
Abstract
Molecular dynamics (MD) simulations were applied to the study of nucleation and growth of pentacene thin films, and stability of clusters (nuclei) comprising standing molecules was investigated. In simulations, the clusters consisting of more than ten standing molecules could stably exist on hydrophobic surfaces, while several tens of molecules were necessary for stabilization on hydrophilic surfaces. Furthermore, the stabilized clusters could grow by incorporating additional molecules in MD simulations. These results suggest that nucleation occurs on hydrophobic surfaces easier than on hydrophilic surfaces and the critical size of the nuclei of "standing" pentacene is about ten molecules on hydrophobic surfaces.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Organic semiconductors are attracting interest due to their promising characteristics such as flexibility and large-area fabrication at low cost. Organic field-effect transistors which are key devices to develop organic electronics are gradually getting improved toward practical applications.1–3) Since one of the biggest advantages of organic semiconductors is that thin films can be easily produced, an ultimate goal in this research field is to establish a technology for making high quality organic thin films and fabricating organic thin film transistors (OTFTs) with high carrier mobility. However, there are many factors which reduce the carrier mobility in polycrystalline thin films of organic semiconductors, such as grain boundary traps,4–6) and we still have a long way to obtain high performance OTFTs enough for practical use. It is quite important to understand the mechanism of nucleation and growth of organic semiconductor thin films to produce high quality thin films and develop high performance OTFTs. In the 1990s and 2000s, a knowledge of the nucleation and growth processes was dramatically accumulated by excellent experimental and theoretical studies.4) However, the mechanisms at the molecular level are still under debate, for example, there is still uncertainty in regarding the questions of why, how, and when molecules stand up on substrates.
There are some universal tendencies in the characteristics of organic semiconductor thin films. Rod-shape molecules in thin films tend to stand on inert substrate surfaces like the surface of amorphous SiO2 when the substrate temperature is relatively high and the growth rate is relatively low.7,8) The molecular axes of such molecules are almost perpendicular to the surface or tilted a little from the normal direction according to the results of experimental studies, for example, analyses using X-ray diffraction. Pentacene, which is focused on in this study, is a promising organic semiconductor with high carrier mobility and one of the typical molecular materials which form thin films comprising standing molecules on inert substrates.9) This standing manner is simply explained by the assumption that (001) plane has the lowest surface energy and leave the lowest energy surface exposed.9) The mechanism of such universal tendency is also explained by the theoretical studies based on thermodynamics and kinetic analyses.10,11)
Although it can be understood that the (001) plane possessing the lowest surface energy appears on the top of the thin film, some questions remain, for example, when the molecules begin standing. The purpose of this study is to investigate the behavior of the molecules on substrate surfaces, at the molecular level, using molecular dynamics (MD) simulations and pentacene as the typical rod-shape organic semiconductor molecule. Some papers on MD simulations of pentacene have been published.12–16) Muccioli et al.15) carried out MD simulations of the deposition of pentacene molecules on the (001) surface of a C60 crystal and observed the transition from flat-lying manner to standing manner of pentacene molecules on the C60 crystal. Their result is probably the sole example succeeded in looking at the spontaneous transition to standing manner by MD simulations to the best of the author's knowledge. However, the tilt angle of the molecular axis from the surface normal direction is much larger than the experimentally observed one, and therefore it is premature to conclude that the mechanism has been fully understood.
Before entering the core of this study, the author would like to show the results of a preliminary MD simulation (the supplementary material "Supplementary movie 1" is available online at stacks.iop.org/APEX/13/015508/mmedia to let readers understand the problem of MD simulations of the deposition process of rod-shape molecules). In the movie, isolated molecules adsorbed on the substrate show flat-lying manner because of the strong interaction between atoms of the adsorbed molecules and atoms of the substrate. The other molecules deposited later are combined with the existing (flat-lying) molecules and the molecular clusters keep the flat-lying manner. According to this result, it seems hard to observe the process in which molecules spontaneously stand up on substrates by MD simulations. We need to take the other approach to deal with this problem by MD simulations.
Nucleation includes stochastic processes. Somewhere on the substrate, by chance, some molecules may stand up. If the molecular clusters which stand up by chance are thermodynamically stable, such clusters (nuclei) might grow further by incorporating the surrounding molecules. Such process in which molecules stand up by chance is not dealt with in this study. Larger computational capabilities which allow us to deal with longer time scale processes and larger size systems are probably required to simulate such stochastic processes. In this study, the author focuses on the stability of the clusters which comprise the standing molecules (the molecules which have already stood up) and investigates the stability of such clusters by MD simulations. Some years ago,17) the author started MD simulations to investigate the mechanism of graphoepitaxy18–20) of organic semiconductor molecules and published the results of the MD simulations.17) In this study, the technique of MD simulations established in Ref. 17 was utilized.
The details regarding the models of the amorphous SiO2 substrates whose dimensions were 12.8 nm (X) × 12.8 nm (Y) × 1.6 nm (Z) and a pentacene molecule used for MD simulations in this study are shown in the supplementary material. The Si atoms on the outermost surface of amorphous silica substrates were replaced by Si–OH or Si–OSi(CH3)3 to prepare the hydrophilic surface or hydrophobic surface, respectively. In actual experiments, the hydrophobic functional group (trimethylsiloxy group, –OSi(CH3)3) is produced by hexamethyl-disiloxane (HMDS) treatment.
To calculate intramolecular and intermolecular interactions, in this study, the DREIDING force field,21) a simple generic set of force equations and parameter values, was utilized. The author performed preliminary test simulations to check on the appropriateness of the empirical parameters mounted in the DREIDING force field by varying them widely and, consequently, came to use the original set of parameters. Buckingham potential22) was used to describe intermolecular interactions (van der Waals forces) between pentacene molecules, while the Lennard–Jones potential was used for other intermolecular interactions, for example, between a pentacene molecule and functional groups which modify the substrate surface. In both cases, the Buckingham potential and the Lennard–Jones potential, Coulomb interactions working between two charges qiqj/(ε0r) were all integrated and added to the intermolecular interaction. Periodic boundary conditions (for X, Y, and Z-directions in the simulations investigating the stability of pentacene clusters and for the X and Y-directions in the simulation of pentacene deposition) were applied to all simulations. The Ewald method was not used for calculating Coulomb interactions this time, because it was found that charges in the substrate which are periodically (virtually) placed upper and lower parts (along the Z-direction) influenced the arrangement of pentacene molecules when the Ewald method was used. All interactions including Coulomb interactions between atoms within the cut-off distance Rc (3.0 nm) were calculated and used for solving the equation of motion. In the MD simulations of the deposition of pentacene molecules, the cut-off distance Rc was decreased to 2.0 nm to reduce the calculation time. Please refer to Ref. 17 for more details about the mathematical expressions of interaction potentials (force field).
The Si and O atoms in amorphous SiO2 substrates were not moved during all the simulations carried out in this study, by fixing the velocity of these atoms as zero. The functional groups placed on the substrate surfaces, Si–OH and Si–OSi(CH3)3, were freely moved during the simulations. In order to prevent these functional groups from detaching from the surface, Si atoms placed at the bottom of these functional groups were fixed by giving the Si atoms a huge amount of mass. The shape of each pentacene molecule was also flexibly changed by calculating the intramolecular interactions expressed by the four energy terms, bond stretch, angle bend, torsion, and inversion (out-of-plane) angle.17,21) The modeling of the substrates and molecules and all MD simulations in this study were carried out using SCIGRESS ME, a software package for materials exploring produced by Fujitsu Limited, and calculations were performed by parallel computing using two cores of Xeon® processor. The NTV ensemble was applied to all the simulations, and temperature (average velocity) of pentacene molecules and functional groups was kept at 370 K. The time of each step to solve the equation of motion in MD simulations was set as 0.2 femtosecond because the time scale of the stretching vibration of hydrogen atoms in molecules is about or less than 0.2 femtosecond.23) The calculation time needed for each simulation ranged from one day to several weeks depending on the number of steps, the number of atoms and molecules, the cut-off distance Rc, and so on.
As mentioned above, it seems quite hard to reproduce the transition from flat-lying manner to standing one by MD simulation. Therefore, in this study, the author focused on the stability of the clusters comprising the pentacene molecules which stood from the beginning. To examine the stability of the cluster made up of standing pentacene molecules, the initial structure was prepared as follows; the clusters comprising 4, 8, 15, 28, and 60 standing pentacene molecules (and also 112 molecules for the hydrophilic substrate) were prepared on substrate surfaces. As an example, three snapshots of the MD simulation dealing with 15 standing molecules are shown in Fig. 1. As the initial condition, pentacene molecules were arranged making the molecular planes parallel (not herringbone packing but brick-wall arrangement) as shown in the 0 picosecond's snapshot of the top view. Within several picoseconds, a herringbone packing structure appeared and the molecules kept standing manner up to 100 picoseconds without collapse in the case of 15 molecules (movies are provided to support the figures as online supplementary movies).
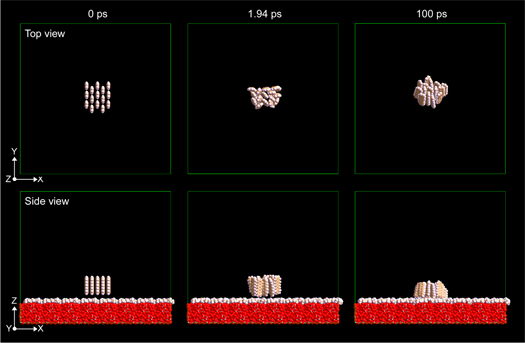
Fig. 1. (Color online) One example (side view and top view) of a MD simulation to examine the stability of the cluster comprising 15 standing pentacene molecules on the surface modified by trimethylsiloxy group (HMDS-treated) showing the hydrophobic nature. The snapshots at 0 ps, 1.94 ps, and 100 ps (total 500 000 steps) are shown. All the atoms of the substrate have been removed from the top view to make pentacene molecules easy to see. The movie related to this figure [Supplementary movie 2 (for Fig. 1)] is provided in the online supplementary data.
Download figure:
Standard image High-resolution imageFigure 2 and the supplementary movie for Fig. 2 show the results of the MD simulations of the clusters comprising 4, 8, 15, 28, and 60 molecules placed on the hydrophobic surface. In the cases of the clusters comprising 4 and 8 molecules, the standing molecules in the clusters easily fell down and never stood up again. However, in the case of 15 molecules, the pentacene molecules kept a standing manner though the cluster showed some fluctuation and still some unstableness, as already shown in Fig. 1 and the supplementary movie for Fig. 1. In the cases of 28 molecules and 60 molecules, the clusters seem stable during the simulation.

Fig. 2. (Color online) Results of MD simulations investigating the stability of the clusters comprising standing pentacene molecules. In the cases of 4, 8, 15, 28, and 60 molecules on the surface modified by trimethylsiloxy group showing hydrophobic nature. All the figures show the snapshots at 100 ps (the 500 000th step). The movie related to this figure [Supplementary movie 3 (for Fig. 2)] is provided in the online supplementary data.
Download figure:
Standard image High-resolution imageThe result largely changed when the hydrophilic substrate was used as shown in Fig. 3 and the supplementary movie for Fig. 3. In the hydrophilic case, the cluster fell down even in the case of 28 molecules. In the cases of 60 molecules and 112 molecules, the clusters seemed stable, but some molecules of outer periphery of the clusters fell down during the simulation. It is also interesting that the cluster of 8 molecules almost collapsed into isolated molecules on the hydrophilic surface (Fig. 3) though the herringbone packing was rigidly kept even in the case of four molecules on the hydrophobic surface (Fig. 2). The result of unstableness on hydrophilic surfaces suggests that the dipole moment of the hydroxyl group had an effect which weakened or detuned the intermolecular interaction between pentacene molecules. Comparing the results of the MD simulations using hydrophobic and hydrophilic models, it was revealed that clusters comprising standing molecules are more stable on hydrophobic surfaces than on hydrophilic surfaces.
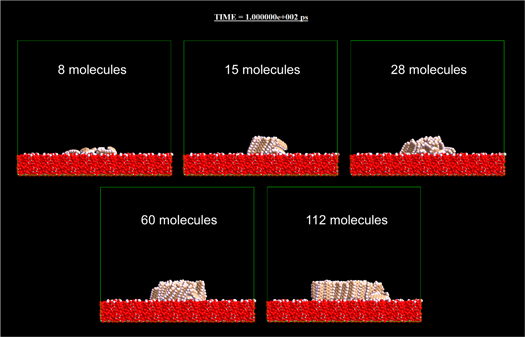
Fig. 3. (Color online) Results of MD simulations investigating the stability of the clusters comprising standing pentacene molecules. In the cases of 8, 15, 28, 60, and 112 molecules on the surface modified by hydroxyl group showing hydrophilic nature. All the figures show the snapshots at 100 ps (the 500 0000th step). The movie related to this figure [Supplementary movie 4 (for Fig. 3)] is provided in the online supplementary data.
Download figure:
Standard image High-resolution imageHere, the knowledge with respect to the stability of molecular clusters comprising standing pentacene molecules is obtained. The next step of the study is to know whether such cluster comprising standing molecules can grow further to a thin film. On the hydrophobic surface, a cluster comprising 15 standing pentacene molecules kept a standing manner though some fluctuation was observed. If the cluster comprising 15 standing molecules can grow by incorporating additional molecules and become bigger, the cluster will become more stable and continue to grow further, keeping the standing manner. Figure 4 and the supplementary movie for Fig. 4 show the growth of the cluster comprising 15 standing molecules on hydrophobic substrate. The initial condition of this simulation is the final state of the simulation shown in Fig. 1. For two nanoseconds, 200 pentacene molecules were deposited on the surface where the standing pentacene cluster already existed (although this high deposition rate is unrealistic, there was nothing for it but to do so under the present limitation of computational capabilities). The cluster comprising 15 standing pentacene molecules incorporated 26 additional molecules and further grew up to be the cluster composed of 41 standing molecules. This result of the simulation suggests that, on the hydrophobic surface, if the cluster comprising 15 molecules appears by chance, the cluster can grow further and can become a stable crystalline island and then a thin film by incorporating additional molecules. The author also confirmed that the clusters comprising 60 and 112 molecules on the hydrophilic surface could grow further by the similar simulations though the data are not shown here.

{kind=link}
{kind=link}
{kind=link}
Fig. 4. (Color online) Growth of an existing cluster consisting of 15 standing pentacene molecules on the surface modified by trimethylsiloxy group. The snapshots at 0 ns, 1 ns (the 5 000 000th step), and 2 ns (the 10 000 000th step) are shown. The initial condition of the calculation (the snapshot at 0 ns) corresponds to the atomic arrangement obtained by the simulation of 15 molecules (at 100 ps), shown in Figs. 1 and 2. All the atoms of the substrate have been removed from the top view to make pentacene molecules easy to see. The movie related to this figure [Supplementary movie 5 (for Fig. 4)] is provided in the online supplementary data.
Download figure:
Standard image High-resolution image{kind=link}
It has been reported that the nucleation density of pentacene is larger on the HMDS-treated hydrophobic surface (amorphous SiO2) than on the clean hydrophilic surface.24) Also in the experimental study carried out by the author himself using sexithiophene (6 T), the nucleation density was larger on the HMDS-treated hydrophobic surface than on the clean (UV/ozone-treated) hydrophilic surface;19) the tendency was the same as the case of pentacene. These experimental results are consistent with the results of MD simulations performed in this study, that is, the clusters made up of standing pentacene molecules are more stable on the hydrophobic surface than on the hydrophilic surface. Since the probability that about 15 molecules have a chance to collectively hold a standing manner together on the (hydrophobic) surface is thought to be higher than the probability that several tens of molecules stand on the (hydrophilic) surface collectively, it is expected that nucleation takes place easier on hydrophobic surfaces than on hydrophilic surfaces.
The results of the MD simulations obtained in this study can also be applied to the discussion about the size of critical nuclei of pentacene. Some previous experimental studies suggest that the number of molecules of critical nuclei of pentacene is four or less than four.25,26) Also in this study, the pentacene cluster comprising four molecules, shown in Fig. 2 (and the supplementary movie for Fig. 2), kept herringbone packing even after falling down on the hydrophobic substrate. However, the result of this study also suggests that four or eight molecules are not enough to keep the standing manner. This means that the number of molecules which completes the "standing" critical nuclei of pentacene on the hydrophobic surfaces is more than 8 and less than 15. The author performed additional MD simulations of 10 molecules and 12 molecules to find the critical size (the results can be seen in the supplementary material "Supplementary movie 6"). In the case of 12 molecules, the molecular cluster could keep the standing manner in all three runs; though, one molecule detached itself from the main cluster in two runs of the three. In the case of 10 molecules, however, the cluster sometimes fell down and sometimes kept a standing manner. These results suggest that the number of molecules "10" almost corresponds to the peak position of the free energy curve which will be discussed later and the number of pentacene molecules forming critical nuclei is about 10. The size of the critical nuclei on hydrophilic surfaces is rather big, probably several tens molecules based on the results of the simulations.
In the classical nucleation theory,27) Gibbs free energy of a spherical nucleus with radius r is composed of two terms; the first term is a negative one which is proportional to the volume (∝ r3) of the nucleus and stabilizes the solid state, and the second term is a positive one which is proportional to the surface area (∝ r2) of the nucleus and prevents the nucleus from growing further. The free energy shows a curve in upward convex shape against the radius, r, and has a maximum point (peak) which corresponds to the radius of a critical nucleus, rc. If the radius exceeds rc, the nucleus is thermodynamically stabilized by growing further. MD simulations have been applied to investigate the nucleation processes of some inorganic materials and revealed the microscopic kinetic processes as well as the thermodynamic properties.28,29) On the other hand, in the thin film growth of organic semiconductors such as pentacene on flat substrate surfaces, nucleation and growth generally occur in a monolayer state before the second layer starts to form (two-dimensional layer-by-layer growth).4,24–26) Therefore, the first term of the above classical nucleation theory is not proportional to the cube of r but is proportional to the square of r.
Kubono and Akiyama11) suggested, based on their theoretical analysis of nucleation of long-chain organic compounds, that the cluster comprising flat-lying (lateral) molecules is thermodynamically more stable than that comprising standing (normal) molecules within the range of less than 30 molecules. However, the free energy curve of the cluster comprising standing molecules shows a peak around 15 molecules though the number depends on the assumed chemical potential change for nucleation, Δμ. This means that the number of molecules for critical nuclei is around 15 if the molecules have a chance to stand up. Although their theoretical study did not focus on a specific compound such as pentacene, the parameters such as surface energy used in their calculation were roughly common to various organic compounds and their result can be used for the discussion about the nucleation of pentacene.
In this study, the author also performed theoretical investigation by modeling a simpler model and summarized the results in the supplementary material. Despite using a simplified model, the analysis gave a result similar to that reported by Kubono and Akiyama.11) Clusters comprising standing molecules are thermodynamically unstable compared to those comprising flat-lying molecules when the number of the molecules is less than several tens. However, the free energy curve has a peak around ten molecules even in the case of a standing manner. If a cluster comprising about ten standing molecules appears on the surface by chance, the cluster can be stabilized and grow further.
In this study, the author focused on the stability of clusters comprising standing pentacene molecules and investigated the stability and subsequent growth of the molecular clusters by MD simulations. This is a new approach to study of nucleation and growth processes of organic semiconductor thin films. The consistency between the results of MD simulations and the previous experimental studies concerning nucleation ensures the usefulness of this approach. Furthermore, the MD simulations suggest the size of the critical nuclei comprising "standing" molecules, quantitatively. Although pentacene molecules have strong tendency to form crystalline state (herringbone packing) just after molecules meet together in MD simulations, crystal embryo and critical nucleus do not necessarily have crystalline state and they can be amorphous. Actually, 6T could not crystallize and displayed a glassy structure in the early stages of deposition in the author's previous MD simulations.17) Therefore, we must make more of an effort to extend the approach proposed in this study to universalize the early stage of thin film growth of organic semiconductors. However, the author believes that MD simulations are important to elucidate microscopic mechanism of nucleation and growth of organic semiconductor thin films and will surely contribute to developing organic electronic devices such as high performance OTFTs.
Acknowledgments
This research was supported by JSPS KAKENHI Grant Number JP15K04674 and JP18K05253, Japan, and the World Premier International Research Center Initiative (WPI), MEXT, Japan. The author would like to thank anonymous referees for their helpful and constructive comments which greatly improved the manuscript.