

Abstract
To improve the performance of energy storage devices, research into anode materials is essential. This study explores the potential of two-dimensional (2D) materials, particularly silicon carbide (Si2C), to enhance the efficacy of lithium-ion batteries (LIBs), sodium-ion batteries (SIBs), and potassium-ion batteries (KIBs). Our first-principles calculations indicate that Si2C achieves storage capacities of 174.7 mAh g−1 for LIBs, 436.8 mAh g−1 for SIBs, and 349.4 mAh g−1 for KIBs. The exceptional performance of Si2C comes from its high conductivity, large surface area, high capacitance, synergistic atomic radius and electronegativity effects. Furthermore, this study delves into the diffusion kinetics of Li/Na/K-ions in Si2C, revealing extremely low energy barriers and uncovering the fundamental principles behind its superior electrochemical performance. This research emphasizes Si2C's potential in energy storage, highlighting its capacity and diffusion advantages for Li/Na/K-ion batteries.
Export citation and abstract BibTeX RIS
1. Introduction
Efficient energy storage systems have always been a core topic of global interest systems have profoundly impacted daily life, with applications ranging from portable devices, electric and hybrid vehicles, to large power grids and various electronic equipment [1, 2]. In the energy storage market, rechargeable lithium-ion batteries (LIBs) dominate due to their high energy storage efficiency, long cycle life, and portability [1]. Since the successful exfoliation of graphene in 2004 [3], there has been widespread interest in two-dimensional (2D) materials [4], which have become a focal point of research, particularly in the field of battery materials, garnering significant attention from many researchers [5–12]. Anode materials, as a crucial component of LIBs, significantly influence battery performance [13]. However, despite their role in commercializing energy and promoting sustainable development, these batteries still face challenges such as insufficient theoretical specific capacity, safety issues, and the gradual depletion of lithium resources [14]. Due to the chemical similarity of sodium and potassium with lithium, Na/K-ion batteries have become a focal point of research. The abundance of these two elements contributes to the substitution of scarce lithium and improves dendrite issues in LIBs, while also offering high charging and discharging efficiency [15]. Research on anode materials for Li/Na/K-ion batteries encompasses non-metals [16, 17], transition metal compounds [18, 19], and heterostructures [20]. MXenes, known for their high specific surface area, conductivity [21], hydrophilicity [22], and flexibility [23] receive particular attention in battery anode applications.
Studies by Liu et al found that three halogenated forms of MXene, Ti3C2Tx (T = F, Cl, Br) can significantly impact lithium-ion storage capacity. Especially for Ti3C2Fx and Ti3C2Clx , achieving maximum specific capacities of 123 and 138 mAh g−1, respectively, highlighting the importance of halogen surface terminations [24]. However, the theoretical capacity of these materials for Li remains low. In research conducted by Zhou et al first-principles calculations were used to predict the energy storage performance of WCrC and WCrCO2 sodium-ion batteries (SIBs) based on W2C and W2CO2, finding improvements in theoretical capacity while maintaining electronic characteristics and stability, with WCrC and WCrCO2 capacities of 175.91 mAh g−1 and 154.98 mAh g−1, respectively [25]. Although there is an improvement over W2C and W2CO2, the capacities are still relatively low. Additionally, previous studies theoretically designed MXene material Mo2CrC2, achieving a theoretical capacity for K of 154.88 mAh g−1 [26]. Despite having a lower diffusion barrier, the theoretical capacity of this material remains a key issue. Therefore, the low theoretical capacities of many MXene materials are attributed to the inclusion of transition metal atoms, leading to a higher molar mass, which poses a limitation in enhancing battery capacity.
This study builds on the observation that lightweight anode materials typically exhibit higher capacities. Specifically, research indicates that non-metallic materials similar to MXenes, especially those with electronic structural similarities, can effectively enhance specific capacity. Our work maintains the MXenes structure while achieving a complete replacement of transition metal elements, selecting silicon (Si) as the substitute. For instance, we replaced the transition metals W and Cr in the MXenes material WCrC with silicon, forming another type of 2D material. This material shows tremendous potential in high-performance SIBs, with a theoretical capacity significantly higher than that of WCrC. First principles were applied to investigate the potential of a single layer of Si2C as an anode material for Li/Na/K-ion batteries, and its structural and electronic characteristics were also comprehensively analyzed. The electrochemical performance of the Si2C single layer as an anode, found that due to its low diffusion barrier, ideal open-circuit voltage (OCV), and high rate capability, the Si2C single layer is an excellent choice for Li/Na/K-ion battery anodes, particularly excelling as a sodium-ion battery anode material. Through this study, we have not only expanded the range of choices for battery elements and structures but also provided a significant reference for future experimental research.
2. Computational method
To probe the attributes of Si2C, first-principles methods were utilized, grounded in the principles of density functional theory (DFT). Our investigation hinged on the Quantum Espresso software suite, a comprehensive computational code package tailored for DFT-based electronic structure calculations and material modeling [27]. Our approach involved employing the generalized gradient approximation (GGA) exchange-correlation functional as per the Perdew–Burke–Ernzerhof (PBE) functional, coupled with ultrasoft pseudopotentials for Si, C, and alkali metal atoms (Li/Na/K) [28, 29]. Following extensive convergence tests, we determined wave function cut-off configurations for our calculations. The wave function cut-off energy was set at 50 Ry, and the charge density cut-off energy was set at 500 Ry. A vacuum layer of 25 Å was chosen to reduce the effects of periodic boundary conditions on the z-axis. Force convergence thresholds were precisely set at  Ry/Bohr, while the total energy convergence threshold was established at
Ry/Bohr, while the total energy convergence threshold was established at  Ry. Recognizing the significance of long-range van der Waals interactions, the semi-empirical Grimme-d2 method was incorporated for adjustments [30]. For K-point grids sampling of the primitive unit cell, the Monkhorst–Pack scheme was employed, opting for a
Ry. Recognizing the significance of long-range van der Waals interactions, the semi-empirical Grimme-d2 method was incorporated for adjustments [30]. For K-point grids sampling of the primitive unit cell, the Monkhorst–Pack scheme was employed, opting for a  configuration. Moreover, the climbing image nudged elastic band (CI-NEB) method was utilized to further validate the diffusion barriers [31].
configuration. Moreover, the climbing image nudged elastic band (CI-NEB) method was utilized to further validate the diffusion barriers [31].
3. Results and discussion
3.1. Structural and electronic properties of Si2C
Following precise structural optimization, the structure of the  Si2C monolayer was comprehensively detailed, as shown in figure 1. This Si2C monolayer is composed of 18 Si atoms and 9 C atoms, totaling 27 atoms. It exhibits a hexagonal structure within the P3m1 space group, with C atoms situated between two Si atoms. After the optimization, the C–Si bond length in Si2C was determined to be 2.03 Å, while its lattice constants are a= b = 2.87 Å. To investigate the stability of the Si2C monolayer structure more thoroughly, we utilized DFT for the calculation of its phonon spectrum, and conducted ab initio molecular dynamics simulations (AIMD) as a complementary analysis. The phonon spectrum of the Si2C monolayer, as depicted in figure 2(a), showed no imaginary frequencies, indicating its dynamical stability. Concurrently, as figure 2(b) illustrates, both the energy and structural variations of the Si2C monolayer were relatively minor, thereby affirming its thermodynamic stability. Furthermore, previous research on the electronic band structure of Si2C reveals a bandgap of 0.08 eV when employing the PBE exchange-correlation functional, while adjustments using the HSE06 functional to address the underestimation by the GGA approximation yield a bandgap of 0.46 eV, suggesting the semiconducting characteristic of Si2C [32].
Si2C monolayer was comprehensively detailed, as shown in figure 1. This Si2C monolayer is composed of 18 Si atoms and 9 C atoms, totaling 27 atoms. It exhibits a hexagonal structure within the P3m1 space group, with C atoms situated between two Si atoms. After the optimization, the C–Si bond length in Si2C was determined to be 2.03 Å, while its lattice constants are a= b = 2.87 Å. To investigate the stability of the Si2C monolayer structure more thoroughly, we utilized DFT for the calculation of its phonon spectrum, and conducted ab initio molecular dynamics simulations (AIMD) as a complementary analysis. The phonon spectrum of the Si2C monolayer, as depicted in figure 2(a), showed no imaginary frequencies, indicating its dynamical stability. Concurrently, as figure 2(b) illustrates, both the energy and structural variations of the Si2C monolayer were relatively minor, thereby affirming its thermodynamic stability. Furthermore, previous research on the electronic band structure of Si2C reveals a bandgap of 0.08 eV when employing the PBE exchange-correlation functional, while adjustments using the HSE06 functional to address the underestimation by the GGA approximation yield a bandgap of 0.46 eV, suggesting the semiconducting characteristic of Si2C [32].

Figure 1. Top and side views of the Si2C monolayer.
Download figure:
Standard image High-resolution image
Figure 2. Si2C monolayer (a) phonon spectrum. (b) AIMD.
Download figure:
Standard image High-resolution image3.2. Cation adsorption and diffusion on Si2C
After confirming the electronic characteristics of the Si2C monolayer, we further investigated the adsorption behavior of Li/Na/K-ions on the monolayer material. To determine the most stable adsorption sites, the adsorption energy was calculated using the following equation:

where  represents the adsorption energy,
represents the adsorption energy,  is the total energy of the Si2C configuration with adsorbed alkali metal atoms,
is the total energy of the Si2C configuration with adsorbed alkali metal atoms,  is the energy of the Si2C monolayer without any adsorbed atoms, and
is the energy of the Si2C monolayer without any adsorbed atoms, and  is the energy of a single alkali metal atom in a bulk metal. According to equation (1), a positive adsorption energy suggests that the adsorption process is endothermic, indicating that the reaction is non-spontaneous, whereas a negative adsorption energy indicates an exothermic reaction, implying that the reaction can occur spontaneously. Utilizing this method, researchers can accurately assess the adsorption stability of different alkali metal atoms on the Si2C monolayer, providing crucial information for optimizing the performance of Si2C monolayers as anodes in Li/Na/K-ion batteries.
is the energy of a single alkali metal atom in a bulk metal. According to equation (1), a positive adsorption energy suggests that the adsorption process is endothermic, indicating that the reaction is non-spontaneous, whereas a negative adsorption energy indicates an exothermic reaction, implying that the reaction can occur spontaneously. Utilizing this method, researchers can accurately assess the adsorption stability of different alkali metal atoms on the Si2C monolayer, providing crucial information for optimizing the performance of Si2C monolayers as anodes in Li/Na/K-ion batteries.
To gain a comprehensive understanding of the adsorption of Li/Na/K-ions on Si2C, multiple configurations and different adsorption sites were considered. Figures 3(a)–(c) showcase the adsorption models of Li/Na/K-ions on three potential adsorption sites: site-a, site-b, and site-c. Specifically, site-a is located above the empty space of a C atom, site-b above the empty space of a Si–C bond, and site-c above the empty space of a Si atom. Our computational results indicate that the adsorption of Li-ions and Na-ions at site-c is unstable, as they naturally migrate to site-a, similarly, the adsorption of K-ions at site-b and site-c also exhibits instability, ultimately moving to site-a. The adsorption energy results, displayed in figure 4, clearly demonstrate that for Li/Na/K-ions, the most stable adsorption site is site-a. Specifically, the adsorption energies at the most stable positions for Li and Na are approximately −1.41 eV, and for K, it is around −1.76 eV. These values are lower than the corresponding adsorption energies of Si3C, which are −0.74 eV for Li-ion, −0.72 eV for Na-ion, and −1.14 eV for K-ion [33]. Of particular significance is the fact that a higher absolute value of negative adsorption energy corresponds to greater thermodynamic favorability, suggesting increased thermodynamic benefits for the adsorption process. This observation indicates that Si2C exhibits more stable adsorption when dealing with single atoms.
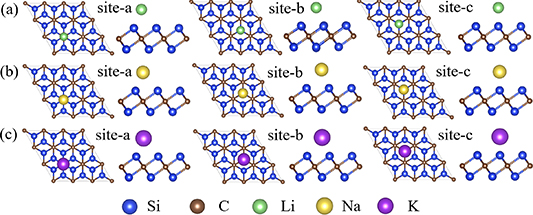
Figure 3. The adsorption sites considered on the monolayer of Si2C (a) Li (b) Na (c) K.
Download figure:
Standard image High-resolution image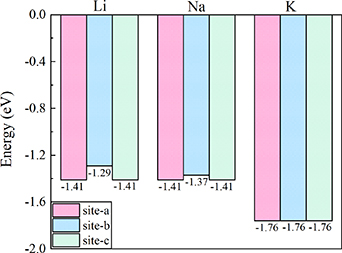
Figure 4. Adsorption energy of Li/Na/K-ions at different sites.
Download figure:
Standard image High-resolution imageIn this study, we conducted a comprehensive investigation of the adsorption behavior and charge transfer phenomena of Li/Na/K atoms on the Si2C monolayer using Bader charge analysis and the charge density difference (CDD) method [34]. The CDD  is calculated according to equation (2):
is calculated according to equation (2):
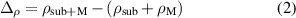
where  represents the charge density of the Si2C system with adsorbed alkali metal atoms,
represents the charge density of the Si2C system with adsorbed alkali metal atoms,  is the charge density of the Si2C monolayer without adsorbed alkali metal atoms, and
is the charge density of the Si2C monolayer without adsorbed alkali metal atoms, and  is the charge density of the isolated alkali metal atoms. This methodology allows us to observe and analyze the charge transfer between the Si2C monolayer and Li/Na/K atoms, thereby gaining a deeper understanding of their interactions and impact on electrochemical properties.
is the charge density of the isolated alkali metal atoms. This methodology allows us to observe and analyze the charge transfer between the Si2C monolayer and Li/Na/K atoms, thereby gaining a deeper understanding of their interactions and impact on electrochemical properties.
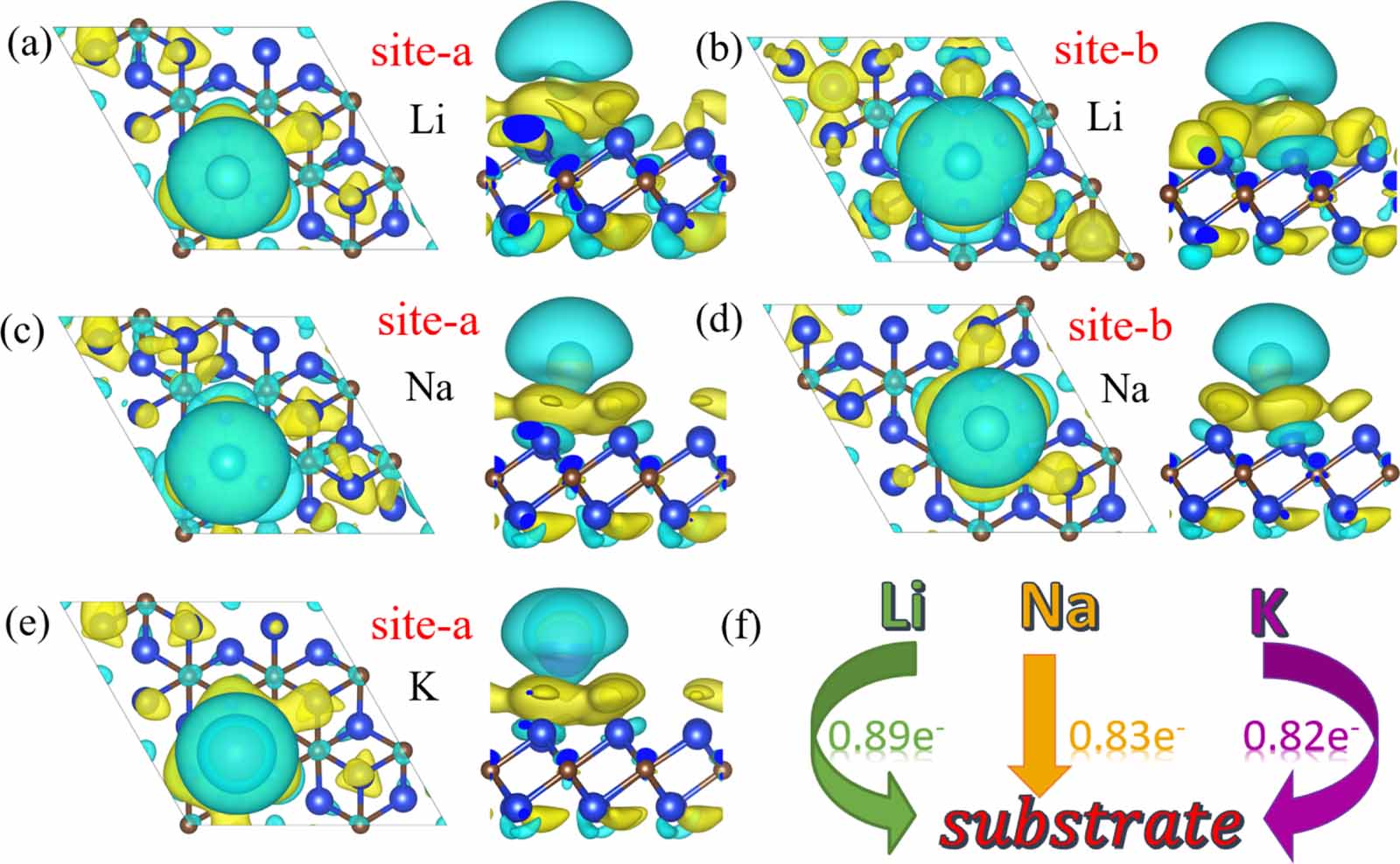
As shown in figure 5, the differential charge images indicate that the charge is primarily concentrated between the monolayer and the Li/Na/K atoms. This pattern of charge distribution suggests that the interaction between the sodium atoms and the monolayer exhibits characteristics of chemical adsorption. Through an in-depth analysis of Bader charge, we further understood the charge transfer between different cations and the Si2C monolayer. Specifically, each lithium atom transfers about 0.89e worth of charge to the Si2C monolayer, sodium transfers 0.83e, and potassium transfers 0.82e. These results reveal significant electron exchange between the alkali metal atoms and the Si2C monolayer. The degree of charge transfer directly affects the material conductivity and electrochemical reaction activity. This comprehension is crucial for understanding the electrochemical performance and ion adsorption behavior of the material. This research carries substantial implications for assessing the potential of the Si2C monolayer as an anode material.

Figure 5. Overview of cation CDD and charge transfer in the Si2C monolayer. (a) and (b) show the CDD of Li at site-a and site-b, respectively. (c) and (d) display the CDD of Na at site-a and site-b, respectively. (e) reveals the CDD of K at site-a. 6(f) Presents in detail the charge transfer of Li/Na/K-ions to the Si2C monolayer.
Download figure:
Standard image High-resolution imageAccording to symmetry considerations, there should be three diffusion pathways on the surface of Si2C. Ultimately, we identified three possible migration pathways for Li/Na/K-ions in Si2C: path1 (site-a1  site-b
site-b  site-a2), path2 (site-a1
site-a2), path2 (site-a1  site-a2) and path3 (site-a1
site-a2) and path3 (site-a1  site-c
site-c  site-a2). These paths involve migration from one most stable position to another, potentially passing through one or two intermediate positions.
site-a2). These paths involve migration from one most stable position to another, potentially passing through one or two intermediate positions.
As depicted in figure 6, on the Si2C monolayer, the diffusion energy barriers for Li/Na/K-ions are 0.14 eV, 0.065 eV, and 0.041 eV, respectively. Considering the influence of electronegativity on an atom's ability to attract electron pairs and consequently affect ion behavior, the strength of the ionic bonds formed between Li, Na, K, and the Si2C substrate follows an order of bLi > bNa > bK to some extent (bLi, bNa, bK respectively represent the bonding strengths of Li/Na/K-ions with the substrate). Therefore, in terms of diffusion barriers, the order would be dLi > dNa > dK. (dLi, dNa,dK respectively represent the diffusion barriers of Li/Na/K-ions on the Si2C monolayer).
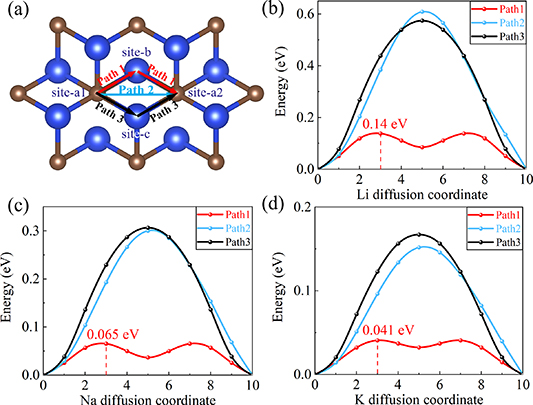
Figure 6. (a) Possible diffusion paths of Li/Na/K on the Si2C monolayer. Diffusion barrier (b) Li-ions. (c) Na-ions. (d) K-ions.
Download figure:
Standard image High-resolution imageMoreover, compared to materials explored by other researchers, such as MoS2 (0.27 eV) [35], InSe (0.23 eV) [36], Ti2CS2 (1.51 eV) [37], SnSe (1.65 eV) [38], SnO2 (1.94 eV) [38], single layer graphene (0.31 eV) [39], the diffusion barrier for Li-ions in Si2C, which is 0.14 eV, is relatively low. For Na-ions, their diffusion barrier in Si2C is 0.065 eV, which is also lower than traditional 2D materials such as MoS2 (0.22 eV) [40], Ti2CC2 (0.16 eV) [41], Ti2CS2 (0.095 eV) [41], CoS2 (0.15 eV) [42], C-silicyne (0.57 eV) [43]. For K-ions, the energy barrier in Si2C is 0.041 eV, which is lower than previously studied materials such as SrSnO3 (0.44 eV) [44], C-silicyne (0.24 eV) [43], T-graphene (0.25 eV) [45], super-borophene (0.44 eV) [46] and graphite nanomesh with the hole density of 46% (0.19 eV) [47]. These comparisons highlight the advantages of the Si2C monolayer as a material for Li/Na/K-ion batteries.
3.3. Theoretical capacity and OCV of Si2C
To establish Si2C as an effective battery anode material, the key lies in its superior theoretical capacity and OCV performance, which are determining factors for battery efficiency. Therefore, this study focuses on evaluating the performance of the Si2C monolayer in terms of theoretical capacity and OCV. Specifically, the theoretical capacitance is determined by the maximum adsorption concentration, while the OCV is calculated based on adsorption energy. The half-cell reaction can be represented by the following chemical equation:
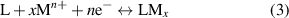
where L represents the monolayer anode material, x indicates the concentration of adsorbed atoms, M is the type of adsorbed atoms, and n is the number of electrons transferred to the anode during atom adsorption. By calculating the maximum capacity of adsorbed cations, we determined the concentration x of adsorbed cations. We found that alkali metal atoms could be adsorbed on both sides of the Si2C monolayer. To assess the effectiveness of one-by-one adsorption, we used the formula for the average adsorption energy  :
:
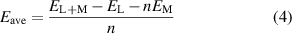
where  is the total energy of the system after atom adsorption,
is the total energy of the system after atom adsorption,  is the total energy of the initial substrate,
is the total energy of the initial substrate,  is the energy of each adsorbed alkali metal atom in bulk metal, and n is the total number of adsorbed atoms.
is the energy of each adsorbed alkali metal atom in bulk metal, and n is the total number of adsorbed atoms.
In exploring the theoretical capacity of the Si2C monolayer, we observed that the maximum adsorption quantity of Li, Na, and K atoms on the Si2C monolayer is limited to a single layer. As shown in figures 7(a)–(c), the Si2C monolayer can adsorb up to 5 Li atoms, 8 K atoms, and a maximum of 10 Na atoms. The interlayer adsorption energies for these different atoms on the Si2C monolayer are −1.12 eV for Li, −0.62 eV for Na, and −0.83 eV for K. Notably, whether it is double-sided adsorption or the adsorption of additional Li/Na/K-ions on a single side, the adsorption energy becomes positive. Figures 7(d)–(f) show the electron localization function (ELF) maps of the Si2C model with the maximum adsorbed states of Li/Na/K atoms. Here, '1' and '0' represent fully localized and fully delocalized electrons, respectively. Typically, ELF values below 0.5 suggest the presence of metallic or ionic bonding, whereas ELF values above 0.5 indicate the formation of covalent bonds. These images clearly demonstrate that covalent bonds are formed between Si and C in the Si2C monolayer, while ionic bonds are formed between the alkali metal ions and the Si2C monolayer.
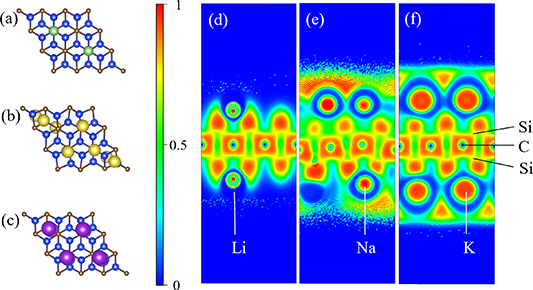
Figure 7. Final adsorption models and ELF maps, (a) Li adsorption model, (b) Na adsorption model, (c) K adsorption model, (d) Li adsorption ELF map, (e) Na adsorption ELF map, (f) K adsorption ELF map.
Download figure:
Standard image High-resolution imageUpon determining the maximum adsorption capacity of the Si2C monolayer, we proceeded to calculate its maximum theoretical capacitance using the following formula:

where n represents the number of adsorbed atoms per unit cell, F is the Faraday constant (26 801 mAh g−1), and M is the molar mass of the Si2C monolayer. The results, as shown in figure 8, provide the maximum theoretical capacitance of the Si2C monolayer. This calculation is crucial for evaluating the energy storage potential of Si2C as an anode material in Li/Na/K-ion batteries, offering insights into its effectiveness in energy storage application.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. OCV of the Si2C monolayer under different concentrations of alkali metal ion adsorption (a) Li-ions. (b) Na-ions. (c) K-ions.
Download figure:
Standard image High-resolution image{kind=link}
In LIBs applications, the Si2C monolayer exhibits a high maximum theoretical capacitance of 174.7 mAh g−1. This figure is notably superior compared to other materials such as Ti3C2Fx (123 mAh g−1) [24], Ti3C2Clx (138 mAh g−1) [24], and Ti2NSSe (121.39 mAh g−1) [48]. For potassium-ion batteries, the Si2C monolayer's maximum theoretical capacitance reaches an impressive 349.4 mAh g−1, significantly higher than Mo2CrC2 (154.88 mAh g−1) [26], K2C6O6 (217.8 mAh g−1) [49], WSSe (156.0 mAh g−1) [50], graphite (279 mAh g−1) [51], and GeC (320 mAh g−1) [51]. In the case of SIBs, the Si2C monolayer stands out with an exceptional theoretical capacity of 436.8 mAh g−1, showcasing a significant capacitance advantage compared to common materials like Na2Ti3O7 (177 mA h g−1) [52], MoS2 (146 mA h g−1) [53], Ti2NS2 (84.76 mA h g−1) [54], V2NS2 (99.8 mA h g−1) [54], WCrC (176 mA h g−1) [25], and WCrCO2 (155 mA h g−1) [25]. Through these comparisons, the Si2C monolayer demonstrates tremendous potential as an anode material for Li-ion, Na-ion, and K-ion batteries, marking an important direction for future research and development in battery materials.
By conducting detailed calculations of the OCV for Li/Na/K-ions in the Si2C monolayer material, we also closely examined the voltage safety during the discharge process. The OCV can be calculated using the following formula:
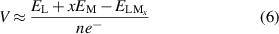
where  is the energy of the substrate with adsorbed atoms,
is the energy of the substrate with adsorbed atoms,  is the energy of the substrate without adsorbed atoms,
is the energy of the substrate without adsorbed atoms,  is the energy of a single alkali metal atom in bulk metal, and x is the number of adsorbed atoms. An OCV below 1 V helps prevent the formation of dendrites within the battery, thereby enhancing its safety, and also implies a lower charging voltage requirement for the battery. As shown in figure 8, we observed different trends in OCV as the concentration of Li/Na/K-ions on the monolayer increases. In figure 8(a), with Li-ions concentration increasing from 1 to 4 atoms, the OCV ranges from 0.78 V to 1.41 V, with an average potential of about 0.95 V. In figure 8(b), as Na-ions concentration increases from 1 to 10 atoms, the OCV is between 0.22 V and 1.33 V, with an average potential of about 0.62 V. In figure 8(c), for K-ions concentration increasing from 1 to 8 atoms, the OCV ranges from 0.21 V to 1.68 V, with an average potential of about 0.83 V.
is the energy of a single alkali metal atom in bulk metal, and x is the number of adsorbed atoms. An OCV below 1 V helps prevent the formation of dendrites within the battery, thereby enhancing its safety, and also implies a lower charging voltage requirement for the battery. As shown in figure 8, we observed different trends in OCV as the concentration of Li/Na/K-ions on the monolayer increases. In figure 8(a), with Li-ions concentration increasing from 1 to 4 atoms, the OCV ranges from 0.78 V to 1.41 V, with an average potential of about 0.95 V. In figure 8(b), as Na-ions concentration increases from 1 to 10 atoms, the OCV is between 0.22 V and 1.33 V, with an average potential of about 0.62 V. In figure 8(c), for K-ions concentration increasing from 1 to 8 atoms, the OCV ranges from 0.21 V to 1.68 V, with an average potential of about 0.83 V.
For the Si2C monolayer, when adsorbing Li-ions, the average potential slightly exceeds 1 V but remains within a controllable range, sufficient to prevent lithium dendrite formation to some extent. In contrast, the average potentials for adsorbing Na-ions and K-ions are both less than 1 V, significantly reducing the risk of sodium and potassium dendrite formation. Moreover, the intercalation voltages of Li/Na/K-ions in the Si2C anode exhibit a stable trend, further evidencing its safety during discharge.
Finally, we also calculated the changes in lattice constants of the Si2C monolayer after adsorption of Li/Na/K-ions, to assess its structural stability during the charge-discharge processes of batteries. The results showed that after adsorption of Li/Na/K-ions, the lattice constant changes of the 3  3 supercell of the Si2C monolayer were extremely minimal, indicating good structural stability during battery operation. Specifically, upon adsorption of Li-ions to their maximum capacity, the lattice constants a and b were both 8.59(6) Å, a change of about 0.21% compared to before adsorption. For Na-ions adsorbed to their maximum capacity, the lattice constants changed to a = b = 8.58(3) Å, a change of about 0.36%. And for K-ions adsorbed to their maximum capacity, the lattice constants were a = b = 8.60(1) Å, a change of about 0.15%. These results indicate that the lattice changes of the Si2C monolayer are very small, whether lithium, sodium, or K-ions can be adsorbed. This suggests that even after adsorption of the maximum number of ions, the Si2C monolayer maintains good structural integrity, which is a desirable characteristic for anode materials in battery applications.
3 supercell of the Si2C monolayer were extremely minimal, indicating good structural stability during battery operation. Specifically, upon adsorption of Li-ions to their maximum capacity, the lattice constants a and b were both 8.59(6) Å, a change of about 0.21% compared to before adsorption. For Na-ions adsorbed to their maximum capacity, the lattice constants changed to a = b = 8.58(3) Å, a change of about 0.36%. And for K-ions adsorbed to their maximum capacity, the lattice constants were a = b = 8.60(1) Å, a change of about 0.15%. These results indicate that the lattice changes of the Si2C monolayer are very small, whether lithium, sodium, or K-ions can be adsorbed. This suggests that even after adsorption of the maximum number of ions, the Si2C monolayer maintains good structural integrity, which is a desirable characteristic for anode materials in battery applications.
4. Conclusions
Based on DFT, our comprehensive evaluation of the Si2C monolayer as an anode material for Li/Na/K-ion batteries in this study covered its key characteristics, including electronic properties, ion adsorption and diffusion kinetics, theoretical capacity, and OCV. Our study observes negative adsorption energies of Li/Na/K-ions on monolayer Si2C, indicating spontaneous adsorption. The adsorption is most stable above the carbon atoms, with the adsorption energy of Li-ions approximately equal to that of Na-ions, both higher than that of K-ions. Li/Na/K-ions share the same diffusion path on the surface of the Si2C monolayer, with diffusion barriers of 0.14 eV, 0.065 eV and 0.041 eV respectively. In terms of battery applications, the theoretical specific capacities of the Si2C monolayer are 174.7, 436.8, and 349.4 mAh g−1 for Li-ion, Na-ion, and K-ion batteries, respectively. Under conditions of maximum capacity, the lattice changes in Si2C are very minimal, indicating excellent structural stability. Furthermore, calculations of OCV demonstrate safety of the Si2C monolayer during discharge. Overall, the monolayer Si2C exhibits outstanding performance as an anode material for Li/Na/K-ion batteries, with high theoretical capacity and low diffusion barriers. These achievements open new perspectives for experimental and theoretical research on 2D materials, holding significant implications.
Acknowledgments
The research is supported by National Natural Science Foundation of China (Nos. 52227813, 11204053, and 11074059). We acknowledge the computing resources operated by the Laboratory for Space Environment and Physical Sciences, Harbin Institute of Technology.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Conflict of interest
The authors declare no competing financial interests.