



Abstract
Twisted two-dimensional materials have recently attracted tremendous interest owing to their unique structures and fantastic electronic properties. However, the effect of interlayer twisting on the phonon transport properties is less known, especially for the twist-angle-dependent lattice thermal conductivity ( ). Using the emerging Janus SnSSe bilayer as a prototypical example, we develop an accurate machine learning potential, which is adopted to efficiently predict the
). Using the emerging Janus SnSSe bilayer as a prototypical example, we develop an accurate machine learning potential, which is adopted to efficiently predict the  at a series of twist angles via iterative solution of the Boltzmann transport equation. It is found that the
at a series of twist angles via iterative solution of the Boltzmann transport equation. It is found that the  exhibits a distinct non-monotonous dependence on the twist angle, which can be traced back to the bonding heterogeneity between high-symmetry stacking regions inside the moiré unit cell. In contrast to the general belief, the optical phonons make a major contribution toward the
exhibits a distinct non-monotonous dependence on the twist angle, which can be traced back to the bonding heterogeneity between high-symmetry stacking regions inside the moiré unit cell. In contrast to the general belief, the optical phonons make a major contribution toward the  of the twisted structures. Moreover, we demonstrate that four-phonon scattering can significantly reduce the
of the twisted structures. Moreover, we demonstrate that four-phonon scattering can significantly reduce the  of SnSSe bilayer at higher temperatures, which becomes more pronounced by interlayer twisting. Our work not only highlights the strong predictive power of machine learning potential, but also offers new insights into the design of thermal smart materials with tunable
of SnSSe bilayer at higher temperatures, which becomes more pronounced by interlayer twisting. Our work not only highlights the strong predictive power of machine learning potential, but also offers new insights into the design of thermal smart materials with tunable  .
.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Since the breakthrough discovery of unconventional superconductivity in twisted bilayer graphene [1], interlayer twisting has been widely used to tune the electronic properties of two-dimensional (2D) materials, such as the ferromagnetic behavior, the Mott insulator state, the electronic correlations, and so on [2–7]. However, compared with significant advances in twistronics, the effect of interlayer twisting on the lattice thermal conductivity ( ) is less known. The possible reason is the difficulty in the evaluation of
) is less known. The possible reason is the difficulty in the evaluation of  for twisted systems, where a large number of atoms are usually involved in the moiré unit cell. Although the classic molecular dynamics (MD) simulations can give a rapid prediction on the
for twisted systems, where a large number of atoms are usually involved in the moiré unit cell. Although the classic molecular dynamics (MD) simulations can give a rapid prediction on the  of large systems, the using of empirical potentials may lead to insufficient accuracy [8, 9], which inevitably hinders their widespread applications. In fact, recent MD studies on twisted systems are severely limited to bilayer/multilayer graphene and MoS2 [10–16], leaving a much larger 2D materials space unexplored. As an alternative, the solution of phonon Boltzmann transport equation (BTE) can accurately predict
of large systems, the using of empirical potentials may lead to insufficient accuracy [8, 9], which inevitably hinders their widespread applications. In fact, recent MD studies on twisted systems are severely limited to bilayer/multilayer graphene and MoS2 [10–16], leaving a much larger 2D materials space unexplored. As an alternative, the solution of phonon Boltzmann transport equation (BTE) can accurately predict  [17, 18]. Such an approach generally requires density function theory (DFT) calculations to obtain interatomic force constants (IFCs), which are however very time-consuming for twisted systems owing to the large size and low symmetry of moiré unit cells.
[17, 18]. Such an approach generally requires density function theory (DFT) calculations to obtain interatomic force constants (IFCs), which are however very time-consuming for twisted systems owing to the large size and low symmetry of moiré unit cells.
As an important data-driven technique, machine learning (ML) has been successfully employed to accelerate the discovery of new materials with desired properties. In particular, ML can be utilized to establish accurate interatomic potentials, which are then implemented into MD simulations or phonon BTE to efficiently determine the  of complex structures [19–28]. For example, Qian et al developed a Gaussian approximation potential (GAP) for amorphous silicon, which outperforms empirical potential in the evaluation of atomic forces, and the predicted
of complex structures [19–28]. For example, Qian et al developed a Gaussian approximation potential (GAP) for amorphous silicon, which outperforms empirical potential in the evaluation of atomic forces, and the predicted  agrees well with that measured experimentally [20]. Besides, Mangold et al proposed a neural network potential (NNP) trained on a small set of crystalline configurations, and demonstrated its good transferability in evaluating the
agrees well with that measured experimentally [20]. Besides, Mangold et al proposed a neural network potential (NNP) trained on a small set of crystalline configurations, and demonstrated its good transferability in evaluating the  of Mnx
Gey
compounds over a broad range of chemical compositions [22]. In the case of twisted systems, Sun et al established a moment tensor potential (MTP) for the layered Bi2O2Se with twist angle of 36.87°, and found that the out-of-plane
of Mnx
Gey
compounds over a broad range of chemical compositions [22]. In the case of twisted systems, Sun et al established a moment tensor potential (MTP) for the layered Bi2O2Se with twist angle of 36.87°, and found that the out-of-plane  is reduced by 83% at 300 K as compared with that of untwisted structure [29]. By adopting the anharmonic IFCs extracted from a force constants potential (FCP), Chen et al investigated the effect of twisting on the phonon transport properties of bilayer penta-graphene [30]. It should be noted that, although the
is reduced by 83% at 300 K as compared with that of untwisted structure [29]. By adopting the anharmonic IFCs extracted from a force constants potential (FCP), Chen et al investigated the effect of twisting on the phonon transport properties of bilayer penta-graphene [30]. It should be noted that, although the  of several twisted systems have been predicted in the above-mentioned works, they are however limited to a specific twist angle. To date, it remains a very challenging task to evaluate the
of several twisted systems have been predicted in the above-mentioned works, they are however limited to a specific twist angle. To date, it remains a very challenging task to evaluate the  of systems at arbitrary twist angle, both rapidly and accurately.
of systems at arbitrary twist angle, both rapidly and accurately.
In recent years, 2D Janus materials have attracted increasing attention from the science community due to their intriguing electronic and optical properties [31, 32]. However, the phonon transport properties of Janus systems are less known, especially for those with twisted structures. In this work, we propose an ML-based MTP for the emerging Janus SnSSe bilayer, which allows ready prediction on the  at arbitrary twist angle. The high accuracy and good transferability of the established MTP are demonstrated by checking the twisted systems both inside and beyond the training set. Interestingly, there is a non-monotonous dependence of
at arbitrary twist angle. The high accuracy and good transferability of the established MTP are demonstrated by checking the twisted systems both inside and beyond the training set. Interestingly, there is a non-monotonous dependence of  on the twist angle, where two local minimum values can be observed. The underlying physical mechanisms are explored in terms of the structural and anharmonic properties of moiré unit cells. Moreover, we highlight the vital importance of four-phonon scattering in accurately predicting the
on the twist angle, where two local minimum values can be observed. The underlying physical mechanisms are explored in terms of the structural and anharmonic properties of moiré unit cells. Moreover, we highlight the vital importance of four-phonon scattering in accurately predicting the  of twisted Janus SnSSe bilayer.
of twisted Janus SnSSe bilayer.
2. Methodology
The optimized structure of Janus SnSSe bilayer can be obtained by performing DFT calculations, as implemented in the Vienna ab-initio simulation package (VASP) [33]. The exchange-correlation energy is described by the Perdew–Burke–Ernzerhof (PBE) functional under the generalized gradient approximation (GGA) [34]. The energy cutoff is set as 500 eV and the van der Waals (vdW) interactions are treated via the DFT-D3 exchange functional [35]. To eliminate interactions between the bilayer and its periodic images, a vacuum distance of 30 Å is adopted along the normal direction of the unit cell. The Brillouin zones are sampled by using Monkhorst–Pack
k
-meshes of  for the bilayers with twist angles of 0° and 60°, and
for the bilayers with twist angles of 0° and 60°, and  for other configurations. The systems are fully relaxed until the magnitude of the force acting on each atom becomes less than 10−3 eV Å−1, which is accurate enough to obtain optimized crystal structures.
for other configurations. The systems are fully relaxed until the magnitude of the force acting on each atom becomes less than 10−3 eV Å−1, which is accurate enough to obtain optimized crystal structures.
As a class of machine learning interatomic potentials, MTP simultaneously exhibits good computational efficiency and first-principles accuracy, and has been successfully exploited for materials design in recent years [36, 37]. The basic information about the MTP is provided in the supplementary material. To establish a reliable dataset for training the MTP of Janus SnSSe bilayer, we perform ab-initio molecular dynamics (AIMD) simulations at 300 K. For the bilayers with twist angles of 0°, 21.79°, 32.20°, and 60°, we respectively adopt  ,
,  ,
,  , and
, and  supercells. The canonical ensemble is selected to run 2000 steps with a time step of 1 fs. The initial dataset contains 800 atomic configurations sampled from the AIMD trajectories every ten steps, which is randomly divided into the training (80%) and validation (20%) sets. With the extracted energies, forces, and stresses from the AIMD simulations, the MTP is trained by solving the minimization problem, as implemented in the so-called MLIP package [38]. In the training process, the weights of energies, forces, and stresses are set as 1, 0.1, and 0.001, respectively. To achieve balance between the prediction accuracy and computational efficiency of MTP, the maximum level of moments (
supercells. The canonical ensemble is selected to run 2000 steps with a time step of 1 fs. The initial dataset contains 800 atomic configurations sampled from the AIMD trajectories every ten steps, which is randomly divided into the training (80%) and validation (20%) sets. With the extracted energies, forces, and stresses from the AIMD simulations, the MTP is trained by solving the minimization problem, as implemented in the so-called MLIP package [38]. In the training process, the weights of energies, forces, and stresses are set as 1, 0.1, and 0.001, respectively. To achieve balance between the prediction accuracy and computational efficiency of MTP, the maximum level of moments ( ), the number of radial basis functions (
), the number of radial basis functions ( ), and the cutoff distance (
), and the cutoff distance ( ) are respectively set to be 20, 8, and 13 Å (see figure S1 of the supplementary material for details).
) are respectively set to be 20, 8, and 13 Å (see figure S1 of the supplementary material for details).
The developed MTP can be leveraged to quickly predict the energies of supercell configurations generated by the finite displacement method, which enable us to readily obtain the second-, third-, and fourth-order IFCs [39, 40]. The codes are respectively embedded in the Phonopy [41], thirdorder.py [18], and fourthorder.py [42] scripts. For the second- and third-order IFCs, the sizes of supercells (cutoff interactions) are set to be  (the 8th nearest neighbors) for the bilayers with twist angles of 0° and 60°,
(the 8th nearest neighbors) for the bilayers with twist angles of 0° and 60°,  (the 4th nearest neighbors) for those with twist angles of 21.79°, 32.20°, and 13.17°, and
(the 4th nearest neighbors) for those with twist angles of 21.79°, 32.20°, and 13.17°, and  (the 3rd nearest neighbors) for other configurations. In the case of the fourth-order IFCs, the sizes of supercells are the same as those of the second- and third-order IFCs, while the cutoff interactions are set to be the 3rd nearest neighbors. The
(the 3rd nearest neighbors) for other configurations. In the case of the fourth-order IFCs, the sizes of supercells are the same as those of the second- and third-order IFCs, while the cutoff interactions are set to be the 3rd nearest neighbors. The  can be obtained by solving the phonon BTE with these IFCs as inputs, which is embedded in the ShengBTE package [18]. By default, the isotope scattering is explicitly considered in the calculations. Moreover, the
q
-mesh is carefully tested to ensure the convergence of
can be obtained by solving the phonon BTE with these IFCs as inputs, which is embedded in the ShengBTE package [18]. By default, the isotope scattering is explicitly considered in the calculations. Moreover, the
q
-mesh is carefully tested to ensure the convergence of  , as shown in figure S2 of the supplementary material.
, as shown in figure S2 of the supplementary material.
3. Results and discussion
Due to the inversion-asymmetric structure of Janus SnSSe monolayer, the bilayer systems exhibit three chalcogen orderings, namely, the SSnSe–SSnSe, the SSnSe–SeSnS, and the SeSnS–SSnSe. For each of them, there exist many high-symmetry stacking configurations, such as the AA, AB, and AC. Among them, the AA-stacking of SSnSe–SeSnS (space group of  ) was demonstrated to be the most stable one [43–45], as illustrated in figure 1(a). The optimized lattice constant and interlayer distance are respectively 3.762 Å and 3.140 Å, which agree well with those reported previously [43–45]. For comparison, we also show the AB and AC stacking configurations of the SSnSe–SeSnS in figure S3 of the supplementary material. As mentioned above, to establish a reliable dataset for training the MTP, we perform AIMD simulations which suggests that the unit cell cannot be too large. We thus choose four typical bilayers with twist angles of 0°, 21.79°, 32.20°, and 60°, where the unit cell respectively contains 6, 42, 78, and 6 atoms, as shown in figure 1(b). Similar to that of the untwisted structure, the system with twist angle of 60° exhibits a high-symmetry stacking configuration (space group of
) was demonstrated to be the most stable one [43–45], as illustrated in figure 1(a). The optimized lattice constant and interlayer distance are respectively 3.762 Å and 3.140 Å, which agree well with those reported previously [43–45]. For comparison, we also show the AB and AC stacking configurations of the SSnSe–SeSnS in figure S3 of the supplementary material. As mentioned above, to establish a reliable dataset for training the MTP, we perform AIMD simulations which suggests that the unit cell cannot be too large. We thus choose four typical bilayers with twist angles of 0°, 21.79°, 32.20°, and 60°, where the unit cell respectively contains 6, 42, 78, and 6 atoms, as shown in figure 1(b). Similar to that of the untwisted structure, the system with twist angle of 60° exhibits a high-symmetry stacking configuration (space group of  ). In contrast, the bilayers with twist angles of 21.79° and 32.20° have lower symmetry (space group of
). In contrast, the bilayers with twist angles of 21.79° and 32.20° have lower symmetry (space group of  ). Moreover, the interlayer distances are 3.418 Å, 3.421 Å, and 3.832 Å for the systems with twist angles of 21.79°, 32.20°, and 60°, respectively. These values are obviously higher than that of the untwisted bilayer (3.140 Å), suggesting that the interlayer twisting can be also used to manipulate the vdW interactions. On the other hand, we calculate two independent elastic constants
). Moreover, the interlayer distances are 3.418 Å, 3.421 Å, and 3.832 Å for the systems with twist angles of 21.79°, 32.20°, and 60°, respectively. These values are obviously higher than that of the untwisted bilayer (3.140 Å), suggesting that the interlayer twisting can be also used to manipulate the vdW interactions. On the other hand, we calculate two independent elastic constants  and
and  of the bilayer systems by using the energy-strain method [46]. As listed in table S1 of the supplementary material, the elastic constants satisfy the Born–Huang criteria (
of the bilayer systems by using the energy-strain method [46]. As listed in table S1 of the supplementary material, the elastic constants satisfy the Born–Huang criteria ( and
and  ), which suggests that all the twisted systems are mechanically stable.
), which suggests that all the twisted systems are mechanically stable.

Figure 1. (a) Side-view of the Janus SnSSe bilayer with AA-stacking. (b) Top-views of the SnSSe bilayers with twist angles of 0°, 21.79°, 32.20°, and 60°, which are used to construct the dataset for training the MTP. The corresponding twist angle ( ) and primitive cell (dashed lines) are indicated.
) and primitive cell (dashed lines) are indicated.
Download figure:
Standard image High-resolution imageAs mentioned above, the AIMD simulations are performed to construct the initial dataset that is randomly divided into the training and validation sets. By using the moment tensor to describe the local atomic environment, we develop an accurate MTP to determine the potential energy surface of twisted Janus SnSSe bilayer. Figures 2(a) and (b) show the quantitative comparisons of the MTP-predicted and DFT-calculated energies for the training and validation sets, respectively. It can be found that all the data points are located around the solid line representing equality, suggesting high prediction accuracy of our MTP. Specifically, for both the training and validation sets, high Pearson correlation coefficient (PCC) of 99% can be found between the MTP-predicted and DFT-calculated energies. Moreover, the root mean squared error (RMSE) is as small as 0.78 and 0.75 meV/atom for the training and validation sets, respectively. Similar picture can be found for the atomic forces indicated in figures 2(c) and (d). Collectively, all these findings demonstrate that the proposed MTP can accurately reproduce the energies and atomic forces obtained from first-principles calculations.
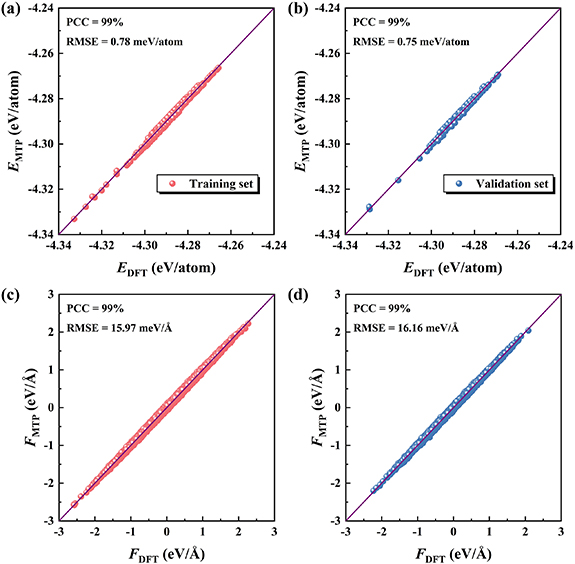
Figure 2. The intuitive linear correlations between the MTP-predicted and DFT-calculated energies ( ) as well as atomic forces (
) as well as atomic forces ( ) for the training and validation sets, where the PCC and RMSE are explicitly indicated.
) for the training and validation sets, where the PCC and RMSE are explicitly indicated.
Download figure:
Standard image High-resolution imageThe reliability of our MTP can be also confirmed by checking the phonon dispersion relations of the twisted bilayers inside the training set. As shown in figures 3(a)−(c), the MTP-predicted phonon dispersions agree well with those obtained from DFT calculations, and the absence of imaginary frequency indicates the dynamical stability of these twisted bilayers. Beyond the initial dataset, we consider a Janus bilayer with twist angle of 13.17° and find that the MTP can reproduce the phonon dispersions calculated by DFT, as indicated in figure 3(d). To have a further check, our MTP is adopted to predict the lattice constants and interlayer distances of the Janus SnSSe bilayers with twist angles of 9.43°, 13.17°, 38.21°, 42.10°, and 46.83°. As listed in table 1, the MTP-predicted results agree well with those obtained from DFT calculations. Collectively, all these observations substantiate the strong predictive power and good transferability of our developed MTP for the twisted Janus SnSSe bilayers.

Figure 3. The phonon dispersions of the Janus SnSSe bilayers with twist angles of (a) 21.79°, (b) 32.20°, (c) 60°, and (d) 13.17°. The results obtained from DFT calculation (solid lines) and MTP prediction (dashed lines) are both shown for comparison.
Download figure:
Standard image High-resolution imageTable 1. The lattice constants ( ) and interlayer distances (
) and interlayer distances ( ) calculated by using DFT and MTP for some twisted Janus SnSSe bilayers beyond the training set.
) calculated by using DFT and MTP for some twisted Janus SnSSe bilayers beyond the training set.
| MTP | DFT | |||
|---|---|---|---|---|
 (Å) (Å) |
 (Å) (Å) |
 (Å) (Å) |
 (Å) (Å) | |
| 9.43° | 22.859 | 3.255 | 22.862 | 3.248 |
| 13.17° | 16.379 | 3.338 | 16.380 | 3.335 |
| 38.21° | 17.219 | 3.400 | 17.222 | 3.404 |
| 42.10° | 20.922 | 3.383 | 20.916 | 3.384 |
| 46.83° | 28.370 | 3.332 | 28.371 | 3.326 |
By implementing the established MTP into the phonon BTE, we can readily predict the  of Janus SnSSe bilayer at arbitrary twist angle. As an example, figure 4(a) shows the room temperature
of Janus SnSSe bilayer at arbitrary twist angle. As an example, figure 4(a) shows the room temperature  at twist angles of 0°, 9.43°, 13.17°, 21.79°, 32.20°, 38.21°, 42.10°, 46.83°, and 60°, where the number of atoms is less than 400 in the moiré unit cell (see figure 4(b)) so that the
at twist angles of 0°, 9.43°, 13.17°, 21.79°, 32.20°, 38.21°, 42.10°, 46.83°, and 60°, where the number of atoms is less than 400 in the moiré unit cell (see figure 4(b)) so that the  can be efficiently predicted by solving phonon BTE. Note that all the
can be efficiently predicted by solving phonon BTE. Note that all the  are calculated with respective to the effective thickness, which is defined as summation of the bucking height of the bilayer and the vdW radii of two outermost S atoms (see table S1 of the supplementary material). It is interesting to find that the
are calculated with respective to the effective thickness, which is defined as summation of the bucking height of the bilayer and the vdW radii of two outermost S atoms (see table S1 of the supplementary material). It is interesting to find that the  exhibits a strong dependence on the twist angle in the ranges from 0° to 21.79° and from 38.21° to 60°, where two local minimum values can be observed at twist angles of 9.43° and 46.83°. In contrast, there is a weak dependence of
exhibits a strong dependence on the twist angle in the ranges from 0° to 21.79° and from 38.21° to 60°, where two local minimum values can be observed at twist angles of 9.43° and 46.83°. In contrast, there is a weak dependence of  on the twist angle in the range from 21.79° to 38.21°. Similar picture can be found at other temperatures, as plotted in figure S4 of the supplementary material. It should be mentioned that the temperature-dependent
on the twist angle in the range from 21.79° to 38.21°. Similar picture can be found at other temperatures, as plotted in figure S4 of the supplementary material. It should be mentioned that the temperature-dependent  of untwisted bilayer agrees well with previously reported results by DFT calculations [45], which once again confirms the reliability of our developed MTP. In general, the
of untwisted bilayer agrees well with previously reported results by DFT calculations [45], which once again confirms the reliability of our developed MTP. In general, the  of a given system is mainly determined by acoustic phonons. However, we see from table 2 that the optical branches contribute nearly 50% to the
of a given system is mainly determined by acoustic phonons. However, we see from table 2 that the optical branches contribute nearly 50% to the  of the Janus SnSSe bilayers with twist angles of 0° and 60°. For the other configurations (moiré unit cells) with lower
of the Janus SnSSe bilayers with twist angles of 0° and 60°. For the other configurations (moiré unit cells) with lower  , the contributions from optical branches become even higher. In particular, at the twist angle of 9.43°, the optical phonon contribution can reach as high as 82.12%. The possible reason is that the moiré unit cells exhibit reduced Brillouin zone, which leads to the folding of phonon branches and thus strong hybridization between the optical and acoustic branches. It should be mentioned that such an effect could also reduce both the phonon group velocity and relaxation time (figure S5 of the supplementary material), so that the twisted bilayers exhibit lower
, the contributions from optical branches become even higher. In particular, at the twist angle of 9.43°, the optical phonon contribution can reach as high as 82.12%. The possible reason is that the moiré unit cells exhibit reduced Brillouin zone, which leads to the folding of phonon branches and thus strong hybridization between the optical and acoustic branches. It should be mentioned that such an effect could also reduce both the phonon group velocity and relaxation time (figure S5 of the supplementary material), so that the twisted bilayers exhibit lower  compared with the untwisted systems. In figure S6 of the supplementary material, we plot the normalized cumulative
compared with the untwisted systems. In figure S6 of the supplementary material, we plot the normalized cumulative  as a function of mean free path (MFP). It is found that the moiré unit cells exhibit significantly lower cutoff MFPs as compared with that of untwisted structure, which is also consistent with their lower
as a function of mean free path (MFP). It is found that the moiré unit cells exhibit significantly lower cutoff MFPs as compared with that of untwisted structure, which is also consistent with their lower  .
.
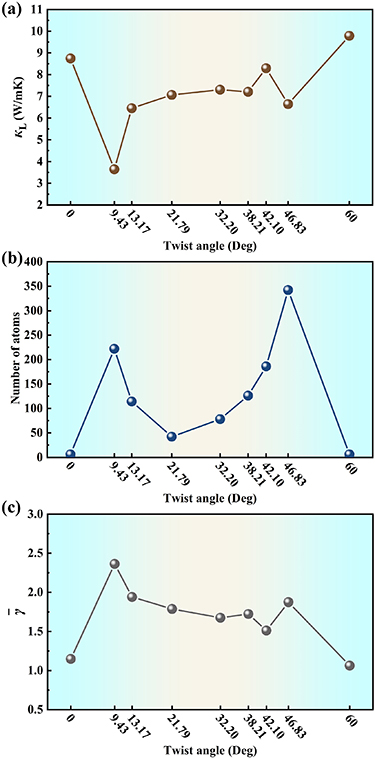
Figure 4. (a) The room temperature lattice thermal conductivity, (b) number of atoms in the moiré unit cell, and (c) the average Grüneisen parameter of the Janus SnSSe bilayer, plotted as a function of twist angle.
Download figure:
Standard image High-resolution imageTable 2. The contributions of ZA, TA, LA and optical branches to the room temperature lattice thermal conductivities of the Janus SnSSe bilayers with different twist angles.
| ZA (%) | TA (%) | LA (%) | OP (%) | |
|---|---|---|---|---|
| 0° | 8.01 | 26.86 | 20.16 | 44.97 |
| 9.43° | 0.05 | 4.80 | 13.03 | 82.12 |
| 13.17° | 5.12 | 9.68 | 17.61 | 67.59 |
| 21.79° | 8.93 | 7.33 | 11.20 | 72.54 |
| 32.20° | 13.21 | 10.29 | 7.53 | 68.97 |
| 38.21° | 7.25 | 8.58 | 8.05 | 76.12 |
| 42.10° | 1.51 | 10.28 | 11.41 | 76.80 |
| 46.83° | 4.56 | 7.07 | 8.29 | 80.08 |
| 60° | 26.77 | 9.70 | 14.27 | 49.26 |
To have a better understanding of the twist-angle-dependent  , we first calculate the average Grüneisen parameter (
, we first calculate the average Grüneisen parameter ( ) of all the phonon modes, as shown in figure 4(c). It is obvious that the variation of
) of all the phonon modes, as shown in figure 4(c). It is obvious that the variation of  with the twist angle shows an opposite trend to that of
with the twist angle shows an opposite trend to that of  , which is consistent with the general belief that stronger anharmonicity leads to lower
, which is consistent with the general belief that stronger anharmonicity leads to lower  . In principle, the bonding characteristic plays an important role in governing the phonon transport, which can be identified by checking the corresponding electron localization functions (ELFs). As an example, figure 5 plots the ELF diagram for the bilayer system with twist angle of 9.43°. It is clear that electrons are localized between the Sn and S (Se) atoms for both the top and bottom layers, suggesting the in-plane covalent bonding of the twisted bilayer. As known, the moiré unit cell can be characterized by the orderly distributed high-symmetry stacking regions [47], which are marked as Ⅰ, Ⅱ, and Ⅲ in figure 5 for our case. Here, the regions Ⅰ, Ⅱ, and Ⅲ are defined as the stackings of the bilayer with the Sn, Se, and S atoms in the top layer directly located above the Sn, Se, and S atoms in the bottom layer, respectively. Similar picture can be found for the other moiré unit cells, as shown in figure S7 of the supplementary material. To compare the bonding strengths between these high-symmetry stacking regions, we calculate the ELF values at the midpoint of all the involved covalent bonds, as listed in table S2 of the supplementary material. It is found that the moiré unit cells exhibit strong bonding heterogeneity, which can be quantified by the standard deviation (
. In principle, the bonding characteristic plays an important role in governing the phonon transport, which can be identified by checking the corresponding electron localization functions (ELFs). As an example, figure 5 plots the ELF diagram for the bilayer system with twist angle of 9.43°. It is clear that electrons are localized between the Sn and S (Se) atoms for both the top and bottom layers, suggesting the in-plane covalent bonding of the twisted bilayer. As known, the moiré unit cell can be characterized by the orderly distributed high-symmetry stacking regions [47], which are marked as Ⅰ, Ⅱ, and Ⅲ in figure 5 for our case. Here, the regions Ⅰ, Ⅱ, and Ⅲ are defined as the stackings of the bilayer with the Sn, Se, and S atoms in the top layer directly located above the Sn, Se, and S atoms in the bottom layer, respectively. Similar picture can be found for the other moiré unit cells, as shown in figure S7 of the supplementary material. To compare the bonding strengths between these high-symmetry stacking regions, we calculate the ELF values at the midpoint of all the involved covalent bonds, as listed in table S2 of the supplementary material. It is found that the moiré unit cells exhibit strong bonding heterogeneity, which can be quantified by the standard deviation ( ) of the ELF values between high-symmetry stacking regions. In figure 6, we plot the
) of the ELF values between high-symmetry stacking regions. In figure 6, we plot the  as a function of twist angle for both the Sn–S and Sn–Se bonds. It is interesting to find that the variation of
as a function of twist angle for both the Sn–S and Sn–Se bonds. It is interesting to find that the variation of  shows the same trend as that of
shows the same trend as that of  , which consequently leads to a distinct non-monotonous dependence of the
, which consequently leads to a distinct non-monotonous dependence of the  on the twist angle.
on the twist angle.

Figure 5. The ELF diagram of the Janus SnSSe bilayer with twist angle of 9.43°. The dashed lines indicate the moiré unit cell, where the atomic configurations of high-symmetry stacking regions Ⅰ, Ⅱ, and Ⅲ are illustrated.
Download figure:
Standard image High-resolution image
Figure 6. The standard deviation of ELF values between high-symmetry stacking regions for the Sn–S and Sn–Se bonds in the Janus SnSSe bilayer, plotted as a function of twist angle.
Download figure:
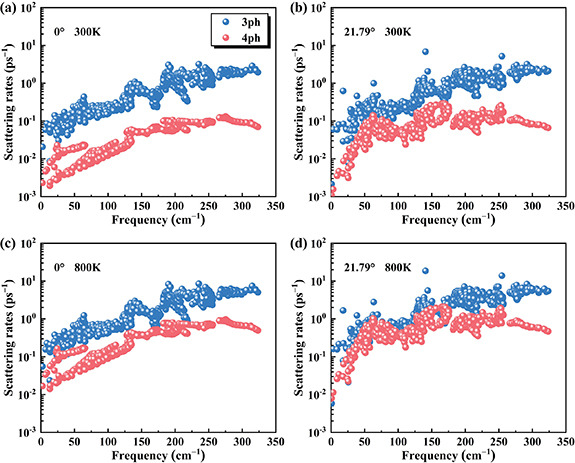
Standard image High-resolution imageUp to now, we are dealing with three-phonon scattering process. Unlike time-consuming or even prohibitive DFT calculations, the established MTP can be used to readily derive the fourth-order IFCs from numerous supercell configurations, which provides a good opportunity to investigate the effect of four-phonon scattering on the  . As an example, we consider the Janus SnSSe bilayers with twist angles of 0° and 21.79°. At room temperature, we see from figures 7(a) and (b) that the four-phonon scattering rates are nearly one order of magnitude lower than the three-phonon scattering rates for the untwisted bilayer, while they become close to each other at twist angle of 21.79°. Such a finding suggests that the interlayer twisting could enhance the fourth-order anharmonic effect and thus decrease the
. As an example, we consider the Janus SnSSe bilayers with twist angles of 0° and 21.79°. At room temperature, we see from figures 7(a) and (b) that the four-phonon scattering rates are nearly one order of magnitude lower than the three-phonon scattering rates for the untwisted bilayer, while they become close to each other at twist angle of 21.79°. Such a finding suggests that the interlayer twisting could enhance the fourth-order anharmonic effect and thus decrease the  . Indeed, we see from table 3 that if both three- and four-phonon scatterings are considered, the
. Indeed, we see from table 3 that if both three- and four-phonon scatterings are considered, the  of SnSSe bilayers are reduced by 13.84% and 22.77% at twist angles of 0° and 21.79°, respectively. At increased temperature of 800 K, such an effect become even pronounced. As can be found from figures 7(c) and (d), the three- and four-phonon scattering rates are comparable for the untwisted bilayer, and almost coincide with each other for the twisted system. As a consequence, we see from table 3 that the
of SnSSe bilayers are reduced by 13.84% and 22.77% at twist angles of 0° and 21.79°, respectively. At increased temperature of 800 K, such an effect become even pronounced. As can be found from figures 7(c) and (d), the three- and four-phonon scattering rates are comparable for the untwisted bilayer, and almost coincide with each other for the twisted system. As a consequence, we see from table 3 that the  are reduced by 27.41% and 43.41%, respectively. In a word, our findings indicate that the effect of four-phonon scattering plays an important role in accurately predicting the
are reduced by 27.41% and 43.41%, respectively. In a word, our findings indicate that the effect of four-phonon scattering plays an important role in accurately predicting the  of Janus SnSSe bilayer, especially for those with twisted structures at elevated temperature.
of Janus SnSSe bilayer, especially for those with twisted structures at elevated temperature.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. The three- and four-phonon scattering rates with respect to the phonon frequency for the Janus SnSSe bilayers with twist angles of 0° and 21.79°. The results at 300 K (a), (b) and 800 K (c), (d) are both shown for comparison.
Download figure:
Standard image High-resolution image{kind=link}
Table 3. The lattice thermal conductivities of the Janus SnSSe bilayers with twist angles of 0° and 21.79°, calculated with ( ) and without (
) and without ( ) consideration of four-phonon scattering at 300 and 800 K.
) consideration of four-phonon scattering at 300 and 800 K.
| 0° | 21.79° | ||
|---|---|---|---|
| 300 K |
 (W m−1 K−1) (W m−1 K−1) | 8.74 | 7.07 |
 (W m−1 K−1) (W m−1 K−1) | 7.53 | 5.46 | |
| 800 K |
 (W m−1 K−1) (W m−1 K−1) | 3.21 | 2.58 |
 (W m−1 K−1) (W m−1 K−1) | 2.33 | 1.46 | |
4. Summary
In summary, we demonstrate that the MTP trained on a computationally feasible DFT dataset can be utilized to realize ready and accurate prediction of the  of twisted Janus SnSSe bilayers, which exhibits unique twist-angle dependence characterized by bonding heterogeneity inside the moiré unit cells. It should be emphasized that although we are dealing with the suspended systems, they are both mechanically and dynamically stable, which provides a good platform to explore the effect of interlayer twisting on the
of twisted Janus SnSSe bilayers, which exhibits unique twist-angle dependence characterized by bonding heterogeneity inside the moiré unit cells. It should be emphasized that although we are dealing with the suspended systems, they are both mechanically and dynamically stable, which provides a good platform to explore the effect of interlayer twisting on the  . In principle, the strategy established here should be applicable to many other 2D materials, which enables efficient evaluation of their
. In principle, the strategy established here should be applicable to many other 2D materials, which enables efficient evaluation of their  at arbitrary twist angle. Our work therefore provides an accelerated route to discover twisted moiré materials with tunable
at arbitrary twist angle. Our work therefore provides an accelerated route to discover twisted moiré materials with tunable  , which is very beneficial for the effective thermal management of micro- and nano-electronic devices.
, which is very beneficial for the effective thermal management of micro- and nano-electronic devices.
Acknowledgments
We thank financial support from the National Natural Science Foundation (Grant No. 62074114). The numerical calculations in this work have been done on the platform in the Supercomputing Center of Wuhan University.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Supplementary data (0.9 MB PDF)