


ABSTRACT
Propene (CH3CHCH2), detected in the cold core TMC-1, is a surprisingly saturated (H-rich) species for observation in such regions. In a recently proposed gas-phase formation mechanism, interstellar propene is produced from its protonated precursor C3H+7 (CH3CHCH+3) via a dissociative recombination process. The precursor ion C3H+7 is itself produced via two consecutive radiative association reactions involving H2 starting from the isomer of C3H+3 with a linear carbon backbone (CH2CCH+). Initial calculations showed that the radiative association reactions are efficient enough to allow the production of an abundance of propene equal to that observed. However, a combination of experiments and more refined quantum chemical ab initio calculations reported here does not corroborate the initial result. Indeed, from both of these approaches, we have learned that the radiative association reactions leading to protonated propene do not occur efficiently at interstellar temperatures due to activation energy barriers. The result is that propene cannot be produced efficiently by the suggested gas-phase synthetic route. It is still difficult to say, however, that no suitable gas-phase syntheses for propene can occur in cold cores such as TMC-1.
Export citation and abstract BibTeX RIS
1. INTRODUCTION
For the most part, the large molecules detected in cold cores such as TMC-1, with a temperature of 10 K and a density of 104 cm−3, are carbon chains, a term that implies that the structures are mainly linear and rather unsaturated, a chemical term meaning that there are few hydrogen atoms attached to the heavy-atom structure. This result was at first surprising given the large abundance of molecular hydrogen in dense clouds compared with any other molecule. But it was gradually discerned by measurement and theory that reactions of the type
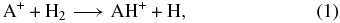
where A+ stands for a molecular ion, do not occur rapidly for ions with more than a few hydrogen atoms in most instances (McEwan et al. 1999). Thus, molecular ions with many hydrogen atoms are not predicted to be abundant, so that the neutral fragments formed from dissociative recombination reactions between these ions and electrons are themselves not rich in hydrogen atoms, especially since dissociative recombination reactions tend to prefer channels with significant breakdown of the parent ionic structure. Although this explanation works in general, there are some exceptions; small abundances of partially saturated species such as methanol (CH3OH) and acetaldehyde (CH3CHO) have been detected in cold cores. Recently, Bacmann et al. (2012) have also detected the organic molecules dimethyl ether (CH3OCH3) and methyl formate (HCOOCH3) in the gas phase of the cold prestellar core L1689B. Even more recently, Cernicharo et al. (2012) have discovered a wide array of oxygen-containing organic molecules in the cold core B1-b. It is now assumed, especially for the case of methanol, that these species are formed by hydrogenation processes on the icy mantles of interstellar grains, with atomic hydrogen the important hydrogenating reactant. Thus the formation of methanol has been assumed to occur as CO accretes from the gas and is sequentially hydrogenated by surface reactions with H atoms to form first formaldehyde (H2CO) and finally methanol (Watanabe & Kouchi 2002). The more complex species may be formed by surface reactions involving other atoms or even radical–radical reactions (Herbst & van Dishoeck 2009). Desorption can then occur by non-thermal processes such as photodesorption, as has been studied recently in the laboratory (Öberg et al. 2009).
Another partially saturated molecule, propene, also known as propylene, was detected in TMC-1 by Marcelino et al. (2007) and found to have a reasonably large fractional abundance with respect to total hydrogen of ≈2 × 10−9. The millimeter-wave rotational spectrum of the molecule had been studied in the laboratory (Pearson et al. 1994). In addition, it is now known that propene reacts rapidly with atomic oxygen at low temperatures, a process likely to destroy it rapidly in cold cores (Sabbah et al. 2007). In retrospect, the detection of propene with its small dipole moment and expected low abundance is remarkable and suggests that its synthesis is far more efficient than that of other partially saturated species in the cold core gas. The obvious gas-phase synthesis, in which the precursor ion C3H+7 is produced via a series of reactions of the form shown in Equation (1), does not occur (McEwan et al. 1999). A surface synthesis analogous to the formation of methanol might be possible although there is no obvious evidence that the H-atom-based synthesis could produce a molecule with six hydrogen atoms, close to the saturated limit of propane, C3H8.
Among the various suggestions for gas-phase syntheses put forward by Marcelino et al. (2007), one is based on radiative association reactions. In a previous paper (Herbst et al. 2010), we investigated the rate coefficients for two ion-molecule association reactions that produce the ionic precursor to propene from the starting propargyl ion H2CCCH+, which is the non-cyclic isomer of the lowest energy form c-C3H+3. According to previous simulations, the abundance of this "linear" isomer is not much reduced with respect to its cyclic counterpart (Herbst et al. 2010). For the first of the two radiative association reactions,
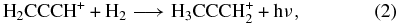
we calculated via ab initio techniques at the CCSD level using a triple-zeta basis set extended by polarization and diffuse functions that there is no activation energy barrier if the product is not the lowest energy isomer of C3H+5, which has the allenic form H2CCHCH+2, but rather is a metastable isomer (H3CCCH+2). We then calculated that a subsequent radiative association reaction,
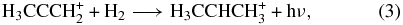
to form the isopropyl radical ion (a form of protonated propene) can also occur without a barrier. (More recent calculations show also that such collisions do not convert the metastable isomer to the lower energy form.) The situation for the lower energy form of C3H+5 is different; here a significant barrier is calculated with this ion as a reactant (Almlöf et al. 1984). The calculated rate coefficients for the association reactions shown in Equations (2) and (3), if truly barrierless, are rather large, especially at low temperatures. Specifically, at 10 K, the calculated rate coefficient for the first reaction is 1.2 × 10−9 cm3 s−1, which is near the Langevin limit, while the second one occurs with the slightly slower rate coefficient of 3.5 × 10−10 cm3 s−1. Both reactions are rate-determining destruction steps for the reactant ions given the high abundance of H2, so that essentially every propargyl ion is converted into the ionic precursor of propene. Once this ion is formed, the dissociative recombination has been shown to produce propene with an unusually substantial branching fraction (Florescu-Mitchell & Mitchell 2006):
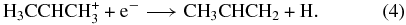
A conclusion of Herbst et al. (2010) was that the production of propene via this mechanism might be efficient enough to explain its observed abundance, although detailed chemical simulations were not undertaken. An early simulation based on estimated rate coefficients had shown that a sufficient amount of propene could be produced (Roueff & Herbst 2009).
The theoretical results obtained for the radiative association reactions are in partial disagreement with earlier experimental results on the analogous three-body association processes measured by McEwan et al. (1999), which were found to be very slow, although it was not clear which isomers of C3H+3 and C3H+5 were used in the experiments. To improve the situation, we performed some detailed experiments on the association reactions and undertook refined calculations for possible activation energy barriers. In this paper, we report that the net result from both detailed experiment and refined theory is that the radiative association reactions proposed to produce protonated propene from the propargyl ion both have sufficiently high barriers that they are unimportant in the cold interstellar medium. Thus our models cannot produce propene via this once promising sequence of reactions, although other efficient gas-phase syntheses may indeed exist.
The remainder of this paper is organized as follows. In Section 2, we discuss the detailed experiments and new refined ab initio calculations. Calculated results for the abundance of propene are presented in Section 3, and the results are discussed in Section 4.
2. EXPERIMENTAL
Rate coefficients for the reactions of C3H+3 and C3H+5 with molecular hydrogen were determined using the FA-SIFT (Flowing Afterglow-Selected Ion Flow Tube) at the University of Colorado, Boulder. Experimental details are described elsewhere (Van Doren et al. 1987; Bierbaum 2003; Snow & Bierbaum 2008). In general, the FA-SIFT is divided into four regions responsible for ion production, mass selection, reaction, and detection. The ions of interest, here C3H+3 and C3H+5, are produced using an electron ionization source (EI, 70 eV) on propene (Airgas, 99.9%) seeded in helium buffer gas (Airgas, 99.997%), with the gas being further purified by passage through a molecular sieve trap immersed in liquid nitrogen. The pressure in the ion production region is about 0.25 torr helium and about 0.01 torr propene. The ions of interest are mass-selected using a quadrupole mass analyzer. The pressure in the mass-selection region is about 10−6 torr. In the experiments, C3H+3 and C3H+5 are guided by ion lenses and injected through a Venturi inlet into the reaction flow tube where the ions are thermalized to room temperature (300 K) through collisions with helium. The pressure in the flow tube is about 0.50 torr (flow = 220 std cm3 s−1). Molecular hydrogen (H2, Airgas, 99.999%) is purified by passage through a molecular sieve trap immersed in liquid nitrogen and is subsequently added into the flow tube at a known reaction distance. The reagent ion intensity is monitored as a function of the concentration of H2 using an electron multiplier, and a reaction rate coefficient is obtained using pseudo first-order kinetic analysis.
The relative populations of the two isomers of C3H+3 can be traced by reaction with the benzene molecule (C6H6). Neutral benzene was added at different inlets along the reaction flow tube, and the ion intensity of C3H+3 was recorded as a function of reaction distance, which is proportional to reaction time, as shown in Figure 1. The plot shows an initial steep fall-off indicating fast reaction, before it levels off for larger reaction distances. The non-reactive component represents about 1850 counts s−1 of the initial 7300 counts s−1 of C3H+3. Thus, about 25% of the C3H+3 ions are non-reactive.
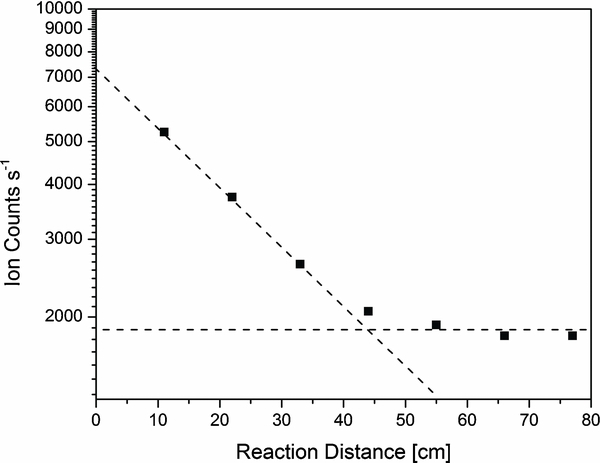
Figure 1. The reaction of C3H+3 with benzene. The reaction is used as a tracer of the isomeric ratio for cyclic/linear C3H+3. In our experiment, the cyclic isomer was determined to be between 20% and 30% of the C3H+3, dependent upon the experimental conditions.
Download figure:
Standard image High-resolution imageCalculations show that the most stable isomer of C3H+3 is the cyclic form whereas in our experiments the higher energy linear form of C3H+3 (propargyl, CH2CCH+) was also produced. Lossing (1972) reports that the cyclic isomer is more stable by 105 kJ mol−1; our calculations show that the cyclic isomer is more stable by 136 kJ mol−1 at the MP2(full)/aug-cc-pVTZ level of theory. The cyclic isomer of C3H+3 does not show any reaction with benzene, but the linear propargyl form reacts readily with benzene at the collision rate via the following mechanism (Ausloos & Lias 1981):
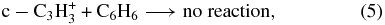
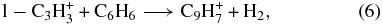
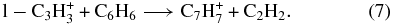
Thus, the 25% of C3H+3 ions that are non-reactive are cyclic in nature, and both cyclic and linear ions are present in the flow tube.
The relative populations of the C3H+5 isomers have been determined by previous collision-induced dissociation studies. Bowers et al. (1980), using the same experimental conditions as our present study (70 eV electron ionization of propene), measured the formation of a mixture of 65:35 H2CCHCH+2:H3CCCH+2. Thus, both of these isomers are expected to be present in our experiments.
2.1. Reactions of C3H+3 and C3H+5 with H2
The C3H+3 and C3H+5 ions containing assorted isotopomers were separately mass-selected and injected into the reaction flow tube, and H2 was added at a fixed inlet. The concentration of H2 was varied during the experiment in order to determine the reactivity of the assorted isomers of C3H+3 and C3H+5with molecular hydrogen to form associated products; i.e.,
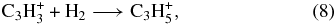
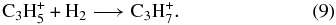
Confirming the results of McEwan et al. (1999), no reaction of H2 was observed in our experiments; i.e., the reaction rate coefficients are below our detection limit (k8 < 1 × 10−13 cm3 s−1, k9 < 1 × 10−13 cm3 s−1). The upper limits here refer to either the ternary regime or the saturated binary regime of association reactions. In the radiative regime, which pertains at low interstellar densities, the radiative association rate coefficients are likely to be lower still. We can estimate these rate coefficients in the following manner (Herbst et al. 2010). If the measured upper limits pertain to the ternary regime, and the rate of collisional stabilization of the unstable collision complex is assumed to be 10−10 cm3 s−1, the upper limit to the radiative rate coefficient at room temperature is ≈10−17 cm3 s−1, assuming a radiative stabilization rate of 102 s−1. With an inverse temperature dependence for the rate coefficient of (T/300 K)−r/2, where r is the number of classical rotational degrees of freedom for the reactants, we get that at 10 K, the upper limit for the radiative association mechanism for reactions (8) and (9) is approximately 10−14 cm3 s−1 in the absence of potential barriers. These small values may still lead to the production of some propylene. It is thus necessary to use theory to determine if they are accurate or if potential barriers exist to make the rate coefficients much smaller at low temperatures. That potential barriers do exist will be discussed in the next section for the two isotopomers (H2CCCH+ and H3CCCH+2) that were once thought to lead, via reactions (8) and (9), to the isopropyl radical ion form of protonated propene.
2.2. New Ab Initio Calculations
Potential energy surfaces for reactions (8) and (9) have been optimized at the CCSD/6-311++G(d, p) level with the propargyl ion and with the metastable isomer for C3H+5. Activation energy barriers were found for both association reactions in the entrance channel to form the unstabilized complex. They have been recalculated at the CCSD(T)/6-311++G (3df, 2pd) level. For reaction (8), the calculated barrier is 25.5 kJ mol−1 (3100 K) and for reaction (9) the barrier is 24.7 kJ mol−1 (3000 K). These high barriers close any remaining chance that protonated propene could be formed from H3CCCH+2 + H2. This ion cannot be produced from the propargyl ion without a barrier and it does not react with H2 without a barrier.
The new ab initio results differ strongly from those of our previous calculations (Herbst et al. 2010) in which we found no transition states, or potential maxima leading to experimental activation energy barriers, in these reactions. Failing to locate transition states involving complex molecules does not always guarantee their absence. Indeed, searching for transition states, which correspond to saddle points on a potential energy surface, is always a challenge. Transition state searches rely on maximizing the energy of the system along one mode while minimizing energy in all others. A successful search should start off in a region where the reaction coordinate already has a negative curvature, which is always a difficult guess for complex molecules with many degrees of freedom.
The main difference between our previous calculations (Herbst et al. 2010) and the present one lies in the version of Gaussian8 used to investigate the potential energy surfaces leading from reactants to products. In the present calculations, we used the latest version of the Gaussian computational chemistry code (Gaussian 09), which differs from the previous one (Gaussian 03) in the default algorithms for longer jobs, implying a number of basis functions as large as 100 and higher (Frisch et al. 2003, 2009). The resulting algorithms for longer jobs such as ours facilitated our search for transition states.
3. CHEMICAL SIMULATIONS
Given the failure of the radiative association synthesis of propene, it is unlikely that enough propene can be formed to explain its observation in TMC-1. Nevertheless, we carried out two different chemical simulations. In one, we used the Ohio State University (OSU) gas-phase network (version 01.2009) with additional reactions to support the formation and depletion of propene and related species. In the second, we used a network of Evelyne Roueff constructed in a similar manner, except that additional larger carbon chains are included and formed mainly by radiative association with H2 at an assumed rate coefficient of 10−13 cm3 s−1. The reaction lists for each calculation can be obtained from the authors. The parameters in the time-dependent augmented OSU simulation and the steady-state simulation of Roueff are the same. The temperature and density are T = 10 K and nH = 2 × 104 cm3 s−1, respectively. The cosmic ray ionization rate is  s−1, while low-metal (oxygen-rich) elemental abundances are utilized (Graedel et al. 1982). In addition to the use of low-metal elemental abundances, we lowered the elemental oxygen abundance to achieve a C/O abundance ratio of 1.2 and utilized these so-called carbon-rich abundances with both networks in further simulations. For the augmented OSU calculation, the initial abundances are atomic in nature except for the case of molecular hydrogen, which is completely molecular.
s−1, while low-metal (oxygen-rich) elemental abundances are utilized (Graedel et al. 1982). In addition to the use of low-metal elemental abundances, we lowered the elemental oxygen abundance to achieve a C/O abundance ratio of 1.2 and utilized these so-called carbon-rich abundances with both networks in further simulations. For the augmented OSU calculation, the initial abundances are atomic in nature except for the case of molecular hydrogen, which is completely molecular.
In the expanded OSU-based network, the synthetic routes that dominate propene production at its peak abundance are (Anicich 2003; Canosa et al. 1997; Roueff & Herbst 2009)
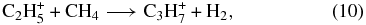
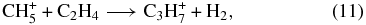
where the production of protonated propene (likely to be the lower energy isopropyl radical ion) is followed by dissociative recombination (Florescu-Mitchell & Mitchell 2006). Neither ion-neutral reaction is well-studied experimentally; reaction (10) has been studied but with a variety of differing results. Moreover, it may be slightly endothermic. Reaction (11) does not appear to produce protonated propylene as a major product. Removal of both of these reactions leads to a one to two order of magnitude drop in the peak abundance of propene.
In the steady-state calculation of Roueff, the main production routes to propene depend upon whether C-rich or O-rich conditions are assumed. In the O-rich case, propene is produced mainly by dissociative recombination of the ion C3H+8 and by the reaction between atomic hydrogen and the radical C3H7:
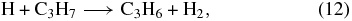
while in the C-rich case, formation occurs via the dissociative recombination of C4H+10 and C4H+11.
In both networks, propene is destroyed in the O-rich case by rapid reaction with atomic oxygen (Sabbah et al. 2007):
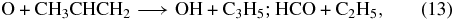
where the products shown here are merely suggestive. In the C-rich case, the same reaction is the principal route of destruction in the augmented OSU calculation, while reactions with the radical CN and atom C are the principal destruction routes in the Roueff calculation. This latter difference is doubtless caused by the different times for the two calculations reported here, viz., early-time for the OSU calculation and steady-state for the Roueff calculation.
3.1. Fractional Abundance of Propene
The calculated fractional abundance of propene with respect to nH as a function of time with the expanded OSU network is shown in Figures 2 and 3, along with fractional abundances of related ions. Figure 2 is obtained with the normal oxygen-rich low-metal abundances, whereas Figure 3 is obtained with carbon-rich abundances, which are often used for TMC-1 (Wakelam et al. 2006). As can be seen in the figures, the calculated peak ("early-time") abundance of propene is not greater than 10−11, which is two orders of magnitude below the observed value. This value is achieved only with C-rich abundances; the peak value with O-rich abundances is lower by two orders of magnitude. The steady-state fractional abundances of propene using the Roueff network are in reasonable agreement with the augmented OSU results at steady state for both O-rich and C-rich conditions. In particular, the calculated abundance for O-rich conditions is 2 × 10−18, and that for C-rich conditions is 1 × 10−12 with the Roueff network, while these numbers are 3 × 10−16 and 8 × 10−12 for the augmented OSU network. Thus, neither model obtains results in which the propene abundance is anywhere near as large as observed.

Figure 2. The fractional abundances with respect to nH of CH3CHCH2 (propene) and related ions are plotted vs. time using low-metal oxygen-rich elemental abundances and the expanded OSU network.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
Figure 3. The fractional abundances with respect to nH of CH3CHCH2 (propene) and related ions are plotted vs. time using low-metal carbon-rich elemental abundances and the expanded OSU network.
Download figure:
Standard image High-resolution image{kind=link}
4. DISCUSSION
We have shown by a combination of experiments and quantum chemical calculations that the abundance of the partially saturated hydrocarbon propene (CH3CHCH2), previously observed in the cold core TMC-1 to be ≈2 × 10−9 with respect to nH, cannot be accounted for by a gas-phase synthesis based on radiative association reactions leading to the precursor ion C3H+7 because the association reactions needed to have large activation energy barriers. Our failure does not prove, however, that there are no alternative efficient gas-phase syntheses. To try to answer this question, we have run two model calculations which are expanded to consider other methods of formation of propene in addition to its destruction. Discussed earlier in the text, these models are based on different networks, viz., an augmented OSU network and the network of Roueff. In these approaches, dissociative recombination reactions dominate the formation of propene. The two calculations yield rather similar and low abundances for propene. We thus find that we cannot produce sufficient propene to explain its abundance in TMC-1, even if C-rich elemental abundances are utilized. Specifically, with the augmented OSU simulation, we showed that the calculated abundance of propene as a function of time does not exceed 10−13 with O-rich elemental abundances and 10−11 with C-rich abundances, both of which fall dramatically below the observed abundance of propene in TMC-1. With the network of Roueff, we find a similar result to the augmented OSU result at steady state, despite the fact that this network has significantly more radiative association reactions than the augmented OSU network. Note, however, that the rate coefficients of many of these, e.g.,
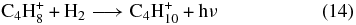
and
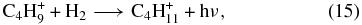
are estimated. Some of these reactions do not possess measured ternary association equivalents in the experiment of McEwan et al. (1999) suggesting that perhaps barriers exist as in the reactions studied here in some detail, suggesting that the reactions are much slower than estimated. We conclude that it is likely that propene cannot be formed via gas-phase reactions with the parameters normally used to reproduce the observed abundances of most other species detected in cold dense cores. This conclusion, however, must remain tentative because we cannot prove that we have exhausted the possible routes of formation of propene in the gas.
Recent observational work on the detection of methanol in the cold core TMC-1 confirms that this species does not peak at the well-known CP peak of unsaturated organic molecules (S. Yamamoto 2012, private communication). This difference suggests a different formation mechanism, such as one that occurs on dust particles, a conclusion that fits well with gas-grain models in which methanol is formed by surface hydrogenation of CO.
It is also likely that grain surface reactions followed by non-thermal desorption play at least a partial role in the synthesis of gas-phase propene in cold clouds. One possibility is that successive surface association reactions with atomic hydrogen starting from C3H2 or even C3 can lead to propene. Knowledge of the activation barrier for the initial reactions in the series—H + C3, H + C3H, and H + C3H2—is lacking, although the reaction with the radical C3H is likely to be barrierless. We do know the barrier for H + C3H4, which is around 1000 K from gas-phase measurements, a value that is not excessively large for surface processes (Herbst & Millar 2008). We also expect the reactions between H and the radicals C3H3 and C3H5 to be barrierless. Measurements of the reaction between H and propene show a significant barrier in the gas phase for the production of the "normal" C3H7 (CH3CH2CH2) radical, but less of a barrier for iso-C3H7 (CH3CHCH3), so it is unclear whether propene is destroyed rapidly on grains via reaction with atomic hydrogen.
Even if propene cannot be produced efficiently on grains, there is the possibility that ethane can be produced in this manner, which might boost its abundance sufficiently in the gas phase so that it could be a precursor of propene via reactions such as
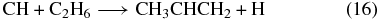
(Canosa et al. 1997; Roueff & Herbst 2009). The production of ethane via successive association reactions with atomic hydrogen on surfaces starting from acetylene (C2H2) likely proceeds efficiently. From gas-phase measurements, barriers under 1000 K exist for H + C2H2 and H + C2H4, while the reactions between H and the radicals C2H3 and C2H5 are expected to be barrierless. Indeed, new measurements on cold water ice appear to confirm the existence of this mechanism (Kobayashi et al. 2012).
We thank J. Cernicharo for suggesting this project. E.H. acknowledges the support of the National Science Foundation for his astrochemistry program, and the support of NASA for studies in the evolution of pre-planetary matter. Support of the experimental research of the Boulder group by NASA and NSF (CHE-1012321) is gratefully acknowledged. E.R. acknowledges the support of the Observatoire de Paris and the Programme National du CNRS-INSU "PCMI." D.T. also acknowledges the support of the CNRS-INSU "PCMI." The ab initio calculations were done using HPC resources from GENCI-[CCRT/CINES/DRIS] (Grant 2011-[x20011085116]).