble MICB is not affected by light. Thus, drug-loaded liposomes (i.e. Lip-SB3CT, Lip-Ce6, and Lip-SC) can inhibit the expression of MMP-2, increase the expression of ULBP2, and, simultaneously, inhibit the production of soluble MICA.

Figure 6. In vitro mechanism study of nanoliposomes. (a) Western blotting was used to detect the effects of nanoliposomes on the expression of MMP-2, MMP-9, and NKG2DLs (i.e. MICA, MICB, ULBP1, and ULBP2). (b) ELISA detects the influence of nanoliposomes on the production of soluble MICA and MICB. (c) After exposure to 660 nm laser for 5 min, ELISA detects the effects of nanoliposomes on soluble MICA and MICB. * Was compared with group of CON. *p < 0.05, **p < 0.01, and ***p < 0.001.
Download figure:
Standard image High-resolution image3.6. NK cell-mediated cytotoxicity
Though Lip-SC improved NKG2DL expression on the surface of A375 cells, NK cell-mediated cytotoxicity assays were performed to directly verify the enhancement of NK-related immunotherapy by Lip-SC. When A375 cells were treated with Lip-SB3CT and Lip-SC, significant enhancements of NK cell-mediated killing were observed, whereas for cells treated with Lip-Ce6 without irradiation, no enhancements of NK cell-mediated killing were observed (figure 7), which is in line with the ULBP-2 and MICA/MICB data in figure 5. However, cells exposed to 660 nm laser were significantly more sensitive to NK cell lysis (figure 7(b)). Approximately 85.67% ± 2.46% of cells treated with Lip-SC plus irradiation were killed by NK cells, whereas only 62.24% ± 3.43% of cells without treatment were killed. Together, these results suggest that Lip-SC sensitizes A375 cells that are targets of NK cell-mediated killing.

Figure 7. NK cell-mediated cytotoxicity to A375 cells without irradiation (a) and after irradiation for 5 min under 660 nm laser (b). Results are expressed as means of triplicate culture ± standard deviation. * Was compared with group of cells. *p < 0.05, **p < 0.01, and ***p < 0.001.
Download figure:
Standard image High-resolution image3.7. Tumor imaging and biodistribution of Lip-SC nanoliposomes in vivo
In order to investigate the anticancer feasibility of Lip-SC in vivo, we explored the biodistribution of nanoliposomes by tumor imaging using A375 tumor-bearing nude mice as animal models. As shown in figure 8, distinctly fluorescent signals were observed in tumor areas after an intravenous injection. Lip-SC nanoparticles were accumulated in tumor 8 h post-injection and sustained inside tumor for 24 h (figure 8(a)). As to free Ce6 treated mice, little to no tumor accumulation was observed. It is clearly shown that there are still strong fluorescence signals at the time of 24 h, which suggested that the Lip-SC could be retained for a long time after accumulation in tumor areas.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. In vivo fluorescence imaging and biodistribution analysis of nude mice bearing tumors after tail vein injection of Ce6 and Lip-SC. (a) Tumor imaging of A375-bearing mice after intravenous injection of Ce6 and Lip-SC nanoparticles at different times. (b) Fluorescence images and (c) mean fluorescence intensity of Lip-SC nanoparticles in tumor and other major organs 24 h after intravenous injection.
Download figure:
Standard image High-resolution image{kind=link}
24 h after intravenous injection, the mice were removed and the biodistribution of Lip-SC in the tumor and other tissues was studies. The main fluorescence was observed in the tumor, while some fluorescence existed in the liver and lung. On the contrary, there is little fluorescence in the heart, spleen, and kidneys. The mean fluorescent signal of Lip-SC in tumor tissue and other organs is demonstrated in figure 8(b). From the results, Lip-SC treated mice indicated higher tumor uptake than free drugs. Other signals were observed mainly in liver due to drug metabolism. In a word, lip-SC were able to efficiently accumulate and be retained in tumor areas after intravenous injection, whose outcome is beneficial for anticancer applications in vivo.
4. Discussion
Conventional PDT is often insufficient to treat metastatic tumors, such as melanoma. Increasing the efficiency of PDT using smart nanoparticles is a promising optional strategy to activate the immune system and achieve stronger immune responses [30]. In this study, we demonstrated that a combination of Ce6-mediated PDT and SB-3CT, an MMP inhibitor, induced the synergistic photodynamic–immunologic response with local intervention to suppress the treated melanoma cells and control their migration by inducing the expression of NKG2DLs on tumor cell membrane, leading to antitumor immunity against A375 cells.
The NKG2D–NKG2DL pathway plays an important role in cancer immunotherapy [15, 31]. The combination of NKG2D on immune cell membranes (mainly NK cells) and NKG2DLs on tumor cell membranes is one of the keys to spontaneous tumor immune recognition and can mediate a direct killing effect of NK cells on tumor cells without the antigen presentation process. Therefore, local therapies that can effectively activate and induce NKG2D-based responses could quickly improve the therapeutic effects of cancer immunotherapy. Mechanistic studies have shown that there is always an inflammatory response in treated cells after PDT, which reactivates immune surveillance of tumor tissue and provides a good microenvironment for in situ immune intervention [32, 33]. Additionally, cytotoxicity induced by PDT alone was insufficient to eliminate distant metastasis tumors, whereas immunotherapy could control the metastasis of tumor cells to a certain extent [34].
Studies have demonstrated that NK cells can be activated after PDT treatment and may contribute to the immune response triggered by PDT [35]. The results may be attributed to the induction of NKG2DLs in tumor tissues after treatment with PDT, thus increasing the sensitivity of cancer cells to NK cells. Our results suggest that Ce6-based PDT sensitizes A375 cells, which are targeted by NKG2D-based killing, and that combination therapy enhances the immune-sensitizing properties of PDT. Park MJ et al [36] have found that mRNA levels of the MICA/B and ULBP1, 2, and 3 genes in the SW-900 human lung cancer cell line were induced by sublethal doses of hematoporphyrin, which was consistent with our results on Lip-Ce6 treatment. However, in a study by Belicha, MICA expression was significantly induced after treatment with 2-[1-hexyloxyethyl]-2-devinyl pyropheophor-bide-a; however, PDT treatment did not increase the expression of MICB or any ULBP family member [37]. Despite the controversial findings, the common point following PDT treatments comes to the increased sensitivity of tumor cells to NK cell lysis. Thus, one of the most important roles of PDT is the induction of NKG2DLs expression, which can directly regulate the activation of NK cells and enhance susceptibility of cancer cells to NK cells.
MMPs are involved in the shedding of NKG2DLs. The inhibition of MMPs reduced the level of released NKG2DLs, increased cell surface expression, and reversed their immunosurveillance escape properties. Increased MMP expression is strongly associated with tumor aggressiveness, stage, and patient prognosis [38]. MMP-2 levels detected in the serum and cancer tissue could be used as indicators of the severity of breast cancer invasion and tumor size [39]. Shiraishi et al have found that MMP inhibitors could restore the expression of NKG2DLs in clinical gastric tumor samples, thereby improving their susceptibility to NK cells in vitro [40]. In this study, we showed that the inhibition of MMPs could enhance susceptibility to NK cells, along with the upregulated surface expression of MICA/B and ULBP-2 in tumor cells.
In this study, we found that the combination of MMP-inhibitor-related immunotherapy and Ce6-based PDT could upregulate the expression level of NKG2DLs, especially ULBP-2, and inhibit the shedding of soluble MICA and MICB, leading to increased sensitivity of NK92 cells toward melanoma cells and enhancement of NK cell-mediated antitumor immune response compared with immunotherapy alone. According to our previous study, along with PDT, NKG2D-based immunotherapy had a significantly higher cure rate in tumor-bearing animals than PDT alone [41]. Moreover, the combination of adoptive transfer of NK92MI cells and PDT has a significantly improved therapeutic effect compared with PDT alone, indicating that advanced NK cell-based adoptive immunotherapy can be developed as an effective adjuvant for PDT for treating solid malignant tumors. This result may be due to the induction of NKG2DLs in tumor tissues after PDT treatment.
Furthermore, the in vivo imaging observations showed that lip-SC can efficiently accumulate and be retained in the tumor site, which is promising for anticancer treatment. These data further supported the anticancer application of PDT plus NKG2D-based immunotherapy: interventional PDT damaged the primary tumors via ROS; Ce6-based PDT induced the expression of NKG2DLs; and the combination of SB-3CT and Ce6 induced NK cell immunity to control metastases. Although this study showed the potential of combination therapy for A375 cells as a new treatment for melanomas, its antitumor mechanism should be further investigated on animal models in the future.
5. Conclusions
In summary, we constructed a nanovesicle platform (Lip-SC) for enhancing the efficiency of A375 cancer PDT combined with NKG2D-mediated immunotherapy. Liposomes acted as a nanocarrier to encapsulate both Ce6 and SB-3CT and exhibited good stability and effective accumulation at tumor areas. The relatively fast internalization of Lip-SC allowed us to induce severe cytotoxicity due to ROS generation with 660 nm laser irradiation and procedural release of SB-3CT. Due to the in situ immunomodulatory effects of SB-3CT, Lip-SC significantly influenced the expression and shedding of NKG2DLs, increasing the sensitivity of A375 cells to NK cells and subsequently inhibiting melanoma cell proliferation and metastasis. The nanoliposomes under study highlight the importance of rationally designing nanomaterials to combine PDT and immunotherapy and promote a new optional strategy for melanoma treatment.
Acknowledgments
The authors thank Dr Huimin Tong and Dr Yu Wang at Instrument Analysis Center of Xi'an Jiaotong University for their help with Cryo-TEM and DLS analysis, Dr Lan Chen for her help with flow cytometry analysis.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Funding
This work was supported by the National Natural Science Foundation of China under Grant [number 61875159, 61727823 and 61975160], and China Postdoctoral Science Foundation under Grant [number 2018T111036].
Disclosure
The authors have no conflicts of interest to declare.