Introduction to animal tissue culture science
Published March 2019
•
Copyright © IOP Publishing Ltd 2019
Pages 1-1 to 1-30
You need an eReader or compatible software to experience the benefits of the ePub3 file format.
Download complete PDF book, the ePub book or the Kindle book

Permissions
Abstract
Cell culture technology is a potential technology that involves diverse disciplines. Culture media, animal tissue culture facilities and several cell characterization tools have emerged in modern biotechnology, particularly in the area of human health. In this chapter basics of animal tissue culture are discussed with a brief glimpse of the historical background, types of cultures, their maintenance and characterization tools involved in this process. It also includes animal tissue cultures facilities and biosafety guidelines while working on mammalian cells under in vitro conditions. One of the most challenging tasks in animal tissue culture laboratory is to prevent contamination; thus this chapter also involves steps that must be considered to prevent contamination.
1.1. Introduction
Animal tissue culture technology is now becoming a significant model for many scientists in various fields of biology and medicine. Despite the various developments in animal cell and tissue culture since the late 1800s, until the early 1950s progress in animal tissue culture was stalled due to the non-availability of a suitable cell line. In the early 1950s, for the first time, successful growth of cells derived from the cervical cancer of Mrs Henrietta Lacks was demonstrated. This breakthrough using Mrs Henrietta Lacks's cells in culture successfully transformed medical and biological research, allowing numerous cellular, molecular and therapeutic discoveries, including the breakthrough of the first effective polio vaccine [1, 2]. This culture is now called HeLa, on which there were more than 60 000 publications by 2017, and which has been involved in numerous Nobel prize-winning innovations [2–4].
Animal cell culture is a significant tool for biological research. The importance of cell culture technology in biological science was realized a long time ago. Earlier dedifferentiation based experiments of cells due to selective overgrowth of fibroblasts resulted in the enhancement of culture techniques. Animal cell culture involves isolation of cells from a tissue before establishing a culture in a suitable artificial environment. Initial isolation of the cells from the tissues can be achieved by disaggregation using enzymatic or mechanical methods. The source of the isolated cells is usually an in vivo environment, but sometimes cells are also derived from an existing cell line or cell strain. Animal cell culture offers suitable model systems for investigating the following factors:
- Drug screening and development.
- Mutagenesis and carcinogenesis.
- Normal physiology and biochemistry of cells.
- Potential effects of drugs and toxic compounds on the cells.
In addition, it also permits reliable and reproducible results, and is thus considered as a significant model system in cellular and molecular biology. Mammalian cell culture requires an optimal environment for growth. Environmental conditions are divided into nutritional requirements and physicochemical requirements. Nutritional requirements include a substrate or medium that provides support and essential nutrients such as amino acids, carbohydrates, vitamins, minerals, growth factors, hormones and gases (O2, CO2). All these factors control physical and chemical factors such as pH, osmotic pressure and temperature. In animal tissue culture the majority of cells are anchorage-dependent and therefore require a solid or semi-solid support in the form of a substrate (adherent or monolayer culture), whereas others can be cultured in the culture medium, called a suspension culture. Cell culture technologies have emerged as a tool to assess the efficacy and toxicity of new drugs, vaccines and biopharmaceuticals, and also play a major role in assisted reproductive technology. Animal cell culture is one of the more important and diverse techniques in current research streams. Animal, plant and microbial cells are always cultured in predetermined culture medium under controlled laboratory conditions. Animal cells are more complex than micro-organisms. Due to their genetic complexity it is difficult to determine the optimum nutrient requirements of animal cells cultured under in vitro conditions. Animal cells require additional nutrients compared to micro-organisms, and they usually grow only when attached to specially coated surfaces. Despite these challenges, different types of animal cells, including both undifferentiated and differentiated ones, can be cultured successfully.
1.2. Historical background
Tissue culture involves the in vitro maintenance and propagation of cells in optimal conditions. Culturing animal cells, tissue or organs in a controlled artificial environment is called animal tissue culture. The importance of animal tissue culture was initially realized during the development of the polio vaccine using primary monkey kidney cells (the polio vaccine was the first commercial product generated using mammalian cell cultures). These primary monkey kidney cells were associated with many disadvantages [5–8] such as:
- Chances of contamination with adventitious agents (risk of contamination by various monkey viruses is high).
- Most of the cells are anchorage-dependent and can be cultured efficiently only when they are attached to a solid or semi-solid substrate (obligatorily adherent cell growth).
- The cells are not well characterized for virus production.
- A scarcity of donor animals as they are on the verge of extinction.
The foundation of animal tissue culture can be considered to have occurred in 1880, when Arnold showed that leukocytes can divide outside the body [9]. Then, in the beginning of the 19th century, Jolly investigated the behavior of animal cells in serum lymph [9]. The development of animal tissue culture commenced after the breakthrough frog tissue culture technique, which was discovered by Harrison in 1907. Due to this effort Harrison is considered as the father of tissue culture. In his experiment he introduced tissue from frog embryos into frog lymph clots and showed that not only did the tissue survive, but nerve fibers grew out from the cells. During the mid-20th century, human diploid fibroblast cells were established by Hayflick and Moorhead [10]. They named this cell line MRC-5 (a cell line of fibroblasts derived from lung tissue). Later, Wiktor et al (1964) explored the utilization of this cell line in the production of rabies virus for vaccine production [11]. After a couple of years they suggested a large-scale production protocol along with a method for the assessment of purified rabies vaccine immunogenicity. During the same time, BHK-21 (C13) cells (baby hamster kidney cells) were established. These cells are susceptible to human adenovirus D, reovirus 3 and vesicular stomatitis virus. The commercial production of inactivated foot and mouth disease (a viral disease that causes sores in the mouth and a rash on the hands and feet of children) vaccine began using a suspension process [12]. Back in 1914, Losee and Ebeling [13] cultured the first cancer cells and after a few decades the first continuous rodent cell line was established by Earle (1943) [14]. In 1951, Gay established that human tumor cells can give rise to continuous cell lines. The cell line considered as the first human continuous cell line was derived from a cancer patient, Henrietta Lacks, as mentioned above, and HeLa cells are still used very widely. Continuous cell lines derived from human cancers are the most extensively used resource in the modern laboratory. The HeLa discovery was followed by FDA approval for the production of interferon from HeLa cell lines [15]. In addition to the progress in the field of cell culture, different media have been explored, which are typically based on specific cell nutritional requirements, such as serum-free media, starting with Ham's fully defined medium in 1965. In the 1970s, serum-free media were optimized by the addition of hormones and growth factors. Currently, thousands of cell lines are available and for the establishment and maintenance of these cell lines many media are available.
1.3. Types of cell cultures
Broadly, animal tissue culture can be divided into two categories:
- Cultures that allow cell–cell interactions and encourage communication or signaling between cells.
- Cultures in which cell–cell communication or interactions are lost or the signaling between them is missing.
The first category includes three different types of culture systems: organ cultures, histotypic cultures and organotypic cultures. The second category includes cultures in monolayers or as suspensions. Organ culture is a culture of native tissue that retains most of the in vivo histological characteristics, whereas culturing cells for their re-aggregation to yield tissue-like structure is known as histotypic culture. In histotypic cultures, individual cell lineages are initially derived from an organ and then cultured separately to high density in a 3D matrix to study interactions and signaling between homologous cells. In organ cultures, whole embryonic organs or small tissue fragments are cultured in vitro in such a manner that they retain their tissue architecture, i.e. the characteristic distribution of various cell types in the given organ.
In an organotypic culture, cells from different origins are mixed together in specific proportions and spatial relationships so as to re-form a component of an organ, i.e. the recombination of different cell types to yield a more defined tissue or organ. Some terms frequently used in animal tissue culture are as follows.
Cell culture. Cell culture is the process of removing cells from an animal or plant and their subsequent growth in an artificially controlled environment.
Primary cell culture. This is the first culture (a freshly isolated cell culture) or a culture which is directly obtained from animal or human tissue by enzymatic or mechanical methods. These cells are typically slow growing, heterogeneous and carry all the features of the tissue of their origin. The primary objective of this culture is to maintain the growth of cells on an appropriate substrate, available in the form of glass or plastic containers, under controlled environmental conditions. Since they are directly obtained from original tissue they have the same karyotype (number and appearance of chromosomes in the nucleus of a eukaryotic cell) as the original tissue. Once subcultured, primary cell cultures can gives rise to cell lines, which may either die after several subcultures (such cell lines are known as finite cell lines) or may continue to grow indefinitely (these are called continuous cell lines). Usually, normal tissues give rise to finite cell lines, whereas cancerous cells/tissue (typically aneuploid) give rise to continuous cell lines. Nevertheless, there are some exceptional examples of continuous cell lines which are derived from normal tissues and are themselves non-tumorigenic, e.g. MDCK dog kidney, fibroblast 3T3, etc. The evolution of continuous cell lines from primary cultures is assumed to involve mutation, which alters their properties compared to those of finite lines. Serial subculturing of cell lines over time can increase the chances of genotypic and phenotypic variation. Bioinformatic studies based on proteomic phenotypes discovered that the Hepa1–6 cell lines lacked mitochondria, reflecting a rearrangement of metabolic pathways in contrast to primary hepatocytes. With the emergence of newer technologies such as 3D culture, the use of primary cells is becoming increasingly prevalent and achieving improved results. Primary cells which are directly obtained from human or animal tissue using enzymatic or mechanical procedures can be classified into two types:
- Anchorage-dependent or adherent cells. Adherent cells are those cells which require attachment for growth and are also called anchorage-dependent cells. In other words, these cells are capable of attaching on the surface of the culture vessel. These types of cells are often derived from the tissues of organs, for example from the kidney, where the cells are immobile and embedded in connective tissue.
- Anchorage-independent or suspension cells. Suspension cells do not require attachment or any support for their growth and are also called anchorage-independent cells. All suspension cells are isolated from the blood system, for example white blood cell lymphocytes, and are suspended in plasma.
For several reasons cells obtained from primary cultures have a limited life span, i.e. the cells cannot be maintained indefinitely. An increase in cell numbers in a primary culture results in exhaustion of the substrate and nutrients, which can influence cellular activity and lead to the accumulation of high levels of toxic metabolites in the culture. This may ultimately result in the inhibition of cell growth. This stage is called the confluence stage (contact inhibition), when a secondary culture or a subculture needs to be established to ensure continuous cell growth.
Secondary cell culture. This simply refers to the first passaging of cells, a switch to a different kind of culture system, or the first culture obtained from a primary culture. This is usually carried out when cells in adherent cultures occupy all the available substrate or when cells in suspension cultures surpass the capacity of the medium to support further growth, and cell proliferation begins to decrease or ceases completely. So as to maintain optimal cell density for continued growth and to encourage further proliferation, the primary culture has to be subcultured. This process is known as secondary cell culture. Major differences between primary and secondary cell cultures are highlighted in table 1.1.
Table 1.1. Differences between primary and secondary cell cultures.
| Primary cell culture | Secondary cell culture |
|---|---|
| Directly obtained from animal or plant tissue. | Originates from a primary cell culture. |
| Closely resembles the parental tissue. | Does not closely resemble the parental tissue. |
| The biological response of the cell may be closer to that in an in vivo environment. | The biological response of the cell differs from that an in vivo environment. |
| The first culture derived from original cells/tissue (from an in vivo environment). | Derived from an existing culture. |
| Cannot be transformed. | Can be transformed. |
| Less chance of mutation. | Can increase the chance of mutation or genetic alteration of primary cells. |
| Acquired through steps of rinsing, dissection, and mechanical or enzymatic disaggregation. | If the primary culture is an adherent culture, the first step is to detach cells from the attachment (the surface of the culture vessel) by mechanical or enzymatic means. Then, the cells have to be detached from each other to form a single-cell suspension. |
| Finite life span. | Prolongs the life span of cells. Periodic subculturing may produce immortal cells through transformation or genetic alteration of primary cells. |
| The risk of contamination is high. More difficult to maintain. | The risk of contamination is lower. Comparatively easy to maintain. |
Cell line. Once a primary culture is subcultured or passaged it represents a cell line. A cell line that experiences indefinite growth of cells during subsequent subculturing is called a continuous cell line, whereas finite cell lines experience the death of cells after several subcultures.
Cell strain. A cell line is a permanently established cell culture which will proliferate forever if a suitable fresh medium is provided continuously, whereas cell strains have been adapted to culture but, unlike cell lines, have a finite division potential. A cell strain is obtained either from a primary culture or a cell line. This is done by selection or cloning of those particular cells having specific properties or characteristics (e.g. specific function or karyotype) which must be defined.
In summary, the first culture that is established from the in vivo environment is called the primary culture. This primary culture can be subcultured many times to develop cell lines. Cell lines are generally immortalized or transformed cells, i.e. cells that have lost control over division, because of mutations or genetic alterations, or because a primary cell was transfected with some genes that immortalized the cells. Most cell lines are tumorigenic as they originated from tumors. Cells derived from a primary cell line do not have this concern, however, it is challenging to maintain these cells. In usual practice, primary cell cultures require a nutrient medium containing a high amount of different amino acids, micronutrients and, occasionally, some types of hormones or growth factors. Primary cell cultures can be efficiently utilized up to a few passages, about two to four, afterwards their risk of contamination is higher than for cell lines. However, primary cell cultures have their own advantages. The biological response received from a primary culture will be closer to that in an in vivo environment than the response obtained from cell lines. From many years, several cell lines have been established and tested under different environmental conditions. This vast research has resulted in a good amount of data supporting the use of specific cell lines as models of primary cells. It has been suggested that cell lines that have been well tested under different conditions should be used instead of primary cultures, in the case that the latter are expensive.
1.4. Primary cell culture
As discussed above, the primary cell culture is the first culture of cells, tissues or organs derived directly from an organism; in other words it is the culture before the first subculture, whereas the cell line is for maintenance or propagation of a culture after subculture. There are certain techniques available for the development of primary cell cultures, such as:
- Mechanical disaggregation.
- Enzymatic disaggregation.
- Primary explant techniques.
1.4.1. Mechanical disaggregation
It is necessary to disaggregate soft tissues such as soft tumors. The mechanical approach involves slicing or harvesting tissue and subsequent harvesting of spill out cells. This can be achieved by sieving, syringing and pipetting. This procedure is inexpensive, rapid and simple, however, all these approaches involve the risk of cell damage, thus mechanical disaggregation is only used when the viability of the cells in the final yield is not very important.
1.4.2. Enzymatic disaggregation
This approach involves efficient disaggregation of cells with high yield by using enzymes such as trypsin, collagenase and others. Enzyme based disaggregation allows hydrolysis of fibrous connective tissue and the extracellular matrix. Currently, the enzymatic method is extensively used as it offers high recovery of cells without affecting the viability of cells.
1.4.2.1. Trypsin based disaggregation or trypsinization
This allows disaggregation of tissue using trypsin, usually crude trypsin because this trypsin contains other proteases. In addition, cells can tolerate crude trypsin well and the ultimate effect of crude trypsin can easily be neutralized by serum or trypsin inhibitor (supplementation of trypsin inhibitor is required in the case of serum-free media). Pure trypsin can also be utilized for disaggregation of cells, provided that it is less toxic and very specific in its action. An overview of primary cell culture development is shown in figure 1.1. Two common approaches, namely warm and cold trypsinization, are described in the following.

Figure 1.1. Alternative approaches for the preparation of primary cell cultures.
Download figure:
Standard image High-resolution imageWarm trypsinization
This approach is extensively utilized for the disaggregation of cells. During the initial step, sliced tissue is washed with dissection basal salt solution and is subsequently transferred to a container of warm trypsin (37 °C). At regular intervals of 30 min the contents are stirred properly. Then, the supernatant having dissociated, the cells are separated to disperse in a suitable medium. Efficient dispersion of cells can be achieved by placing the container over ice.
Cold trypsinization
This method is also called trypsinization with cold pre-exposure. In this process the chance of cellular damage due to constant exposure to trypsin is reduced, which results in a high yield of viable cells with an improved survival rate for the cells (after 24 h of incubation). Since this method does not involve frequent stirring or centrifugation, it can be conveniently adopted in the research laboratory. During this process, after washing and chopping, tissue pieces are kept over ice in a vial and then subjected to treatment with cold trypsin for 6–24 h. Then, after the cold trypsin treatment the trypsin is removed and discarded. However, the tissue fragments still contain residual trypsin. These fragments are incubated at 37 °C (for 20–30 min) followed by repeated pipetting. This will encourage the dispersion of cells. The fully dispersed cells can be counted using a cell counter and properly diluted, and then further utilized.
Drawbacks of trypsin disaggregation
Trypsinization of cells can damage some cells, such as epithelial cells, and sometimes it is not effective for certain tissues, such as fibrous connective tissue, thus other enzymes are also recommended for dissociation of cells.
1.4.2.2. Collagenase based disaggregation
Collagenase is an enzyme which is responsible for the cleavage of peptide bonds in collagen. Collagen is a structural protein which is abundantly found in higher animals, mainly in the extracellular matrix of connective tissue and muscle. Collagenase, mainly crude collagenase, can be successfully used for the disaggregation of several tissues that may or may not be sensitive to trypsin. Purified collagenase has also been experimented with, but has shown poor results in comparison to crude collagenase. So far collagenase disaggregation has be carried out on several human tumors, epithelial tissues, the brain, lungs and other mammalian tissue. The combination of collagenase with hyaluronidase offers better results in disaggregating rat or rabbit liver, which can be achieved by perfusing the whole organ in situ. Several researchers have also utilized trypsin and collagenase in combination to dissociate cells to develop chick serum.
This process involves an initial transfer of the desired tissue into a basal salt solution which contains antibiotics. This is followed by washing with settling and then transfer into a medium containing collagenase. The solution is incubated for 1–5 days, followed by repeated pipetting for uniform dispersal of cells. Separation of these dispersed cells is encouraged by keeping the solution in a stationary phase to further encourage the settling of cells, as shown in figure 1.2.

Figure 1.2. Standard growth curve of cells in a culture.
Download figure:
Standard image High-resolution image1.4.2.3. Other enzymes
In addition to the above mentioned enzymes, certain other enzymes such as bacterial proteases (e.g. dispase, pronase) have been tested, but unfortunately have not shown significant results. However, enzymes such as hyaluronidase and neuraminidase have received attention due to their significant results, and thus can potentially be utilized in conjugation with the enzymes discussed above.
1.4.3. Primary explant technique
In 1907 Harrison provided the first demonstration of the primary explant technique, which subsequently underwent many modifications. A simple protocol for the primary explant technique is represented in figure 1.1. As in the above procedures, in this process tissue is initially suspended in basal salt solution and then chopped properly and washed by settling. Tissue fragments are uniformly distributed over the growth surface. This is followed by the addition of a suitable medium and then incubation for 3–5 days. Old medium is replaced by fresh medium unless desired growth or considerable outgrowth of the cells is not achieved. Once optimum growth is achieved the explants are separated and transferred to new culture vessels which contain fresh medium.
This technique is mainly used for disaggregation of small quantities of tissue. Mechanical and enzymatic disaggregation are not suitable for small amounts of tissues, as there is a risk of cell damage which can ultimately affect cell viability. A major drawback of this technique is the poor adhesiveness of certain tissues on the growth surface (substrate material), which can create problems in the selection of cells for desirable outgrowth. However, this technique has been utilized frequently for culturing embryonic cells, in particular glial cells, fibroblasts, myoblasts and epithelial cells.
1.5. Segregation of non-viable cells from viable cells
After the development of a primary cell culture, it is essential to remove the non-viable cells from the disaggregated cells, which can be achieved by repeatedly changing the medium. Only a few will be left after dilution of the medium, and finally will gradually disappear when viable cells start proliferating. The alternative approach of centrifugation, mixing cells with ficoll and sodium metrizoate, can also be utilized to remove non-viable cells from the primary cell culture. Dead cells form a pellet at the bottom which can easily be removed from the solution.
1.6. Ethical issues in animal tissue culture
Animal tissue culture techniques involve the frequent utilization of animal or human tissues, which raises the need for safety and ethics guidelines for using animals in research, also known as medical ethics. Handling animals raises numerous issues that are typically not faced when using animal tissue. In addition to the consent of local ethical committees, the consent of the patient or his/her relatives is required to initiate research or to study a human sample in the form of fetal materials or biopsy samples. Samples collected from a human donor should be accompanied by a donor consent form in a prescribed format. When dealing with human tissue, the following issues should be considered [16]:
- The patient's or relatives' consent for using tissue for research purposes.
- Ownership of specimens, in particular cell lines and their derivatives, i.e. the recipient will not trade or transfer the cell lines and their derivatives.
- Consent for genetic modification, in particular in the case of cell lines.
- Patent or intellectual rights for the commercial use of cell lines.
- Guidelines should be refined to meet the requirements of the latest ongoing
developments in animal tissue culture science. These guidelines are framed to
offer knowledge to those new to the field and others involved in training and
instruction, with the data required to improve their awareness of issues and to
allow them to deal with them more efficiently. The primary areas of focus in
guidelines are:
- i.Acquisition of cell line.
- ii.Authentication of cell line.
- iii.Characterization of cell line.
- iv.Cryopreservation of cell line.
- v.Development of cell line.
- vi.Instability of cell line.
- vii.Legal and ethical requirements when deriving cell lines from human and animal tissues.
- viii.Microbial contamination of cell line.
- ix.Misidentification of cell line.
- x.Selection and maintenance of equipment.
- xi.Transfer of cell lines between laboratories.
Generally, when dealing with human tissue, the donor/relative is asked to sign a disclaimer statement in a prescribed format before tissue is collected. Doing this can reduce the chances of legal problems [16].
1.7. Safety considerations in animal tissue culture
Handling human tissue involves a high risk of exposure to various infections, thus it is essential to handle human material in a biohazard cabinet. Before their use, tissues must be screened properly for various infections such as hepatitis, tuberculosis and HIV. In addition, media, apparatus and glass wares should be properly sterilized (autoclaved) to considerably reduce the chances of spreading any infections.
1.8. Cell lines (first subculture or passage)
A cell line can be defined as a permanently established cell culture which will propagate forever, provided the continuous supply of suitable fresh medium and the availability of space for the cells to propagate. Thus, generally, a cell line can be defined as the propagation of a culture after the first subculture. In other words, when primary culture is subcultured it results in the development of a cell line. Cell lines differ from cell strains in that they become immortalized. A cell line contains several cell lineages, either similar or different in their phenotypical characteristics, and such cells can be selected by cloning or cell separation or by any other suitable procedure. The cell line obtained after selection or cloning is called a cell strain, which does not have a infinite life, since they die after a number of divisions.
1.8.1. Types of cell lines
As discussed above, cell lines that lose their ability to divide after a limited period of time are finite cell lines, i.e. these cell lines have a limited life span. Usually, finite cell lines contain cells which can divide 20–100 times (i.e. population doubling by 20–100 times) before losing their capability to divide. The extent of population doubling is dependent on several factors, such as cell lineage, cell type, origin, species, culture environment, etc. It has been noted that population doubling for human cell lines is between 50–100 times, whereas murine cell lines divide 20–30 times before extinction.
In an independent culture, continuous subculturing of cells or treatment of cells with carcinogens (chemicals), oncogenic viruses, etc, results in changes in phenotypical characteristics, in particular morphology, which can alter cells and lead to the development of cells that grow faster than normal cells. Cell lines obtained from these altered cells have infinite life spans. Such types of cell lines are referred to as continuous cell lines. These cell lines are immortal, transformed and tumorigenic (unlike the cell strains from which they were derived). Terms frequently used in animal tissue culture, in particular in the context of cell lines, are defined below:
- Adherent cells. Cells with the potential to adhere to the surface of the culture vessel using the extracellular matrix.
- Immortalization. Achieving a state of cell culture when cells proliferate continuously.
- Attachment efficiency. The proportion of cells that actually adhere to the surface of the culture vessel within a given time after inoculation.
- Passaging. The transfer of cells from one culture vessel to another. A more specific term is subculturing where the cells are first subdivided before being transferred into multiple cell culture vessels. A passage number will refer specifically to how many times a cell line has been subcultured. A number of adherent cell cultures will stop dividing when they become confluent (i.e. the stage when they entirely cover the surface of the cell culture vessel), and a number will die if they are retained in a confluent state for longer periods. Thus adherent cell cultures require repeated passaging, which means that when the cells are at the confluent stage, subculturing is required. Regular passaging is required in the case of suspension cultures, where suspended cells use their culture medium rapidly, particularly when the cell density becomes very high. While repeated passaging is essential to maintain cultures, the process is comparatively traumatic for adherent cells since they need to be trypsinized. Thus passaging of adherent cell cultures more than once every 48 h is not recommended.
- Split ratio. Divisor of the dilution ratio of a cell culture.
- Generation number. The number of doublings that a cell population has undergone. It should be observed that passage and generation number are not the same.
- Population doubling time. The population doubling (PD or pd) number is the estimated number of doublings that the cell population has undergone since isolation.
- Passage number. The number of times the culture has been subcultured.
1.8.2. Standard nomenclature of cell lines
The source and clone number (which represents the number of cell lines derived from the same donor) help in understanding the nomenclature more easily. The basic nomenclature is usually followed by assigning codes or designations to cell lines for their further identification, e.g. HeLa-S3 represents a human cervical tumor cell line, and similarly NHB 2-1 is a cell line derived from normal human brain (NB), followed by cell strain 2 and clone number 1. Another example is the MG-63 cell line. It is the 63rd sample of a tumor that produces a high amount of interferon beta. Therefore, its nomenclature is 'human tumor-63', or in Dutch, 'menselijk gezwell-63' or MG-63. Recently cell lines have transformed scientific study and are used for several purposes, such as:
- Vaccine production.
- Examining drug metabolism.
- Cytotoxicity.
- Antibody production.
- Investigating gene function.
- Development of artificial tissues (e.g. artificial skin).
- Production of biological compounds (e.g. therapeutic proteins).
Cell line requirements can be assessed through recent publications using specific cell lines. The American Type Culture Collection (ATCC) cell biology collection contains information on almost 3600 cell lines derived from 150 species. Although they are a useful tool, researchers must be careful when using cell lines instead of primary cells. The simultaneous use of cell lines and primary cells has been supported recently.
Cell lines should display and maintain functional features as close to the primary cells as possible. This may be particularly difficult to determine, as often the functions of the primary cells are not entirely understood. Since cell lines are genetically manipulated, this may alter their phenotype, native functions and responsiveness to stimuli. Serial passage of cell lines can further cause genotypic and phenotypic variation over an extended period of time, and genetic drift can also cause heterogeneity in cultures. Therefore, cell lines may not adequately represent primary cells and may provide different results. Additional problems include the chance of contamination with other cell lines and mycoplasma. In the early 1970s, cell line (inter- or intraspecies) mediated cross-contamination was explored by Nelson-Rees. Contamination of one cell line with a new one results in mixed cultures or occasionally complete overgrowth of the original cells by the contaminating line, and is an old problem. Nelson-Rees used chromosome banding (a procedure in which condensed chromosomes are stained to produce a visible karyotype) to prove that numerous immortal cell lines, earlier supposed to be unique, were in fact HeLa cell lines. He also demonstrated the fact that contamination with HeLa cells is responsible for the outgrowth of other cell lines [17–19]. Nelson-Rees demonstrated clearly that most of the of cell lines being investigated globally and distributed by cell banks [20] were contaminated with HeLa cells. This is the most considerable challenge for the animal tissue culture industry. During cell line contamination, contaminants, in particular rapidly proliferating cells, take over a whole cell line before its own growth takes place [21, 22]. HeLa cells are a frequent contaminant and, moreover, other contaminants such as mycoplasma can continue undetected in cell cultures for a long period of time. This prolonged exposure to contaminants can cause widespread changes in gene expression and cell behavior. According to certain reports, 15%–35% of cell lines submitted to cell banks were likely to be contaminated with mycoplasma [23, 24]. Thus appropriate precautions must be taken whenever cell lines are investigated.
1.8.3. Cell line selection
Usually the selection of high-producing cell lines is tedious and labor-intensive. High-producing cells are usually selected after transfection by using limiting dilution cloning to avoid non- and low-producing cells from outgrowing high-producing cells. This process usually takes more than three months. During this time, the cells have to be screened occasionally to ensure stability of the selected clone. High-producing mammalian cell line selection is one of the considerable challenges in the production of biopharmaceuticals. Increasing demand for therapeutic proteins requires the urgent development of methods for the selection of mammalian cell lines stably expressing recombinant products at high levels in an efficient, cost-effective and high-throughput manner [25–27]. Numerous approaches for selecting and screening cells have been explored, including flow cytometry, gel microdrop methods (encapsulating the cells in gelatin beads) and matrix based secretion assays. Recently, fluorescence-activated cell sorting has been utilized to estimate the cell-specific productivity (Qp), or the quantity of product produced per cell per day [25–27]. This parameter is utilized in biopharmaceutical cell selection in a cell-specific manner, which allows multi-purpose characterization and isolation of individual cell clones from heterogeneous populations. Several factors are considered during the selection of cell lines, such as [25–27]:
- Origin of the cell line (human or non-human cell line; human cell lines are more vulnerable to different types of contamination).
- Type of cell line (finite or continuous).
- Types of cells (normal or transformed).
- Growth patterns.
- Cloning efficiency.
- Cell number under specified culture conditions (saturation density).
- Population doubling time.
- Availability of cell line.
- Availability of growth factors or media for its maintenance.
- Physical expression of traits, or characteristics.
1.8.3.1. Quarantine
To avoid microbial contamination, new cell lines should be quarantined (kept entirely separate from existing cell line stocks). Usually, an independent quarantine laboratory should established for this purpose. A class-II microbiological safety cabinet (MSC) and an incubator dedicated to quarantine can be considered as an alternative approach.
1.8.4. Verification of a cell line
To confirm the origin of a cell line and to avoid misidentification, cell line authentication is carried out using an established DNA based method. With the advancements in tissue culture science, it is now possible to equate the cell line DNA with that of the tissue of origin, however, this is only possible for a few cell lines that are already available. Short tandem repeat analysis can be used to confirm the origin of a cell line. A short tandem repeat pattern is derived and compared for the cell line and the primary culture. The primary culture should be frozen or processed so that it can clearly be determined that the cell line is obtained from a recognized donor. This method is recommended for purposes of authentication, so that the unique identity of the primary culture is available for the international database (NCBI 2013) [28]. Some other methods, such as genotypic methods (karyotype, copy number variation mapping or even whole-genome sequencing) can also be used to authenticate cell lines.
1.8.5. Characterization of cell lines
Before characterization, the handler first ensures that the cell line obtained is appropriate or acceptable for their designed experiment or purpose. Even after confirmation of the cell line, it is essential to check whether the cell line is still carrying key characteristics after persistent passaging. To reveal changes in a cell line, karyotyping is the most recommended approach. It can demonstrate that a cell line has a normal karyotype, and the cell line can then be used for various research purposes.
Karyotyping is thus a simple test that can reveal changes in a cell line. Indeed, it is routine to demonstrate that a line of embryonic stem cells or induced pluripotent stem cells has a normal karyotype if they are to be used for experiments involving the production of chimeras and germ line transmission. Molecular assays for copy number variation or RNA profiling will also be indicative of changes, but are more costly. Nevertheless, a great deal of time and effort can be saved by confirming the presence of appropriate characteristics before commencing work. It is also advisable to capture an image of the cell line in culture at different cell population densities and perform basic characterization (e.g. calculating the population doubling time for that cell line) soon after arrival. For a newly developed cell line it is imperative to authenticate the origin of the cell line and the extent of variation between cells present in the primary tissue culture.
1.8.6. Misidentification of cell lines
Cross-contamination is considered as the primary cause of misidentification. A high risk of cross-contamination is usually associated with continuous cell lines as they may replace other, slow growing cell lines. The following lists a number of factors that are responsible for misidentification:
- Alterations in cellular behavior or morphological variations.
- Developing two cell lines in an Mesenchymal stem cells simultaneously.
- Failure in maintaining good cell culture practice.
- Liquefying the wrong ampoule.
- Mislabeling a flask or ampoule.
- Persistence of the mitotic activity of feeder cells such as embryonic stem cells because of insufficient irradiation or treatment with mitomycin C.
- Poorly controlled manipulation.
- Unintended transfer of cells to a stock bottle of medium.
- Using unsterilized media (used without suitable filtration to remove cells).
1.8.7. Maintenance of a cell line
Current research practice demands the development of good models, as good science cannot be achieved with bad models. Several cell culture procedures have been developed in the current century which overcome the drawbacks of traditional culture procedures and are more scientifically rigorous, such as stem cell derived human cells, co-cultures of different cell types, scaffolds and extracellular matrices, tissue architecture, perfusion platforms, organ-on-chip technologies, 3D culture and organ functionality. The biological relationships between such models can be further improved by organ-specific approaches, more widespread assessment of cell responses using high-content methods and by using biomarker compounds. These strategies can be utilized to make a microphysiological model system. One of the most significant advantages of this type of model system is that it generates results closer to the in vivo situation, however, controlling multiple parameters is considered a significant challenge for animal tissue culture industries. Cell line maintenance has become a very valuable undertaking, both in academic research and in industrial biotechnology. The following factors should be considered during maintenance of a cell line in culture.
1.8.7.1. Cellular morphological examination
Cells should be examined routinely to check for the presence of any other contaminant. Morphological examination is essential to investigate and differentiate the natural cellular organization and the physiological state of the cells from the contaminated. Therefore, morphological examination is usually used as a qualitative and quantitative measure of various biological assays.
1.8.7.2. Media replacement
Regular changing of the medium is required to maintain cell lines in culture; however, the frequency of changing the medium always varies. For example, proliferating cells require more nutrients in comparison to non-proliferating cells. The rate of cellular growth and metabolism decides the interval between the changing, or addition of fresh, medium. To understand this better, we can consider HeLa, rapidly growing transformed cells. In order to avoid contamination and to meet cell nutrient requirements, HeLa cell medium should be replaced twice in a week, whereas for slow growing cells (non-transformed cells), e.g. IMR-90, the medium can be replaced once a week. Thus, rapidly growing or proliferating cells require more frequent changes of medium than slow growing or non-proliferating cells. Several factors should be considered when changing the medium:
- Cell density. Cultures with a high density of cells use medium faster than those with low density, thus medium need to be changed more frequently for high densites of cells.
- Fluctuations in pH. Changes in pH should be monitored carefully, since a decrease in pH may be associated with a decline in the growth rate of the cells. The optimal pH for the growth the cells is 7, and a decline in pH (6.5) can retard the growth of cells. Further decline in pH (usually to between 6.0–6.5), may stop the growth of the cells, and if the low pH persists cells will start losing their viability. Thus the pH should be carefully monitored for each cell line and controlled with a suitable medium. Medium changing is not required when the pH declines by 0.1 units/day, as such a decline may not harm cells, however, declines of 0.4 pH units/day may affect growth, and eventually the viability of cells, so in this case an immediate medium change is required.
- Type of cell. Feeder cells such as embryonic stem cells and tumorigenic cells such as transformed cell lines (continuous cell lines) grow fast and thus require a greater supply of nutrients. Thus rapidly growing cells require more frequent medium changes than normal cells.
- Phenotypical variations. It is important to examine cell morphology carefully using specific techniques, as any change in morphology could be a sign of contamination or deterioration which can ultimately affect the growth of the cells.
1.9. Subculture
Subculture is defined as the transfer of cells from one culture to start another culture. During this process proliferating cells are subdivided, which allows the development of new cell lines. This step is referred to as a passage, and a passage number is the recorded number of times a cell culture has been subcultured. Numerous adherent cell cultures will stop proliferating when they reach the confluent stage (i.e. when they completely cover the surface of the cell culture vessel), and certainly will die if they are left in the confluent stage for a prolonged period. Thus adherent cell cultures should be regularly passaged, that is, when cells reach the confluent stage a portion of the cells need to be passaged or subcultured to a new cell culture vessel. However, it is not recommended to regularly subculture adherent cells (no more than once every 48 h) as they must be trypsinized. In contrast, suspension cultures with high cell density require routine passaging as they use medium rapidly.
The standard growth curve of cells in a culture is shown in figure 1.2.
During the initial lag phase there is less growth as the cells are not adapted to the environment. Once they start adapting to the environment they proliferate exponentially, which is why this is called the exponential or log phase. This is the time when all cells actively grow and consume medium. During this time the medium should be changed, otherwise growth will stop. As discussed above, the confluent phase is reached when the culture exceeds the capacity of the medium. At this stage the culture has to be divided into subcultures. There are two types of subcultures: monolayer and suspension subcultures.
1.9.1. Monolayer cultures
Monolayer cultures involve anchorage-dependent cells which can be established from human tissue after enzymatic treatment to disperse them into single cells in order to form a monolayer or single-cell continuous layer over the bottom of the vessel. Cellular attachment between cells and with the interface is facilitated by surface glycoproteins and calcium ions. Several quantitative approaches have been explored for examining viable cells in monolayer cultures, such as:
- Microscopic screening to examine morphological changes.
- Cytotoxicity studies.
- Incubation with dye followed by colorimetric analysis.
The initial step in subculturing of monolayers is to remove cells from the interface of the vessel by trypsinization or mechanical means [30]. The final dispersion is then subdivided and transferred to fresh cultures. The growth of the secondary cultures is periodically monitored and further subcultured to produce tertiary cultures, etc. As discussed above, the time interval between subculturings is entirely dependent on the growth rate and varies with the cell line.
1.9.2. Procedures for cell detachment
There are various means of cellular detachment from the culture vessel interface, such as physical and chemical methods (figure 1.3).

Figure 1.3. Methods for dissociation of cells.
Download figure:
Standard image High-resolution imageThe utilization of proteases is not recommended when cultures are loosely adhered, thus mechanical shaking and scraping are more appropriate in such cases. Due to its advantages, trypsin is often used for cell dissociation, however, other enzymes such as pronase, dispase and collagenase are used when monolayers cannot be disaggregated with trypsin. Prior treatment with EDTA is required to remove Ca+2 so that it will not interfere with the action of enzymatic dissociation, and eventually uniform dispersion can be achieved [30]. As one-cell-thick monolayers are the simplest tissues in multicellular organisms, they act as a suitable model for development and normal physiology. It has been determined that the extracellular material (ECM) should be carefully considered when selecting the dissociation approach, as this helps in determining the effects of the dissociating agent on the cytoskeleton, adherent junctions and desmosomes. Usually monolayers can withstand the different mechanical stresses exerted by the interface itself under in vitro conditions, and can shield the internal environment from harmful external elements. Since dissociating elements or external environmental factors can affect ECM synthesis, optimization of the dissociating element is required before treatment in order to estimate a suitable dose for dissociation [30].
As mentioned in figure 1.4, subculturing is usually carried out between the middle log and plateau phases; it is not recommended to start subculturing during the lag phase.

Figure 1.4. Subculturing.
Download figure:
Standard image High-resolution imageUnderstanding of growth patterns is necessary for:
- Designing culture experiments.
- Regular maintenance of a culture.
- Monitoring cell proliferation.
- Evaluating a culture's response to external factors.
There are certain considerations in subculturing monolayers, as follows.
Cellular density
Subculturing time is dependent on the cellular density. Cellular density is usually found to be high at the confluence stage. So, whenever normal or transformed cells reach the confluence stage it is advisable to perform subculturing as this can maintain the balance between nutrient supplementation and consumption by the cells/micro-organisms. In the confluence stage, when all the growth area is utilized and cells start coming closer to each other, growth may be hampered due to the negative force (contact inhibition) developed between the cells striving for nutrients to further meet their energy demands.
Exhaustion of nutrients
Usually, in microbiology a sudden drop in pH represents an increase in cellular density which again signifies the confluence stage, thus a drop in pH often necessitates subculturing.
Reason for subculture
Subculturing is also done in those cases when cells are to be used for any specific purpose other than routine propagation, in order to obtain high yield or stock, or to change the type of medium. In such cases the cell has to be subcultured frequently.
Scheduled timings for subculture
As we know, regular subculturing is generally performed as per a strict schedule to obtain significant results. Seeding density should be increased in the case that the cell will not reach the confluence stage at a suitable time, and seeding density should be reduced when the cells will reach the confluence stage a little too early. It is now possible to determine the correct seeding density and subculture interval by studying standard growth curves. In most cases medium change is performed after 3–4 days and subculturing after 7 days.
The steps involved in monolayer subculture are shown in figure 1.5. Monolayer subculturing involves multiple steps:
- Medium removed and monolayer washed.
- Treatment of cell with trypsin.
- Trypsin removed leaving a residual film.
- Incubation (37 °C for 30 min).
- Cell rounding up after incubation.
- Resuspension of the cells in the medium.
- Reseeding of cells.
- Confluent stage of monolayer.

Figure 1.5. Process of monolayer subculture.
Download figure:
Standard image High-resolution imageFor the majority of continuous cell lines, the seeding concentration for subculturing lies between 1 × 104 to 5 × 104 ml. However, for developing a new culture, the initial concentration should be high and should then be reduced to meet the culture's requirements.
1.10. Suspension cultures
Most cell lines are grown as monolayer adherent cells, which grow only on the surfaces of culture vessels, however, certain cells are not adhesive, such as cells derived from leukemic tissue. Moreover, certain cells do not require support for their growth. These cells can be mechanically kept in suspension, and such cultures are referred to as suspension cultures. Transformed cells are usually subcultured using this method. Suspension culture of animal tissues is similar to the method used to subculture bacteria or yeast. There are a number of advantages to suspension cultures over monolayer cultures:
- Bulk production or production in mass can be achieved.
- The cultured cell has access to nutrition from all directions.
- Easy to maintain.
- Frequent replacement of medium is not required.
- The lag period is short.
- The process of propagation is fast.
- Scale-up is convenient.
- Trypsin treatment, or any other enzyme treatment, is not required.
Similar parameters have been reported for suspension cultures as for monoculture, i.e. culture density, fluctuations in pH, schedule timings, the purpose of the subculture, etc. During this process cells are suspended in a culture flask (figure 1.6) which contains culture medium. In the stirred flask technique, the medium is continuously stirred with the help of a magnetic pendulum, in order to offer homogeneous stirring and to avoid aggregation. This magnetic pendulum is allowed to rotate at the base of the flask and the suspension of cells should be regularly monitored for contamination, aggregate formation or any signs of deterioration.

Figure 1.6. Stirred flask technique for suspension cultures at a large scale.
Download figure:
Standard image High-resolution image1.10.1. Cell synchronization
Synchronized cells have the same growth rate in all generations, whereas unsynchronized growth means different growth rates of cells, as shown in figure 1.7. The cell culture has to be synchronized so that the cells will be at the same phase at the same time, which makes it easier to determine the growth rate. Cell synchrony is essential to study the development of cells through the cell cycle, which needs to be monitored at periodic intervals. Several methods have been introduced to achieve cell synchronization. These approaches are broadly divided into two categories:
- Cell synchronization by physical means.
- Cell synchronization by chemical means.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 1.7. Simple illustration of synchronized (cells are dividing at the same time) and unsynchronized growth (cells are not dividing at the same time).
Download figure:
Standard image High-resolution image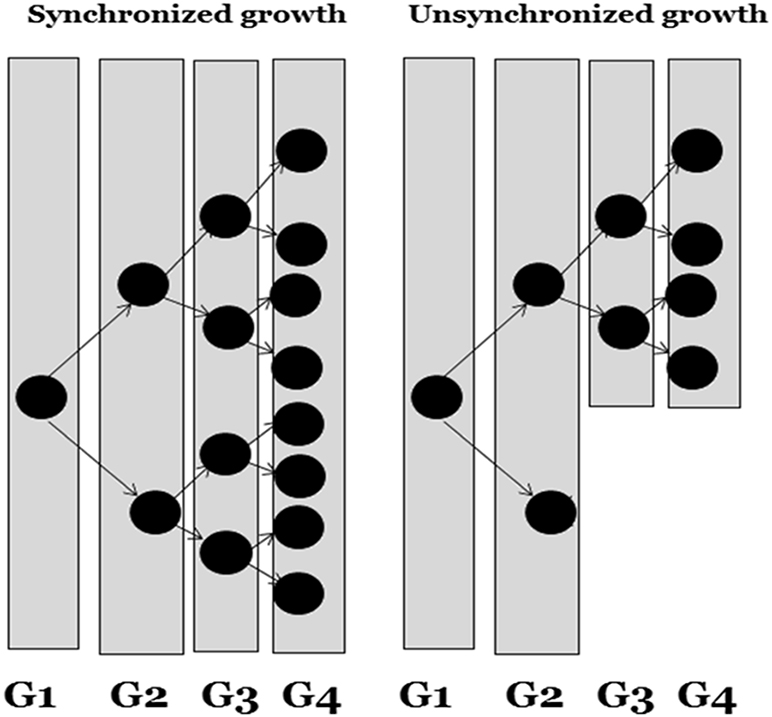{kind=link}
Cell synchronization using physical methods is more effective than using chemical methods, as the latter can cause toxicity to the cells. Deprivation of nutritional resources (part of the chemical approach) cannot be utilized to synchronize transformed cells. As the cell cycle is composed of different development or growth phases, these determine the synchrony of the cells, and more synchrony can be obtained at the first cycle than at the second or third cycles.
1.10.2. Cell synchronization by chemical means
In this approach cells are synchronized by blocking metabolic reactions, which can be achieved by either adding inhibitor substances to the culture medium or by depriving the micro-organisms or cells of nutritional sources.
Inhibitors such as thymidine, aminopterine, hydroxyurea, cytosine and arabinoside, which have variable effects, are utilized to block DNA synthesis during the S phase of the cell cycle, bringing the cells to the same phase.
Removing essential growth substances, such as serum or isoleucine, from the culture medium for almost 24 h leads to the accumulation of cells at the G1 phase. This approach of depriving cells of certain nutritional components exposes the cells to similar types of stress in response to which the cells will present similar adaptations, and thus synchrony can be achieved.
1.10.3. Cell synchronization by physical means
Separation of cells by physical means to achieve synchrony can be done using characteristics such as cell density, affinity against antibodies, light scattering or fluorescent emission by labeled cells. Techniques which can be commonly utilized to separate cells based on their adaptations and phenotypical variations are centrifugal elutriation and fluorescence-activated cell separation.
Centrifugal elutriation is a process to enhance the sedimentation rate to improve the yield of cells. The process is based on cell size and sedimentation velocity. During this process cells in the medium are ejected into the separating chamber such that they will be forced to the edges. This occurs in such a way that the centripetal force will be equivalent to the sedimentation rate of the cells. As the cell present in the culture must have phenotypical variations, such as size, shape, density, cell surface, etc, thus cells at different phases of the cell cycle have a tendency to sediment at different rates and different positions in the chamber. The whole process can be monitored via a porthole, as the chamber is illuminated by stroboscopic light.
1.11. Algal extracts in animal tissue culture
Algal extracts contain high amounts of potential secondary metabolites which can be utilized to either elicit or promote growth of cells under in vitro conditions. These metabolites can be utilized in media to further increase the growth of the cells. Our recent research on the red algae, Porphyra vietnamensis, found among its diverse chemical compounds some with significant pharmacological properties [31–50]. Such types of algae can be utilized to elicit the growth of animal cells. Shinohara et al observed that algal phycocyanins are responsible for the growth of human cells in culture [50]. In their study, growth-promoting substances derived from blue-green algae, Synechococcus elongatus var., were separated to produce a biliprotein fraction that promoted the growth of RPMI 8226 cells; allophycocyanin was found to be more active than phycocyanin.
1.12. Animal and plant tissue culture
Tissue culture is the art of growing cells outside a living body. As we have already discussed the historical background and current innovations of the field of biotechnology in the first two volumes, it is well understood that there are direct and indirect relationships between the developmental biology of plant and animal cells [47]. There are certain challenges involved in culturing both plant and animal cells, such as exhaustion of nutrients in the growth medium, apoptotic/necrotic cell accumulation, cell cycle arrest (or senescence) due to intercellular communication or contact inhibition, etc, which should be investigated further [47]. Various approaches can be utilized to manipulate cell cultures of both plant and animal cells. Subculturing is a common practice which is adopted to replace old medium with new, nutrient enriched, medium. Subculture can also be used to prevent the major problem of senescence. This involves transferring a small number of cells into a new culture dish. The animal–plant co-culture system has not been explored due to its greater vulnerability to contamination of the culture. There is, however, scope to maintain suitable aseptic conditions and encourage a co-culture system to further study the impacts of their growth on each other [47].
The success of animal tissue culture products depends on their efficacy, cost effectiveness and the potential for scale-up. Recent and current advances in tissue culture science have enhanced the complexity in the design of biomaterials which have either been proposed or utilized to grow animal cells [51]. This complexity generally increases the difficulty for manufacturing industries in designing suitable fabrication techniques. It is worth noting that most of the features that are suitable for designing biomaterials originate in the structure and function of plants [51]. Several investigations have shown that decellularized plant tissues can be utilized as a suitable scaffold for the culture of human cells. It has been observed that through an approach of simple biofunctionalization it is possible to achieve the adhesion of human cells on various sets of plant tissues. The increased water transport efficiency and hydrophilicity of plant tissues facilitate increases in cell number over prolonged periods of culture [51]. In addition, animal cells are able to adapt well to the microstructure of plant frameworks without breaking any physiological conditions. This results in perfect cell positioning and formation of a perfect pattern over the feeding layer of plant cells. This supportive plant tissue based micro-framework can be utilized as an alternative potential scaffold for mammalian cells [51].
1.13. Biomaterials and animal tissue culture
Recently, there has been a boom in biopolymers of natural and synthetic origin [51–53]. It is essential to understand the possible interactions between cells and these materials in order to develop new materials [48, 54]. When isolated cells from suitable tissues are cultured on a plastic culture dish, this transition of cells from an in vivo to in vitro environment results in the loss of several functions and cells usually begin dedifferentiation, for unknown reasons. Identification of the microenvironmental signals that are responsible for changes in cellular phenotype and function will help in understanding cell behavior under in vitro conditions. Most of the current research that deals with tissue-engineered constructs involves suitable scaffolds that not only act as anchorage cells, but that also help in studying cell behavior and developmental stages in more detail [48, 54]. Several scaffolds have been developed that offer suitable architecture, in particular initial structural integrity and support or a backbone in the form of a matrix in which the cells arrange themselves and form a mass of functioning tissue [51–53]. Several techniques have also been developed, such as three-dimensional matrices for culturing animal cells, to help in understanding how cells probe their surroundings [51–53]. The biomaterial matrix is designed in such a way that it will be able to control the cell position and function inside artificial environments [48, 54]. For most of the materials developed so far, we lack understanding of the effects of the biomaterial or surrounding microenvironment on cell development, behavior and functions.
Physiologically, cells are always surrounded by a sophisticated and dynamic microenvironment which includes the extracellular matrix, growth factors and cytokines, as well as neighboring cells. The extracellular matrix helps in connecting the cells' extracellular matrix proteins through specific cell surface receptors such as integrins [55–57]. Such receptors are responsible for connecting the intracellular cytoskeleton to the extracellular matrix [48, 54]. It is important to understand the interaction between the ligands present in the extracellular matrix and the receptors of the cell. Such an interaction allows multiple intracellular signaling processes that can result in the alteration of cellular behaviors, such as growth, migration and differentiation. Natural extracellular matrices such as collagen offer natural adhesive ligands that encourage cellular connection with integrins. Such biomaterials can be considered as potential resources for engineering new biomaterials [55–57]. One of the major shortcomings of such natural extracellular matrices is our failure to control their physicochemical properties. Several natural biomaterials have been explored recently. In one of the recent innovations in ligand chemistry, a short peptide sequence (arginine–glycine–aspartic acid) which is responsible for cellular adhesion was discovered [55–57]. This peptide can be conjugated with other biologically inert polymers to study their effect on cultured animal cells [55–57]. These peptides, once conjugated over the matrix of inert biomaterial, can initiate cellular adhesion which can further allow researchers to develop suitable matrices on the surface of which these peptides can be conjugated. This approach allows the development of suitable matrices the adhesion potential and chemistry of which can be controlled. Additionally, the incorporation of growth factors in the matrix facilitates their distribution to cells in a controlled fashion. Therefore, cellular adhesion and growth of the cell can be controlled by changing the chemical nature of the polymeric network. One of the most common approaches is functionalization. Growth factors can also be immoblized over polymeric networks to study and manipulate cells. One of the common examples of such immobilization is used in the study of insulin and epidermal growth factor [55–57].
Pattern-immobilization is a more reliable approach as in this process cells are allowed to culture in a matrix with an architecture that produces a three-dimensional structure mimicking the in vivo environment [55–57]. In such a systematic spatial arrangement, growth factor proteins or other proteins, such as those carrying the ligands, are embedded to encourage interaction between cell receptors and ligands. Such artificial frameworks help in the development of tissue through non-diffusion mechanisms, in which the movement of proteins is not dependent on the concentration gradient. This type of stimulation by immobilized growth factors mimics the in vivo environment of membrane-anchored growth factors such as heparin-binding epidermal growth factor, transforming growth factor and tumor necrosis factor. Furthermore, cellular growth can also be enhanced by co-immobilization with adhesion factors and by means of thermosensitive polymers, which allows the development of cells and, most importantly, allows the recovery of the cell through reducing the temperature [55–57].
1.14. Nanotechnology and biotechnology
Three dimensional biomaterials with large pore size (greater than 100 μm) carry a high number of functional units essential for the regeneration of various tissues. Pore size greater than 100 μm is essential for the cell adhesion and proliferation, whereas biomaterials with 325 μm pore size encourage migration of cells through the scaffolds. Scaffolds with pore size less 85 μm showed lowest intensity of cell adhesion and migration. So far, most tissue engineering studies have focused on macro-sized frameworks for cells greater in size than 100 μm (subcellular size) or cellular arrangements larger than 10 μm (cellular size). Such massive structures are required to produce real-sized organ systems [49]. However, to design functional units of tissue, not only are the subcellular and cellular scales required, but also nanostructures, 1–100 nm in size. This type of structural arrangement is essential to control cell behavior, in particular cell–cell interactions, cell–molecular interactions and the cellular environment [49]. The recovery of cell characteristics, in particular structure and function, can only be achieved by reconstruction of the nanostructures of the tissue itself. The current prospects of tissue engineering are very dependent on understanding the interaction of cells with these nanostructures. These tiny structures with three-dimensional arrangements can directly or indirectly affect the cell functions [49]. The trend to fabricate ever smaller structures (called miniaturization), mainly to regenerate the components inside the targeted tissue, has proven a reliable approach for researchers. Thus far, several nano-materials have been developed to mimic native tissues. These nanostructures are tissue-engineered grafts, biomaterial scaffolds that are engineered and then manufactured at the molecular level [49]. Several techniques are available to optimize materials, even at the level of atoms, molecules and supermolecules, 1–100 nm in scale. By means of nanotechnology, various materials or devices can be fabricated or designed to offer a product with high biocompatibility, and most importantly highly predictable biological and physical properties [49]. Animal biotechnology is a broad discipline that includes DNA science, genetic engineering, transgenic science and stem cell research. DNA research involves DNA isolation and screening methods, whereas genetic engineering involves the manipulation of the genetic makeup of an organism to either synthesize a product or alter the character of the organism [58, 59]. Currently, all these fields are being utilized to derive biopharmaceuticals. Moreover, enzyme research, mainly protein engineering, immobilization and biotransformation, has several applications in animal tissue culture science [58, 59].
References
- [1]Rodríguez-Hernández C O, Torres-Garcia S E, Olvera-Sandoval C, Ramirez-Castillo F Y, Muro A L and Avelar-Gonzalez F J 2014 Cell culture: history, development and prospects Int. J. Curr. Res. Acad. Rev. 2 188–200
- [2]del Carpio A 2014 The good, the bad, and the HeLa Berkley Sci. Rev. 5
- [3]Masters J R 2002 HeLa cells 50 years on: the good, the bad and the ugly Nat. Rev. Cancer 2 315–9
- [4]Schwarz E, Freese U K, Gissmann L, Mayer W, Roggenbuck B, Stremlau A and zur Hausen H 1985 Structure and transcription of human papillomavirus sequences in cervical carcinoma cells Nature 314 111–4
- [5]Wezel A L, Steenis G, Hannik Ch A and Cohen H 1978 New approach to the production of concentrated and purified inactivated polio and rabies tissue culture vaccines Dev. Biol. Stand. 41 159–68
- [6]Stones P B 1977 Production and control of live oral poliovirus vaccine in WI-38 human diploid cells Dev. Biol. Stand. 37 251–3
- [7]Beale A J 1981 Cell substrate for killed poliovaccine production Dev. Biol. Stand. 47 19–23
- [8]Steenis G, Wezel A L, Groot I G M and Kruijt B C 1980 Use of captive-bred monkeys for vaccine production Dev. Biol. Stand. 45 99–105
- [9]Shenoy M 2007 Animal cell culture Animal Biotechnology (New Delhi: Firewall) ch 1, p 3
- [10]Hayflick L and Moorhead P S 1961 The serial cultivation of human diploid cell strains Exp. Cell Res. 25 585–621
- [11]Wiktor T J, Fernandes M V and Koprowski H 1964 Cultivation of rabies virus in human diploid cell strain WI-38 J. Immunol. 93 353–66
- [12]Capstick P B, Telling R C, Chapman W G and Stewart D L 1962 Growth of a cloned strain of hamster kidney cells in suspended cultures and their susceptibility to the virus of foot and mouth disease Nature 195 1163–4
- [13]Losee J R and Ebeling A H 1914 The cultivation of human sarcomatous tissue in vitro J. Exp. Med. 20 140–8
- [14]Earle W 1943 Production of malignancy in vitro. IV. The mouse fibroblast cultures and changes seen in the living cells J. Natl Cancer Inst. 4 165–212
- [15]Pullen K, Johnston M D, Philips A W, Ball G D and Finter W B 1984 Very large scale suspension cultures of mammalian cells Dev. Biol. Stand. 60 175–7
- [16]Geraghty R J et al 2014 Guidelines for the use of cell lines in biomedical research Br. J. Cancer. 111 1021–46
- [17]Gómez-Lechón M J, Donato M T, Castell J V and Jover R 2003 Human hepatocytes as a tool for studying toxicity and drug metabolism Curr. Drug Metab. 4 292–312
- [18]MacDonald C 1990 Development of new cell lines for animal cell biotechnology Crit. Rev. Biotechnol. 10 155–78
- [19]Schurr M J et al 2009 Phase I/II clinical evaluation of StrataGraft: a consistent, pathogen-free human skin substitute J. Trauma 66 866–73
- [20]Nelson-Rees W A, Daniels D W and Flandermeyer R R 1981 Cross-contamination of cells in culture Science 212 446–52
- [21]Capes-Davis A et al 2010 Check your cultures! A list of cross-contaminated or misidentified cell lines Int. J. Cancer 127 1–8
- [22]Jiang L, Zeng X, Wang Z and Chen Q 2009 Cell line cross-contamination: KB is not an oral squamous cell carcinoma cell line Eur. J. Oral. Sci. 117 90–1
- [23]Fleckenstein E, Uphoff C C and Drexler H G 1994 Effective treatment of mycoplasma contamination in cell lines with enrofloxacin (Baytril) Leukemia 8 1424–34
- [24]Hay R J, Macy M L and Chen T R 1989 Mycoplasma infection of cultured cells Nature 339 487–8
- [25]Carroll S and Al-Rubeai M 2004 The selection of high-producing cell lines using flow cytometry and cell sorting Expert Opin. Biol. Ther. 4 1821–9
- [26]Browne S M and Al-Rubeai M 2007 Selection methods for high-producing mammalian cell lines Trends Biotechnol. 25 425–32
- [27]Gallagher C and Kelly P S 2017 Selection of high-producing clones using FACS for CHO cell line development Methods Mol. Biol. 1603 143–52
- [28]NCBI Resource Coordinators 2015 Database resources of the National Center for Biotechnology Information Nucleic Acids Res. 44 D7–19
- [29]Borenfreund E and Puerner J A 1985 Toxicity determined in vitro by morphological alternation and neutral absorption Toxicol. Lett. 24 119–24
- [30]Harris A R, Peter L, Bellis J, Baum B, Kabla A J and Charras G T 2012 Mechanics of cultured cell monolayers Proc. Natl Acad. Sci. 109 16449–54
- [31]Bhatia S, Goli D, Naved T and Sharma A 2018 Nutraceutical properties of Indian seaweed porphyra Adv. Invest. Pharmacol. Ther. Med. 1 47–54
- [32]Bhatia S 2016 Nanoparticles, plants, and algae Natural Polymer Drug Delivery Systems (Cham: Springer) pp 117–27
- [33]Bhatia S and Goli D 2016 Leishmaniasis: Biology, Control and New Approaches for Its Treatment (Boca Raton, FL: CRC Press) pp 164–73
- [34]Bhatia S, Namdeo A G and Nanda S 2010 Factors effecting the gelling and emulsifying properties of a natural polymer Sys. Rev. Pharm. 1 86–92
- [35]Bhatia S 2018 Introduction to Pharmaceutical Biotechnology: Enzymes, proteins and bioinformatics vol 2 (Bristol: IOP Publishing) pp 172–7
- [36]Bhatia S, Garg A, Sharma K, Kumar S, Sharma A and Purohit A P 2011 Mycosporine and mycosporine-like amino acids: a paramount tool against ultra violet irradiation Pharmacogn. Rev. 5 138–46
- [37]Bhatia S, Rathee P, Sharma K, Chaugule B B, Kar N and Bera T 2013 Immuno-modulation effect of sulphated polysaccharide (porphyran) from Porphyra vietnamensis Int. J. Biol. Macromol. 57 50–6
- [38]Bhatia S et al 2008 Novel algal polysaccharides from marine source: porphyran Pharmacogn. Rev. 2 271–6
- [39]Bhatia S, Sharma K, Namdeo A G, Chaugule B B, Kavale M and Nanda S 2010 Broad-spectrum sun-protective action of Porphyra-334 derived from Porphyra vietnamensis Pharmacogn. Res. 2 45–9
- [40]Bhatia S 2018 Introduction to Pharmaceutical Biotechnology: Basic techniques and concepts vol 1 (Bristol: IOP Publishing) pp 167–74
- [41]Bhatia S et al 2014 Significance of algal polymer in designing amphotericin B nanoparticles Sci. World J. 2014 564573
- [42]Bhatia S et al 2015 Investigation of the factors influencing the molecular weight of porphyran and its associated antifungal activity Bioact. Carbohydr. Dietary Fibre 5 153–68
- [43]Bhatia S, Sharma K, Sharma A, Nagpal K and Bera T 2015 Anti-inflammatory, analgesic and antiulcer properties of Porphyra vietnamensis Avicenna J. Phytomed. 5 69–77
- [44]Bhatia S, Sharma K and Bera T 2015 Structural characterization and pharmaceutical properties of porphyran Asian J. Pharm. 9 93–101
- [45]Bhatia S and Bera T 2015 Evaluation of pharmacognostical, phytochemical and anti-microbial properties of Porphyra vietnamensis Int. J. Green Pharm. 9 131–7
- [46]Bhatia S, Sharma K, Sharma A, Namdeo A G and Chaugule B B 2011 Anti-oxidant potential of Indian porphyra Pharmacology 1 248–57
- [47]Bhatia S et al 2015 Modern Applications of Plant Biotechnology in Pharmaceutical Sciences (New York: Academic, Elsevier) pp 164–74
- [48]Bhatia S 2016 Systems for Drug Delivery (Berlin: Springer) pp 122–7
- [49]Bhatia S 2016 Nanotechnology in Drug Delivery: Fundamentals, Design, and Applications (Boca Raton, FL: CRC Press)
- [50]Shinohara K et al 1988 Algal phycocyanins promote growth of human cells in culture In Vitro Cell. Dev. Biol. 24 1057–60
- [51]Khan F and Tanaka M 2017 Designing smart biomaterials for tissue engineering Int. J. Mol. Sci. 19 17
- [52]Lee E J, Kasper F K and Mikos A G 2014 Biomaterials for tissue engineering Ann. Biomed. Eng. 42 323–37
- [53]Knight E and Przyborski S 2014 Advances in 3D cell culture technologies enabling tissue-like structures to be created in vitro J. Anat. 227 746–56
- [54]Bhatia S 2016 Natural Polymer Drug Delivery Systems (Cham: Springer)
- [55]Fontana G et al 2016 Biofunctionalized plants as diverse biomaterials for human cell culture Adv. Healthc. Mater. 6 1601225
- [56]Liua W F and Chen C S 2005 Engineering biomaterials to control cell function Mater. Today 8 28–35
- [57]Ito Y 2002 Cell culture engineering using intelligent biomaterials Animal Cell Technology: Challenges for the 21st Century , ed K Ikura et al (Dordrecht: Springer)
- [58]Bhatia S 2018 Introduction to Pharmaceutical Biotechnology: Basic Techniques and Concepts vol 1 (Bristol: IOP Publishing)
- [59]Bhatia S 2018 History, scope and development of biotechnology Introduction to Pharmaceutical Biotechnology vol 2 (Bristol: IOP Publishing)