
Abstract
In this study, we critically evaluate the performance of various interatomic potentials/force fields against a benchmark ab initio database for bulk amorphous alumina. The interatomic potentials tested in this work include all major fixed charge and variable charge models developed to date for alumina. Additionally, we introduce a novel machine learning interatomic potential constructed using the NequIP framework based on graph neural networks. Our findings reveal that the fixed-charge potential developed by Matsui and coworkers offers the most optimal balance between computational efficiency and agreement with ab initio data for stoichiometric alumina. Such balance cannot be provided by machine learning potentials when comparing performance with Matsui potential on the same computing infrastructure using a single Graphical Processing Unit. For non-stoichiometric alumina, the variable charge potentials, in particular ReaxFF, exhibit an impressive concordance with density functional theory calculations. However, our NequIP potentials trained on a small fraction of the ab initio database easily surpass ReaxFF in terms of both accuracy and computational performance. This is achieved without large overhead in terms of potential fitting and fine-tuning, often associated with the classical potential development process as well as training of standard deep neural network potentials, thus advocating for the use of data-efficient machine learning potentials like NequIP for complex cases of non-stoichiometric amorphous oxides.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Alumina, a glass oxide, is considered one of the most crucial materials in the field of technology. Its application as a coating for aluminum parts is widely recognized for its ability to enhance wear and corrosion resistance. Recently, amorphous oxide glasses have emerged as a promising material for modern electronics, as reported in the study by Frankberg et al [1], and as a strengthening agent in nanoscale multilayered systems, as demonstrated in the work by Edwards et al [2]. The broad application range of amorphous alumina makes its properties highly interesting, including thermal and electrical transport in semiconductor devices. To predict the device's reliability under various loading conditions, understanding the mechanical properties of amorphous oxides is critical. Therefore, atomic-scale simulations become inevitable to control and comprehend the structural changes during mechanical loading. However, classical molecular dynamics (MD) simulations are susceptible to errors, especially with regard to the interatomic potentials/force fields.
To address the limitations of classical MD simulations, ab-inito calculations offer a potential solution. These calculations obtain interatomic forces from highly accurate first-principle density functional theory (DFT) calculations, which adequately describe the chemical bonding of systems and have been shown to provide results in agreement with experimental data [3–6]. Despite their proven usefulness in predicting material properties, such as crystal structures, electronic structures, elastic constants, and energetics, ab-inito calculations have several limitations [7]. The computational speed is by far the most significant drawback allowing for simulating systems composed of a few hundred atoms for a few picoseconds, resulting in insurmountable difficulties in resolving experimental time and length scales. Moreover, these calculations can primarily be used to study the structure and vibrational dynamics of bulk crystalline phases and small clusters.
In the computational material sciences field, (semi-)empirical interatomic potentials and machine learning (ML) interatomic potentials for MD are valuable companions for upscaling DFT calculations. These interatomic potentials can handle structures up to micrometer size, making them indispensable for practical applications. Particularly important is that those interatomic potentials are able to reproduce correctly the aforementioned physical properties.
Since the 1970s many interatomic potentials have been proposed for materials with covalent, ionic, or metallic bonding nature. A recent overview summarizing the various empirical interatomic potentials and methods used in the past to model oxide glasses was published [8]. Therefore, in this paper, we will solely focus on Al2O3 and their limits of applicability. For alumina, several Born–Mayer-Huggins (BMH) potentials were proposed, containing a pairwise term as well as a Coulomb term based on fixed point charges. The BMH potential developed by Alvarez et al, was initially developed for modeling the metastable γ-Al2O3 phase [9, 10], but has proven to be a suitable candidate for modeling size-dependent phase transformations in alumina nanoparticles [11]. The potential of Matsui [12], also follows the BMH formalism and was initially designed to be a transferable potential for modeling crystals and melts. It can model well the structural properties quantified by XRD and neutron diffraction of liquid, amorphous, and crystalline alumina [13, 14] and showed good agreement for elastic properties through extrapolation from experimental data [1]. A BMH potential for modeling the structural and dynamic properties of aluminosilicate melts is the most recent BMH potential that can be used to model amorphous alumina [15]. It was developed by Jakse and coworkers and recently used in a study that compares different potentials for the sintering of two alumina nanoparticles [16], or for studying structural, vibrational, and transport properties of liquid and amorphous alumina [17].
Vashishta et al complemented the two-body interactions with a three-body term [18]. This three-body adds the covalent contribution to a potential, as it is able to describe the angular contributions Al-O-Al and O-Al-O. Due to its three-body term, this potential is computationally more expensive and was not able to represent the particle size-induced phase transformations as well as the Alvarez potential [11]. However, for elastic constants, bulk modulus, and phase stability for the α-phase, which are most important for mechanical properties, the Vashishta potential showed good agreement with experiment [18].
Coming from the metallic description of materials many-body potentials were developed that were able to describe many fundamental properties of metals. This potential family is called the embedded-atom method (EAM) potential, for modeling fcc metals like aluminum. Streitz and Mintmire (SM) were the first to describe the Al2O3 with the metallic EAM formalism. Specifically, they combined a Finnis–Sinclair potential with a charge equilibration (QeQ) algorithm [19] leading to so-called charge transfer ionic potentials (CTIP). This QeQ algorithm allows a variation of charges that depends on the atomic environment by considering the radial distribution of charges instead of point charges [20]. The SM potential now allows us to describe both, Al2O3 and Al, which is extremely important for modeling actual applications of Al2O3. Furthermore, the SM potential shows a transition to the amorphous phase [11], from crystalline γ-phase to amorphous phase with decreasing nanoparticle size. The SM potential becomes unstable for short ion distances and lacks generality when including more than one metal element [21]. To overcome this shortcoming, Zhou et al added charge bounds to the original CTIP model, allowing for high-pressure simulations as a transferability of the procedure to all EAM potential [21]. Unfortunately, the Zhou CTIP model is not available for a broader community as it is not implemented in any of the common MD codes.
Overcoming the limitations of the EAM potentials to fcc metals, Baskes proposed a modified embedded-atom method (MEAM) potential that considers a directionality of bondings along the first neighbors, enabling the description of fcc-, bcc-, hcp-, diamond-structured, as well as gaseous elements [22]. Later, Lee and Baskes extended the MEAM model to the second nearest neighbor (2NN-MEAM) in order to prevent artificial structures in bcc metals [23, 24]. A detailed description of the formalism and different applications for the 2NN-MEAM potentials have been summarized in [25], showing the vast horizon of applicable systems for this potential family going beyond just metallic systems. To further expand its applicability range, the 2NN-MEAM developers proposed a QeQ procedure (2NN-MEAM+QeQ) based on the existing models [19–21] that showed for the representative covalent and metallic oxides of Ti and Si a great performance for structural and mechanical properties [26]. Recently, the potential for alumina was developed by the same group [27] and is currently being tested for the properties of crystalline alumina polymorphs, while we got early access to test potential performance for amorphous alumina structures.
The Second-Moment-Tight-Binding-QeQ (SMTB-Q) also aims to describe the bulk, surfaces, and defects of ion-covalent compounds. The advantage of those potentials is that they can be fitted on the physical properties of one stable phase, whereas the properties of other phases can be obtained without fitting. Due to the analytical expression for ionic charges as well as covalent energy, the potential aims for fast calculations. An SMTB-Q potential for bulk Al2O3 was proposed by Salles et al [28].
Complementary to the metallic description for materials through EAM potentials, bond order or so-called Tersoff potentials were first developed to describe covalent materials this approach incorporates the dependence of bond order on the local environment [29]. Tersoff potentials, therefore, make it possible to model bond-breaking and formation [29]. Finding that the combination of a Tersoff potential with a QeQ procedure for the Si/SiO2 system showed similar charge instabilities as the SM potential [19, 20, 30], Liang et al developed a modified Tersoff+QeQ potential, the charge optimized many-body (COMB) potential. The third generation of the COMB potentials (COMB3) was also optimized for the Al/Al2O3 system. More details on the especially COMB potentials can be found in [31].
Another, even older, many-body potential considering variable charges of ionic bonds initially developed for modeling hydrocarbons is the so-called reactive force field (ReaxFF) developed by van Duin et al [32]. The numerical methods and algorithmic techniques for the charge equilibration in ReaxFF and its implementation in LAMMPs have been developed by Aktulga et al [33]. ReaxFF potentials can be used on a huge variety of materials including, hydrocarbons [32], Si-glasses, and metallic oxides, including the Al2O3/Al system [34]. This potential has been applied to test the mechanical behavior of alumina-coated aluminum nanowires under tension and compression [35]. Cyster et al used it to model the fabrication of am-Al2O3 tunnel junctions, showing agreement with experimental densities in the final junctions. All details on ReaxFF can be found in the recent review article [36]. The review also indicates that the older versions of ReaxFF for the Al2O3/Al system are no longer maintained and available. The formalisms of COMB3 and ReaxFF are of fundamental difference even though the materials that can be modeled are similar, more details about similarities and details of both potentials can be found in [37]. Both COMB3 and ReaxFF have recently been used to model the nanoindentation in Al2O3 coated Al, where COMB3 shows crack initiation and propagation in agreement with experiments, while ReaxFF with no crack is visible but the potential rather displays a pile-up of atoms surrounding the indenter.
As ML became a hot topic in all different branches of digital and technical applications, ML and data-driven approaches and potentials for atomistic modeling of materials are getting more and more popular. The ML potential is then a complex mathematical description of a potential energy surface that has been learned from a reference database [38]. High-dimensional neural networks using atomic-centered descriptors with a certain cut-off are used to describe the total energy of a system. This method is based on the assumption, that the energy of an atom only depends on its atomic and respective environment [8]. Different methods and algorithms are summarized elsewhere [8, 39]. In the case of amorphous alumina, the Behler–Parinello neural network potential was developed in the n2p2 framework [40] and reported in the literature but has been never shared with the research community [41]. Considering other amorphous ionic materials, Batzner et al [42] developed and trained a graph equivariant neural network (NequIP) potential on amorphous Li4P2O7 obtaining very good results in predicting forces, energies, and structures. Moreover, the NequIP framework due to its equivariant construction is superior to n2p2 in many aspects, demonstrating better data efficiency and higher performance at the same levels of accuracy [42, 43]. The biggest downside of ML potentials is their low extrapolation ability due to the lack of physics, so if configurations occur that were not learned by the model, unpredictable results can be produced. To overcome this, active learning methods are developed allowing for training ML potentials on the fly on DFT data computed for new configurations appearing in the simulation [44]. To achieve this, on-the-fly error estimation and uncertainty analysis become essential for identifying when extrapolation occurs. In any case, long-range effects, like Coulomb interactions, are extremely important for the descriptions of partly ionic systems such as Al2O3 and other metal oxides, which can only be covered with extremely high cut-offs, which limit the computational throughput. In particular, liquids and amorphous materials become challenging, as there is no long-range order that can be learned from small DFT supercells in the training dataset. Anyways, several methods have been proposed to handle the long-range interactions in ML potentials [45, 46], while none of them is so far really widely accepted by the community [8].
So even with state-of-the-art methods, we arrive always at the same problem, that potential is only intended to perform well inside the boundaries of the training data or inside boundaries that it was fitted, also limited by potential formulation, amount of physics included, and number of fitting parameters available. For many real applications of interatomic potentials, however, their accuracy should be balanced with the speed: (1) speed of training defined by the time needed to collect the fitting database as well as the time to optimize potential parameters with such database; and (2) speed of use i.e. computational performance of the potential defined by its mathematical complexity and parallelization ability. ML models are well-known to be data-reliant and slow as compared to classical empirical potentials. Thus, taking amorphous oxides as an example, can we define the limits of applicability of classical and ML models and provide some rules of thumb on when to use which model?
To address such question, we evaluated here various Al-O interatomic potentials to determine their effectiveness in modeling amorphous alumina (am-Al2O3) at different densities and stoichiometries, anchoring our approach on extensive ab-initio calculations reported in the literature. As the properties of amorphous materials, in general, are highly dependent on the manufacturing pathway, it makes it hard to compare them with each other. Therefore in this study, we assess the performance of various potentials against ab initio calculations for key properties like energies and forces, focusing on the atomic structure across a broad spectrum of densities and stoichiometries in amorphous alumina. Adopting force- and energy-matching methodologies, commonly employed for fitting interatomic potentials, has been validated as effective for replicating a range of material properties, including structural, vibrational, mechanical, and transport attributes [41–43, 47–50]. It's noteworthy that the n2p2 neural network potential, despite not being publicly accessible, fitted to energy and force data from the same database, has demonstrated remarkable accuracy in emulating these properties in amorphous alumina, aligning well with both experimental and ab initio data [41]. Thus, our research aimes to explore whether physics-based empirical models could rival ML/neural network approaches in specific material categories like metal oxides while offering superior computational efficiency and meaningful physical interpretation of atomic interactions. We contend that this objective has been successfully met, with the boundaries of existing models' applicability being thoroughly established and articulated. Our findings are instrumental in guiding the initial selection and subsequent assessment of interatomic potentials for simulations in real-world scenarios, focusing on particular problems and properties.
Conducting fair comparisons in real-world scenarios within the scope of this work is challenging due to the intrinsic uncertainties in the amorphous structure formation processes used in atomistic simulations, often executed via melt quenching [14, 18, 41]. Such methodologies significantly influence the resulting structure, its formation energy, and various properties, depending on the parameters like annealing temperature, annealing duration, and quenching rate [14, 51–53]. Similarly, uncertainties pervade experimental data for amorphous materials produced through different methods, such as natural oxidation or anodization of metal [54–56], magnetron sputtering [57, 58], or atomic layer deposition of oxide films [59, 60]. These factors limit the ability to perform direct and equitable comparisons with real-world models, thereby necessitating a reliance primarily on comparisons to ab initio data.
Our objective in this comparative study was not just to highlight differences but to provide clear guidance on optimizing MD simulations for material research. The findings are expected to be instrumental in guiding the choice of interatomic potentials in subsequent research involving am-Al2O3. First, we present our methods of data preparation and evaluation of interatomic potentials as well as fitting neural network NequIP potentials in the 'Methods' section. The 'Results and Discussion' section is divided into two primary subsections. The initial subsection delves into the accuracy of the various interatomic potentials when applied to stoichiometric am-Al2O3 samples. The subsequent subsection shifts its lens towards non-stoichiometric am-Al2O3. Such non-stoichiometric samples are particularly intriguing for advanced applications, especially those boasting bulk defects and free surfaces or exhibiting pronounced material inhomogeneities when subjected to external stimuli. Finally, we summarize our findings in the 'Conclusions' section, also sharing our outlook on which applications and material properties can be covered by Pareto-optimal interatomic models identified in this work.
2. Methods
2.1. Selection of the reference data
To assess the accuracy of the empirical interatomic potentials in modeling amorphous alumina, a comparison with ab-inito MD data was performed. In order to reduce computational time, a pre-existing database (https://archive.materialscloud.org/record/2020.89) from a neural network interatomic potential study was selected as the reference for comparison [41]. The database comprises approximately nstructures = 160 000 structures from ab initio MD calculations, and a representative [61] selection of less than 1.0% of these structures were chosen for analysis. Three critical properties, namely energy/atom, stoichiometry, and density, were selected to represent the database's essential features. To ensure the database's integrity, abnormal configurations such as overlapping atoms were removed by limiting the energy range from −7.5 to 0 eV/atom. Additionally, pure Al and O structures were eliminated as the focus of the study is on pure am-alumina.
From this cleaned database the selection process was started, with the goal of having three independent datasets, one for testing classical and neural network potentials, and two for training and validating neural network potentials, with the latter used to avoid potential overfitting. First, the database was split into N bins in each space of energy/atom, stoichiometry, and density. The respective N ( ,
, ,
, ) was selected by using the Freedman-Diaconis rule [62]. After that, approximately
) was selected by using the Freedman-Diaconis rule [62]. After that, approximately  structures were extracted from every bin in each space. The selected structures from every bin, were then split into three datasets in a ratio of 0.4, 0.4, and 0.2 for testing, training, and validation respectively. Structures that were selected twice for example for stoichiometry and density, were only used for stoichiometry. Bins that contained less than three structures were not considered. This resulted in three different datasets of size ntest = 835, ntrain = 835 and nvalidation = 382. The same procedure was used to create a test database for stoichiometric am-Al2O3 with ntest,stoichiometric = 889, in that case it was only binned in density and energy. The normalized energy/atom distribution of the different datasets can be seen in figure 1, for clarity only the full dataset distribution and the ntest and ntrain are shown. It shows that the small datasets resample the same energy/atom space as the full dataset while amplifying underrepresented structures to reduce the bias present in the original full dataset.
structures were extracted from every bin in each space. The selected structures from every bin, were then split into three datasets in a ratio of 0.4, 0.4, and 0.2 for testing, training, and validation respectively. Structures that were selected twice for example for stoichiometry and density, were only used for stoichiometry. Bins that contained less than three structures were not considered. This resulted in three different datasets of size ntest = 835, ntrain = 835 and nvalidation = 382. The same procedure was used to create a test database for stoichiometric am-Al2O3 with ntest,stoichiometric = 889, in that case it was only binned in density and energy. The normalized energy/atom distribution of the different datasets can be seen in figure 1, for clarity only the full dataset distribution and the ntest and ntrain are shown. It shows that the small datasets resample the same energy/atom space as the full dataset while amplifying underrepresented structures to reduce the bias present in the original full dataset.

Figure 1. (a) The stoichiometry and density of the full dataset in a 2D histogram. (b) The stoichiometry and density of the test dataset in a 2D histogram. (c) The normalized energy/atom distribution for the three different datasets and the original cleaned dataset from [41].
Download figure:
Standard image High-resolution imageTo train the NequIP graph neural network potentials [42], we used the version v0.5.5 that is available on GitHub (github.com/mir-group/nequip) (see here the details on the NequIP algorithm [42], underlying E(3)- equivariant graph neural network [63, 64], and the proposed training of the potential [65]). To systematically assess the influence of various hyperparameters, multiple sets of NequIP potentials were trained. Each set corresponds to one hyperparameter change around the default settings to elucidate the effect of each hyperparameter on potential accuracy and performance. Comprehensive details of these hyperparameters and sets of their values are provided in supplementary materials. For the performance and accuracy assessment of the NequIP potentials, we exclusively employed testing functions from the NequIP package. Consequently, these results might not align seamlessly with those obtained using the workflow designated for classical potentials. When considering the computational efficiency of the NequIP potential in terms of time per call, we recognize that this metric omits some computational overhead intrinsic to MD calculations. However, it remains a suitable metric for contrasting the performance across different sets.
In subsequent sections of this paper, we present a specific NequIP set corresponding to default settings. While this chosen potential is neither the pinnacle of performance nor accuracy, its hyperparameters are the ones selected from the NequIP developer's initial training file. The file with the hyperparameters, used for the training can be found here: https://doi.org/10.24435/materialscloud:ya-3k [61].
2.2. Comparison of force and energy
The force and energy comparison between the DFT database and the different interatomic potentials was a critical aspect of this study. The MD calculations were primarily carried out using the open-source simulation software LAMMPS [66], while a Fortran code, namely KISSMD was used for the 2NN-MEAM+QeQ potential https://cmse.postech.ac.kr/home_2nnmeam.
In all calculations, no dynamic steps were executed. To account for the different QeQ procedures, 10 simulation steps without updating atomic positions were performed to converge the charge calculation in order to ensure charge equilibrium with all the different QeQ methods. This is done, as some of the QeQ procedures do not allow adjusting the maximum number of charge equilibration steps manually. Detailed procedure and parameter values are available in the different simulation scripts published on Materials Cloud [61].
Forces and energies were extracted from the MD simulations and the reference database using the Atomic Simulation Environment (ASE) as well as the Open VIsualization TOol (OVITO) [67, 68].
Direct force comparisons are feasible between DFT and MD methods. When assessing energies across varied structures, reference energies become essential for the comparison of DFT and MD. Yet, for ML potentials, direct energy comparison remains applicable. Thus, to establish a coherent comparison, consistent reference energies were ascertained for both DFT and interatomic potentials.
For stoichiometric Al2O3 structures, the reference energy chosen was the energy per atom  for α-Al2O3. Specifically, the DFT energy corresponding to n units of α-Al2O3 in an amorphous structure was subtracted from the DFT energy of the structure of interest. This procedure facilitated a meaningful comparison with the MD results. Consequently, the energy discrepancy was calculated using:
for α-Al2O3. Specifically, the DFT energy corresponding to n units of α-Al2O3 in an amorphous structure was subtracted from the DFT energy of the structure of interest. This procedure facilitated a meaningful comparison with the MD results. Consequently, the energy discrepancy was calculated using:

where n is the number of α-Al2O3 units in a structure. The reference value for α-Al2O3 is given in table 1.
Table 1. The E/N in eV/atom for α, and γ-Al2O3, as well as the type of model, is presented for the different interatomic potentials used in this work. The formation energies Ef
for Al and the oxygen dimer O2 are presented for the tested potential. Note that, SMTB-Q parametrization does not allow for consideration of pure phases of aluminum and oxygen. All energies are denoted in eV/atom.
and the oxygen dimer O2 are presented for the tested potential. Note that, SMTB-Q parametrization does not allow for consideration of pure phases of aluminum and oxygen. All energies are denoted in eV/atom.
| Potential | α-Al2O3 | Al
| O
| Model |
|---|---|---|---|---|
| COMB3 [69] | −6.51 | −3.36 | −2.58 | variable charge |
| ReaxFF [34] | −6.10 | −3.43 | −2.77 | variable charge |
| 2NN-MEAM+QeQ [27] | −6.30 | −3.36 | −2.56 | variable charge |
| SM [19] | −6.39 | −3.42 | −1.56 | variable charge |
| SMTB-Q [28] | −6.40 | — | — | variable charge |
| Matsui [12] | −8.80 | — | — | fixed charge |
| Alvarez [9] | −9.66 | — | — | fixed charge |
| Vashishta [18] | −6.35 | — | — | fixed charge |
| Jakse [70] | −13.10 | — | — | fixed charge |
| NequIP [42] | −7.46 | — | — | machine learning |
| Exp | −6.35 [71] | — | — | — |
| DFT | −7.46 (−7.65 [69]) | −3.75 | −4.39 | — |
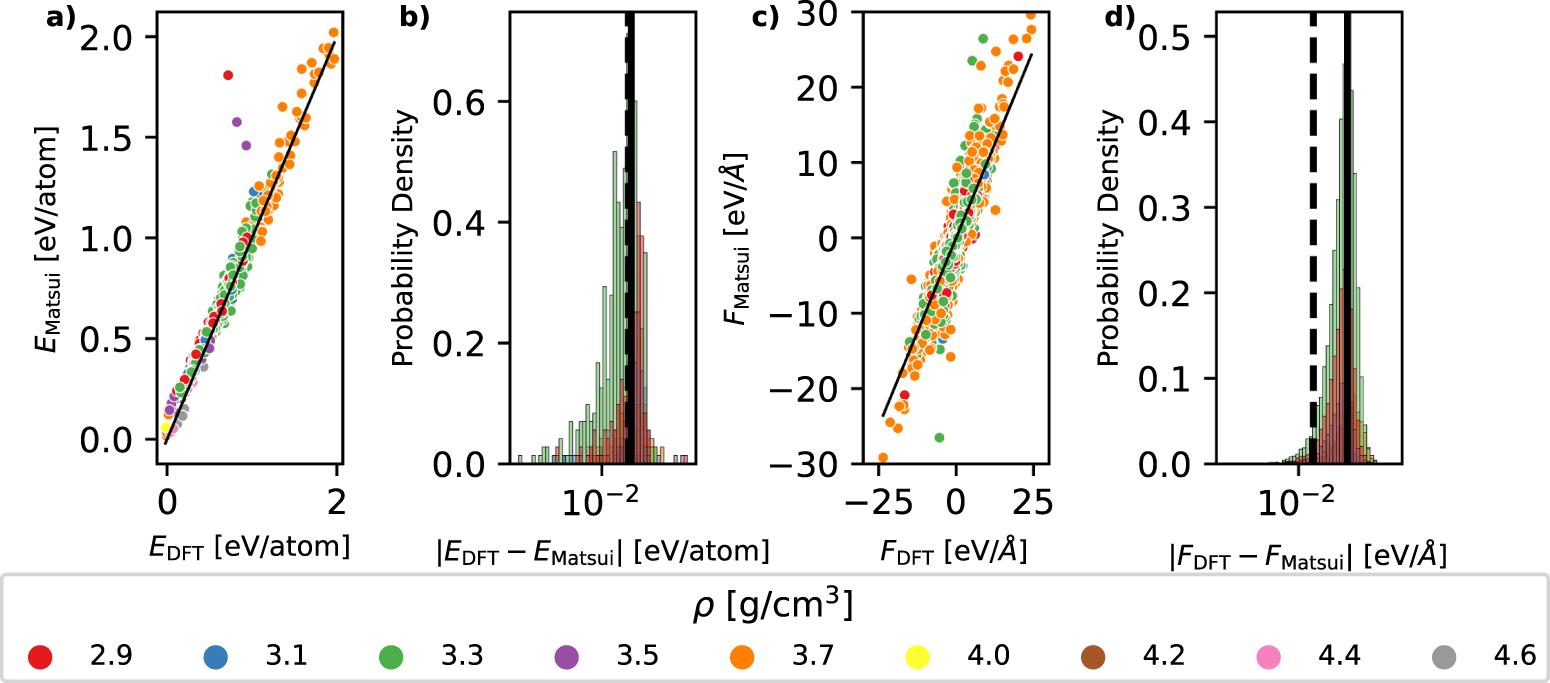
To evaluate the accuracy of interatomic potentials for stoichiometric Al2O3 against the ab initio reference, we employ force and energy matching plots as shown in figure 2. These plots use different colors to represent the structure's densities. Specifically, figure 2(a) depicts the ab initio energies  plotted against the interatomic potential energies EMatsui. Here, equation (1) was used, facilitating a direct comparison of both energies. In figure 2(a), the black line signifies an exact match with the DFT data. Points that deviate from this line indicate higher absolute error values, denoted by
plotted against the interatomic potential energies EMatsui. Here, equation (1) was used, facilitating a direct comparison of both energies. In figure 2(a), the black line signifies an exact match with the DFT data. Points that deviate from this line indicate higher absolute error values, denoted by  . Figure 2(b) displays the probability density of these error values, color-coded by different densities. Both the mean and median values of these distributions are indicated by solid and dashed lines, respectively. Lastly, figures 2(c) and (d) showcase the accuracy for forces in a similar manner, with each point representing a force vector Fi
acting on an individual atom within a structure. All force and energy matching plots for the different interatomic potentials for stoichiometric Al2O3 are given in the supplementary materials.
. Figure 2(b) displays the probability density of these error values, color-coded by different densities. Both the mean and median values of these distributions are indicated by solid and dashed lines, respectively. Lastly, figures 2(c) and (d) showcase the accuracy for forces in a similar manner, with each point representing a force vector Fi
acting on an individual atom within a structure. All force and energy matching plots for the different interatomic potentials for stoichiometric Al2O3 are given in the supplementary materials.

Figure 2. (a) The accuracy of the Matsui potential, in terms of energy, in an energy matching plot with the DFT reference on the left. As reference energy for this the formation energy of an Al2O3 unit in α-Al2O3 is used . (b) the histogram presents the density of absolute errors of (a), with dashed line being the median and the solid black line being the mean value of the distribution. (c) The accuracy of the Matsui potential, in terms of force, in an energy-matching plot with the DFT reference. (d) the histogram presents the density of absolute errors of (c). Colors refer to different densities of the amorphous structures, given in g cm−3.
Download figure:
Standard image High-resolution imageFor non-stoichiometric alumina, an alternative comparison technique is necessitated. The formation energies for the face-centered cubic structure of aluminum Al denoted as
denoted as  and oxygen dimers
and oxygen dimers  were derived as benchmark energies for both DFT and interatomic potentials. Table 1 contains the cohesive energies of various potentials, including experimental benchmarks and DFT references.
were derived as benchmark energies for both DFT and interatomic potentials. Table 1 contains the cohesive energies of various potentials, including experimental benchmarks and DFT references.
Subsequently, the formation energies of diverse structures denoted as  , were calculated, following Hess's law:
, were calculated, following Hess's law:

With these calculations, formation energies computed from both MD and DFT methods are directly comparable.
For the non-stoichiometric am-Al2O3 samples, force, and energy matching plots were produced in a manner analogous to the stoichiometric am-Al2O3 samples. An exemplar of these plots for the ReaxFF interatomic potential is presented in figure 3. In figure 3(a), the comparison was based on equation (2). The color coding represents the varying stoichiometries of the structures. Force and energy matching plots corresponding to all other employed interatomic potentials can be found in the supplementary materials.

Figure 3. (a) The accuracy of the ReaxFF potential, in terms of formation energy per atom, in an energy-matching plot with the DFT reference on the left. (b) The histogram presents the density of absolute errors. Colors refer to different stoichiometries of the amorphous structures, given as a fraction of  . (c) The accuracy of the ReaxFF potential, in terms of force, in an energy-matching plot with the DFT reference on the left. (d) The histogram presents the density of absolute errors. Colors refer to different stoichiometries of the amorphous structures, given as a fraction of
. (c) The accuracy of the ReaxFF potential, in terms of force, in an energy-matching plot with the DFT reference on the left. (d) The histogram presents the density of absolute errors. Colors refer to different stoichiometries of the amorphous structures, given as a fraction of  .
.
Download figure:
Standard image High-resolution imageFor a more detailed quantitative assessment of accuracy, we extract statistical metrics such as RMSE, median, standard deviation, skewness, and the Pearson correlation coefficient r. Summaries of these metrics for energies and forces can be found in supplementary tables S1–S4. As highlighted by Morrow et al [38], the RMSE likely offers the most insightful measure for gauging the overarching accuracy of the potentials as compared to the DFT reference. Accordingly, potentials are ranked and sequenced based on their RMSE values in the tables. Evaluating the median alongside the standard deviation and skewness can pinpoint potential shifts in accuracy distribution. Such a shift can signify a potential's difficulty in reproducing certain densities, an aspect growing in significance when addressing non-stoichiometric am-Al2O3. Finally, r gives the linear relationship between two variables. Its value spans from −1 to 1; an r value of 0 denotes the absence of any linear correlation between the given variables.
2.3. Computational performance assessment
The computational performance assessment of stoichiometric Al2O3 was conducted using the α-Al2O3 simulation cell sourced from the LAMMPS examples, consisting of 2160 atoms. Prior to each performance simulation, the simulation cell was fully relaxed. Five independent runs in the canonical ensemble (NVT) 1000 iterations each were executed on a single core (Intel(R) Xeon(R) CPU E5-2630 v3 @ 2.40 GHz) without GPU acceleration. As the NequIP potentials are optimized on Graphical Processing Units (GPUs) and too slow on a single core, we compare the performance of the Pareto optimal classical potentials with the selected Pareto optimal NequIP potentials on a single GPU node (NVIDIA® Tesla® P100 16GB and Intel(R) Xeon(R) CPU E5-2690 v3 @ 2.60GHz). No MPI parallelization is tested in this work.
Regarding non-stoichiometric Al2O3, a simulation cell randomly selected from the test dataset and multiplied till comprising 4368 atoms (2352 O and 2016 Al) was utilized, maintaining the same simulation protocol as that for stoichiometric Al2O3. Only potentials with available QeQ-procedure were employed for the non-stoichiometric Al2O3 simulations.
3. Results and discussion
It's vital to consider the context of this study when interpreting its results. The test structures utilized here have a relatively uniform distribution, and surface interactions were not considered. Hence, giving an overall ranking of interatomic potentials may be premature. Though the results will be presented in terms of statistically average metrics such as root mean square and median errors, this should not be taken as a final directive. While some potentials may appear prominent overall, their accuracy across different densities and stoichiometries as provided in supplementary materials should not be dismissed. Instead, researchers are encouraged to critically assess the data and tailor their potential choice to their specific application.
Traditionally for potential comparison against ab initio databases, we first assess the accuracy/efficiency trade-offs provided by each potential in Pareto plots, shown in figures 4(a) and (b) for stoichiometric and non-stoichiometric samples, respectively. Taking simultaneously RMSE evaluations of energies and forces, three Pareto optimal models were identified for stoichiometric alumina: Vashishta, Matsui, and ReaxFF.

Figure 4. (a) The computational performance as a function of time for stoichiometric Al2O3. 'x' symbols represent force RMSE whereas dots represent energy RMSE. (b) The computational efficiency P as a function of time for non-stoichiometric Al2O3.
Download figure:
Standard image High-resolution imageThe Vashishta potential exhibited the highest computational efficiency due to the inherent screening in the Coulomb and charge-dipole interactions in the model, though demonstrating a relatively high RMSE. However, as one can see in supplementary figure S1, the potential predictions for both energies and forces are quite good in the experimentally relevant range of densities of amorphous alumina between 2.6 and 3.4 g cm−3 [72]. Thus, we can expect such potential to only fail at high densities of bulk amorphous alumina by overestimating the corresponding energies, and defining the limits of applicability of such potential.
The Matsui potential displayed the most favorable trade-off between computational efficiency and accuracy, providing outstanding accuracy for the classical potential across all investigated densities, see supplementary figure S2. Despite a basic foundational physical model, it rivals advanced models in accuracy. This might explain why it aligns so closely with experimental findings, such as those by Frankberg et al [1]. Numerous studies, reflecting on experimental outcomes like fracture tendencies in amorphous alumina, have leaned on the Matsui potential [1, 73]. Moreover, recent research has employed this potential to study the sintering of Al2O3 nanoparticles [16, 74]. While the Vashishta and many reactive potentials showed misalignment with our databases in many areas, they have exhibited good concurrence with properties of crystalline phases [18, 69]. The inherently unique properties of amorphous materials might mean that the simpler functional form of pair potentials can describe them adequately. Our analysis reveals that the Matsui potential is the most accurate fixed charge potential among the tested ones. As indicated in figure 4(a), the accuracy of the Matsui potential is only negligibly inferior to the ReaxFF, even though the latter demands greater computational resources due to the much more complex functional form and additional charge equilibration procedure. Moreover, Matsui is overperforming ReaxFF in the high-density regime as the latter tends to underestimate the energies, especially for high-energy configurations as one can see in supplementary figure S3.
However, ReaxFF has a substantial advantage over the Matsui potential when moving on to non-stoichiometric configurations: when the structures deviate from stoichiometry, the Ewald summation that addresses long-range Coulombic interactions in fixed charge models is compromised. This is because this method loses its validity for systems that are not charge-neutral [75]. As a result, only variable charge potential models can be used to simulate non-stoichiometric configurations. Figure 4(b) demonstrates the Pareto plot for such models, with a detailed assessment of such potentials provided in supplementary materials. Unlike the previous section, results here will be categorized and presented based on their stoichiometry rather than their density. Based on overall performance over energies and forces, two Pareto optimal potentials were identified: SMTB-Q and ReaxFF. In particular, the SMTB-Q potential demonstrated superior computational efficiency and reasonable accuracy in predicting forces for Al-rich amorphous alumina (see supplementary figure S11), making SMTB-Q a preferable classical potential for accessing the dynamic and mechanical properties of such materials. Besides large computational overhead, ReaxFF provides consistent accuracy in force prediction over the whole range of stoichiometries, though tends to overestimate the energies of O-rich alumina (see supplementary figure S12). Thus, it represents the best choice for accessing the thermodynamic properties of equilibrium and Al-rich amorphous alumina.
As shown in figure 4, RMSE gives a good initial understanding of the overall accuracy of each potential. However, RMSE is particularly sensitive to outliers, which is apparent for the SM potential with notably high RMSEs due to the high number of outliers in O-rich amorphous alumina (see supplementary figure S15). The primary reason for this discrepancy is the presence of closely spaced oxygen atoms in the amorphous structure. As shown by Zhou et al [21], the charge calculation will diverge when considering small atomic distances with the SM potential, leading to unphysically high energies. Note that, Zhou et al proposed a modification of the SM potential that is not implemented in LAMMPS, and therefore not evaluated in this study.
As an alternative to RMSE, the median error is an effective measure of average potential accuracy, especially when dealing with data that might be skewed by extreme values. To provide proper statistical analysis of energy and force error distributions, we start with visualizing error data as standard boxplot figures, shown in figure 5 for stoichiometric and complete datasets, respectively. The box limits correspond to the 25th and 75th percentiles in the data, while the whiskers correspond to the 1.5 interquartile range in each direction. The points outside of whiskers are commonly treated as outliers. In this way, we can compare the number of outliers, while also seeing the typical range of error values for each potential. Due to the large error variation by orders of magnitude, the data is presented as the logarithm of corresponding energy and force errors. The vertical line inside each box represents the median value of error for each potential and metric of interest.

Figure 5. Potential accuracy for (a) forces and (b) energies using structures with (left) 2Al/3 O stoichiometry and (right) all tested stoichiometric. Potential accuracy for (c) forces and (d) energies using non-stoichiometric structures.
Download figure:
Standard image High-resolution imageNotably, the median value of the SM potential in figure 5 align more closely with other variable charge potentials, giving a more accurate representation of its typical accuracy. This potential is one of the extreme examples of mean and median separation in the error distribution, see supplementary figure S14. Similar mean-median separation to some degree is present in all potentials, see tables S1–S4, with median error commonly lower than RMSE. One interesting exception is the energy error of COMB3 potential for stoichiometric alumina, for which RMSE is lower than the median error due to the overall shift of energies while using corundum as a reference structure (see supplementary figure S9). In this case, most of the outliers are accumulated at the bottom of the distribution, skewing it towards zero, which can be also noticed in figure 5(b). This feature of COMB3 disappeared in energy error distribution for the complete dataset while using face-centered cubic Al and dimer oxygen gas as the references. The reason is a substantial overestimation of energies for Al-rich samples as shown in Supplementary figure S13, skewing the error distribution upward so that the outliers are present on both sides (see figure 5(d)).
When determining Pareto-optimal potentials, it might be useful to consider median accuracy to see if some potentials would stand out. For instance, the force error distribution for the complete dataset using 2NN-MEAM+QeQ leads to incrementally lower median error as compared to SMTB-Q, making it Pareto-optimal in both energies and forces. Moreover, the corresponding energy error distribution for 2NN-MEAM+QeQ has one-order of magnitude higher RMSE than the ReaxFF potential but a substantially lower median error (see supplementary table S3). This implies that the 2NN-MEAM+QeQ potential might be better for modeling am-Al2O3 than ReaxFF for certain stoichiometries. And indeed, by comparing supplementary figures S11 and S12, we notice that ReaxFF performs better on Al-rich structures while 2NN-MEAM+QeQ performs better on O-rich structures. In any case, there is no potential that can deliver a similar level of accuracy on the complete dataset that was demonstrated for stoichiometric alumina (compare top performing potentials in supplementary tables S1 and S2 and with the ones in supplementary tables S3 and S4, respectively).
Such inability of classical potentials to capture the physics of chemically diverse systems with complex chemical bonding led to the emergence of ML potentials [43, 76]. While the incorporation of long-range Coulomb interactions into ML interatomic potentials is still in progress, we have sufficient evidence from the literature that such interactions are effectively screened in amorphous metal oxides such as alumina [42, 46].
To demonstrate this, we trained the out-of-box graph neural network interatomic potential NequIP (no Coulomb interactions) with default settings using training, validation, and testing datasets as defined in Methods. The process of training and implementing this potential in LAMMPS was more straightforward than anticipated. Trained on just around 1000 configurations, the NequIP potential displayed much better accuracy on the complete dataset than classical potentials, see figures 5(c) and (d). It was expected to some extent since this potential was trained on similar, though very limited data. As ReaxFF is the frontrunner classical potential over both energies and forces, we further compare NequIP with it. Specifically, ReaxFF still excels in energy accuracy for stoichiometric alumina being marginally better than NequIP, though the latter excels in predicting forces. This outcome aligns with expectations since NequIP was trained with default settings on limited data, mostly for non-stoichiometric alumina. As NequIP was originally designed for extending the timescales of ab initio MD simulations, it is not surprising that default settings favor accuracy in forces over accuracy in energies, as observed in figures 5(a) and (b).
As depicted in supplementary figure S16, the exceptional energy and force predictions of the NequIP ML potential are evident. The energy is consistently matched across the entire spectrum of stoichiometries. While a few outliers are present, their presence could be mitigated by using a larger training dataset and/or fine-tuning training parameters. It's worth noting that optimizing the RMSE was not the central focus of this study, but future refinements might further decrease the RMSE, potentially approaching the accuracy reported by Li et al [41]. For example, we tested different hyperparameters one by one, varying values around the default settings with the results shown in figure 6(a). The meaning of each hyperparameter is given in the caption of the figure, and all the sets tested in this work are listed in supplementary table S5. As one can see, the majority of points are confined to the small region of computational performance with the zoomed-in version shown in figure 6(b). The most substantial role is played by the number of layers in the network, affecting both accuracy and performance. Increasing the number of base functions in the radial basis was shown to improve accuracy without large computational overhead. By switching  and
and  False, the computational performance can be improved without a large increase in RMSE, still doing worse than changing number of layers in the network
False, the computational performance can be improved without a large increase in RMSE, still doing worse than changing number of layers in the network  .
.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. (a) Pareto plot for the force RMSE for different sets of hyperparameters for the NequIP potential. Here, r equals the cutoff radius of the potential, N
equals the cutoff radius of the potential, N is the number of interaction blocks in the neural network, l
is the number of interaction blocks in the neural network, l is the maximum irrep order (rotation order) for the network's features, inv
is the maximum irrep order (rotation order) for the network's features, inv is the number of radial layers of the radial network, inv
is the number of radial layers of the radial network, inv is the number of hidden neurons in the radial function, N
is the number of hidden neurons in the radial function, N is the multiplicity of the features, r
is the multiplicity of the features, r is the p-exponent used in polynomial cutoff function, N
is the p-exponent used in polynomial cutoff function, N number of basis functions used in the radial basis, p decides whether to include features with odd mirror parity, for detailed description please see [42, 65]. The performance is given in time per call. (b) shows the same as in (a) as a zoom between time per call 0.0485 s and 0.0525 s.
number of basis functions used in the radial basis, p decides whether to include features with odd mirror parity, for detailed description please see [42, 65]. The performance is given in time per call. (b) shows the same as in (a) as a zoom between time per call 0.0485 s and 0.0525 s.
Download figure:
Standard image High-resolution image{kind=link}
From the computational performance point of view, the performance of NequIP on a single GPU is substantially slower than fixed-charge models running on the same GPU but is comparable with variable charge models that enable to model of non-stoichiometric structures (see table 2, all identified Pareto optimal classical models are GPU-optimized in LAMMPS). Moreover, the recent works by Musaelian et al [47, 77] suggest that this kind of graph neural network potential can benefit from efficient parallelization on hybrid high-performance computing platforms with near-ideal scaling, not accessible to classical potentials such as ReaxFF [78].
Table 2. The computational performance and accuracy in energy and force for stoichiometric and non-stoichiometric Al2O3 of a small selection of potentials on a single GPU/CPU core, including different NequIP potentials with different sets of hyperparameters. The accuracy was determined using a single consistent workflow for all presented potentials.
| Potential | Time per iteration (s) | stoichiometric RMSE (E) (eV/atom) | stoichiometric RMSE (F) (eV/Å) | non-stoichiometric RMSE (E) (eV/atom) | non-stoichiometric RMSE (F) (eV/Å) |
|---|---|---|---|---|---|
| Vashishta | 0.02 | 0.15 | 1.14 | — | — |
| Matsui | 0.06 | 0.05 | 0.75 | — | — |
| ReaxFF | 0.62 | 0.04 | 0.67 | 0.37 | 1.21 |
| NequiIP (num_layers = 1) | 0.10 | 0.08 | 0.20 | 0.08 | 0.14 |
| NequiIP (default settings) | 0.74 | 0.06 | 0.18 | 0.04 | 0.25 |
| NequiIP (num_base = 11) | 0.78 | 0.05 | 0.10 | 0.04 | 0.03 |
| NequiIP (num_layers = 8) | 1.87 | 0.04 | 0.08 | 0.03 | 0.02 |
4. Conclusion
In conclusion, this study compared the performance of different classical potentials in modeling am-Al2O3 structures using MD simulations. Our research strongly supports the Matsui potential for modeling bulk stoichiometric amorphous alumina materials, given its impressive computational efficiency and near DFT-level precision. The variety of potentials (ReaxFF, COMB3, 2NN-MEAM+QeQ,...) that can be used for non-stoichiometric am-Al2O3, present their own advantages and peculiarities, necessitating critical evaluation before application. Statistical analyses, like RMSE and median error, provide nuanced insights into the accuracy of these potentials. For instance, while the 2NN-MEAM displayed a higher RMSE compared to ReaxFF, its median suggests that it might have advantages in modeling certain configurations of Al2O3.
More importantly, we advocate in this work for applying NequIP to modeling more complex cases due to its performance and ease of implementation within LAMMPS. The potential advancements in the realm of MD simulations, especially with the possibility of benefiting from high-performance computing platforms through the Allegro package as suggested by Musaelian et al [47, 77], marks an exciting frontier for computational materials science. This raises the prospect of achieving simulations that reach DFT accuracy across wider scales without limitations of classical potential. Achieving this upscaling is readily feasible by leveraging existing DFT data, as demonstrated in our study, or by creating a concise training set from AIMD simulations to address varied scientific inquiries.
To effectively evaluate the potentials, both the total energy and force values of structures must be jointly considered. While energy accuracy influences the stability and local stoichiometry of amorphous oxides i.e. their thermodynamics, force accuracy plays a pivotal role in determining dynamic processes related to heat and mass transport as well as the mechanical response of such materials. The choice of potential for atomistic studies of am-Al2O3 largely depends on the specific phenomena under investigation. Beyond guiding potential selection, this study also provides deeper insight into the implications of previously published research, also guiding the development of further research in the field. For example, we highlight that capturing long-range Coulomb interactions is not necessary for accurate modeling of amorphous oxides, even with non-stoichiometric configurations, which was confirmed with fitting ML interatomic potentials.
Acknowledgments
We express our sincere gratitude to Prof. Byeong-Joo Lee for the generous opportunity to visit his lab in Korea, access to computational infrastructure for executing this work, early access to his new Al-O 2NN-MEAM+Qeq potential, invaluable insights, and enriching discussions. We also appreciate enormous support from Prof. Lee's student, Jisu-Jisub, with 2NN-MEAM+Qeq framework and KISSMD software.
We are also deeply thankful to Prof. Noel Jakse for the enlightening scientific discussions and insights on classical and ML interatomic potentials as well as analysis and comparison with ab initio MD simulation results, which have contributed significantly to shaping the research methodology in this work.
This research was supported by the NCCR MARVEL, a National Centre of Competence in Research, funded by the Swiss National Science Foundation (Grant Number 205602). V T thanks the Bern Economic Development Agency for financial support.
Data availability statement
The data that support the findings of this study are openly available at the following URL/DOI: https://doi.org/10.24435/materialscloud:ya-3k [61].
Supplementary material (7.9 MB PDF) The supplementary material features tables of potential accuracy, force and energy plots for each potential, and hyperparameters for Nequip ML potential. Access scripts and data at https://doi.org/10.24435/materialscloud:ya-3k.