



Abstract
Characterizing the geochemistry of Europa and Enceladus is a key step for astrobiology investigations looking for evidence of life in their subsurface oceans. Transition metals with several oxidation states, such as iron, may be tracers of the oxidation state of icy ocean moon interiors. Their detection, as well as the characterization of their oxidation states, on the moons' (plume) ice grains would bring valuable new information about the geochemistry of both the subsurface oceans and surface processes. Impact ionization mass spectrometers such as the SUDA instrument on board Europa Clipper can analyze ice grains ejected from icy moons' surfaces and detect ocean-derived salts therein. Here we record mass spectra analogs for SUDA using the Laser Induced Liquid Beam Ion Desorption technique for Fe2+ and Fe3+ salts (both sulfates and chlorides). We show that impact ionization mass spectrometers have the capability to detect and differentiate ferrous (Fe2+) from ferric (Fe3+) ions in both cation and anion modes owing to their tendency to form distinct ionic complexes with characteristic spectral features. Peaks bearing Fe3+, such as [Fe3+ (OH)2]+ and [Fe3+ (OH)a Clb]−, are particularly important to discriminate between the two oxidation states of iron in the sample. The recorded analog spectra may allow the characterization of the oxidation state of the oceans of Europa and Enceladus with implications for hydrothermal processes and potential metabolic pathways for life forms in their subsurface oceans.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The subsurface oceans of several icy moons (e.g., Enceladus and Europa) are in direct contact with their silicate interiors, making them some of the most likely candidates in the solar system to support habitable conditions (e.g., Chyba 2000; Marion et al. 2003; Barge & Rodriguez 2021), and therefore are compelling targets in the search for life beyond Earth. A better understanding of the geochemistry of icy moons' subsurface oceans is needed for astrobiology investigations looking for traces of life. Icy moons' surfaces hold clues to the composition of their subsurface oceans, as they are shaped by both internal processes (e.g., resurfacing, plume activity) and external processes (e.g., space weathering). Their characterization is key to understanding icy moons' evolution over geological timescales, as well as their current habitability.
The subsurface ocean of Enceladus, one of the moons of Saturn, is currently the best-characterized extraterrestrial ocean in the solar system and is accessible owing to the active plume at its south pole that ejects water vapor and ice grains into space (Spahn et al. 2006; Waite et al. 2006; Postberg et al. 2018a; Villanueva et al. 2023a). Enceladus's ocean is global, with an ice shell decoupled from its core and heated at the ocean–core interface (Lainey et al. 2012; Thomas et al. 2016), where hydrothermal reactions are inferred to occur between the porous chondritic bedrock and percolating oceanic water (Hsu et al. 2015; Sekine et al. 2015; Choblet et al. 2017; Waite et al. 2017). Material derived from the plume (e.g., silica nanoparticles and molecular hydrogen; Sekine et al. 2015; Waite et al. 2017) is consistent with the presence of alkaline, Lost City–type hydrothermal vents, with exothermic serpentinization reactions between sea water and rocks (Kelley et al. 2001). The detected molecular H2 is thought to be a major species of the plume (Waite et al. 2017), suggesting an overall reducing Enceladus ocean. The ocean is rich in salts, including sodium chlorides, carbonates (Postberg et al. 2009), and phosphates (Postberg et al. 2023), and has a pH estimated to be around 8.5–10.5 (Zolotov 2007; Postberg et al. 2009; Hsu et al. 2015; Glein & Waite 2020, Fifer et al. 2022). It also contains a variety of organic molecules, including high molecular mass (>200 u) refractory insoluble organic material (Postberg et al. 2018b) and low molecular mass (<100 u) volatiles, including various moieties bearing oxygen, nitrogen, aryl groups (Khawaja et al. 2019), and tentative alkene and acyl groups (Khawaja et al. 2023).
Europa, one of the Galilean moons, also possesses a global salty ocean (Khurana et al. 1998; Kivelson et al. 2000), which lays beneath a 3–47 km ice shell (e.g., Schubert et al. 2009; Howell 2021) and may be communicating material to the surface through plume activity or other forms of upwelling (Sotin et al. 2002; Roth et al. 2014; Sparks et al. 2017; Jia et al. 2018; Singer et al. 2021; Villanueva et al. 2023b). Europa likely contains a solid iron-rich metal core (Moore & Hussmann 2009). Its subsurface ocean could be anoxic or even significantly oxygenated (Hand et al. 2007), and the surface ice is rich in salts, including chlorides (e.g., Hand & Carlson 2015; Trumbo et al. 2019) and sulfates (e.g., McCord et al. 1998; Dalton 2007; Hibbitts et al. 2019; Mermy et al. 2023). Its young surface age (∼40–90 Myr; Bierhaus et al. 2009) implies that recent endogenic resurfacing events could provide material from the subsurface ocean onto the surface, potentially including organic material. This material may originate from hydrothermal systems, which may be present owing to the considerable heat generated by tidal interactions between Europa, Jupiter, and the other Galilean satellites and transmitted through the liquid ocean (Lowell & DuBose 2005; Běhounková et al. 2021). Hydrothermal plumes may be implicated in the formation of chaos terrains by melting or by inducing convection in the ice shell (Goodman 2004). Chaos terrains such as Thrace Macula are prime targets of the Europa Clipper and JUICE missions, as they may provide the freshest material for sampling by future spacecraft (Matteoni et al. 2023).
The detection and quantification of dissolved salts in icy moons' subsurface oceans can provide information about water–rock interactions, which are necessary for habitability, and the geochemical reactions involved. Enceladus's core is thought to have a composition close to CI or CM carbonaceous chondrites (Sekine et al. 2015), and Europa's core is thought to have a composition close to L/LL-type chondrites (Kuskov & Kronrod 2005). Both moons' interiors should therefore host a range of iron-bearing minerals, including phyllosilicates (e.g., serpentine), olivine, iron oxides (e.g., magnetite, ferrihydrite), pyroxene, and sulfides (Sekine et al. 2015; Ray et al. 2021; Hamp 2022). A variety of Fe2+- and Fe3+-bearing mineral phases are expected to be present (some being formed by hydrothermal water–rock interactions), and their dissolution could supply Fe2+ and Fe3+ ions to the ocean. The oceanic abundances of Fe2+ and Fe3+ ions are likely limited by both the bulk Fe content and the precipitation of ferrous and ferric minerals (i.e., by the solubility of Fe2+ and Fe3+), which is tightly linked to key geochemical parameters of the ocean, notably its oxidation state and pH. While the oceans of Europa and Enceladus may contain several iron species in a variety of oxidation states, concentrations are still unconstrained (Zolotov 2007; Sekine et al. 2015; Glein & Waite 2020). However, under the assumed redox state of Enceladus's ocean, Fe should always be in the form of Fe2+ (Christ 1965). Ray et al. (2021) estimated the concentrations of dissolved ferrous ions in Enceladus's ocean, as determined by the dissolution of stable minerals, to be ∼10−7 to 10−9 mol L−1 for oceanic pH values of 9 and 11, respectively, and Hao et al. (2022) estimated concentrations from 10−5 to 10−8 mol L−1 for pH values ranging from 8.5 to 11.
On Europa's surface, iron (if present) would likely be in its ferric form owing to the intense radiation chemistry that oxidizes the surface ice. Iron compounds have been suggested for Europa's surface (Carlson et al. 2009), particularly ferric oxides and ferric sulfate given the oxidizing nature of the surface, but there is currently no strong evidence for these compounds at observable amounts. Ferrous iron could be oxidized radiolytically (by oxidants originating from the radiation-induced dissociation of water), producing ferric iron. Exchanges between the ice shell and the subsurface ocean (e.g., a recycling of the ice shell) could provide Fe3+ to the ocean and provoke the precipitation of ferric oxyhydroxide and/or a reduction of Fe3+ to Fe2+ if the ocean is sufficiently reduced.
The detection of Fe2+ and/or Fe3+ (in molecular complexes) in fresh material from Europa and Enceladus would bring insight into the geochemistry of their subsurface oceans, thus constraining the concentrations of ionic species and therefore geochemical models. It could allow the evaluation of redox disequilibria, a prerequisite for the origin of life, and may also bring insight into potential metabolic processes that may happen in the oceans (Ray et al. 2021).
On both Enceladus and Europa, regional heating (Soderlund et al. 2014; Choblet et al. 2017) hints toward chemical and thermal disequilibria on a global scale, which is essential for the appearance of life and entails the presence of multiple physicochemical gradients of diverse nature (Shock & Canovas 2010). One example of these are redox gradients, expected to arise between a reducing rocky core and oxidized oceanic water (and on Europa between the ocean and more oxidized surface ice), thus potentially providing abundant chemical energy for life to develop (Chyba 2000; Hand et al. 2007; Barge & White 2017; Deamer & Damer 2017; Angelis et al. 2021). Hydrothermal vents are prime candidates for observing prebiotic chemistry at play, as they gather a large variety of redox gradients and are regarded as the most likely environment for a putative origin of life on both early Earth and icy moons (e.g., Russell & Martin 2010; Russell et al. 2014). Under hydrothermal conditions, iron minerals provide a rich source of both Fe2+ and Fe3+. This redox imbalance can promote chemical reactions among simple carbon-containing substrates found in hydrothermal vents giving rise to molecules relevant for prebiotic chemistry, such as amino acids (Barge et al. 2019). The abundance of iron minerals in hydrothermal systems both on early Earth and currently on icy moons could in both cases have triggered geochemical gradients, perhaps driving prebiotic chemistry leading to more complex organic reaction networks.
On Earth, a major model for the origin of life is that life started on a Fe2+-rich early Earth (e.g., Russell et al. 1989, 1994) within alkaline hydrothermal vents where thin mineral walls containing catalytic Fe(Ni)S minerals offered redox and pH gradients. Such gradients could drive prebiotic chemistry and are comparable to those needed in all cells to function (Sojo et al. 2016), which makes a very strong case for hydrothermal vents being the site for the origin of life. Furthermore, inorganic cofactors utilized in ancient pathways such as the acetyl-CoA pathway (carbon fixation) and the reverse Krebs cycle (energy transduction) in cells share the same composition as catalytic Fe(Ni)S clusters from the redox-active minerals of the vents' walls (Eck & Dayhoff 1966; Russell & Hall 1997; Russell & Martin 2004; Bender et al. 2011). Therefore, iron-based metabolisms could have been essential for the development of primitive life on early Earth and may also be of prime importance on icy moons. Several metabolisms have been proposed to be viable in hydrothermal environments on icy moons, such as methanogenesis (Taubner et al. 2018; Tenelanda-Osorio et al. 2021) and iron-based metabolisms (Ray et al. 2021; Roche et al. 2023). Iron-oxidizing bacteria have also been documented in terrestrial alkaline hydrothermal systems analogous to those expected on Enceladus (Scott et al. 2015). The detection of the oxidation state of metallic elements such as Fe on icy moons' oceans could constrain potential biogeochemical oceanic iron cycles (Zolotov & Shock 2004) and the possible metabolic pathways of putative organisms.
Jupiter's icy moons are the targets of the upcoming JUICE mission from ESA and Europa Clipper from NASA. Several mission concepts have been proposed to explore Enceladus, such as Moonraker (Mousis et al. 2022) and Enceladus Life Finder (Reh et al. 2016). To investigate the geochemistry of extraterrestrial ocean worlds, a powerful technique is impact ionization mass spectrometry (MS). Such instruments include the Cosmic Dust Analyzer (CDA; Srama et al. 2004) on board the past Cassini mission and its successor instruments, the SUrface Dust Analyzer (SUDA; Kempf et al. 2023) on board Europa Clipper, and the proposed High Ice Flux Instrument (HIFI; Mousis et al. 2022) on board Moonraker. Impact ionization mass spectrometers measure the ionized species created by the hypervelocity impact of dust grains onto a metal plate. Around icy ocean moons, they can provide compositional analysis of ice grains ejected from the surface ice by the ambient micrometeorite bombardment (Postberg et al. 2011a; Goode et al. 2021) and of ice grains ejected from subsurface water reservoirs by plume activity (Postberg et al. 2011b).
The performance and data analysis of these instruments require laboratory calibration, which may be done with analog experiments such as the Laser Induced Liquid Beam Ion Desorption (LILBID) technique coupled to time-of-flight MS (ToF MS; Wiederschein et al. 2015; Klenner et al. 2019). This will be demonstrated and showcased in the present paper. Due to the technical difficulties linked to the hypervelocity acceleration of water-ice grains in laboratory experiments (Adamson et al. 2017; Belousov et al. 2021; Miller et al. 2022; Spesyvyi et al. 2023), LILBID is still the only currently available method to accurately reproduce impact ionization mass spectra of water-ice grains from icy moons. Various LILBID analog experiments have already been performed with salts (Postberg et al. 2009; Napoleoni et al. 2023a, 2023b; Postberg et al. 2023), organic material (Postberg et al. 2018b; Khawaja et al. 2019, 2022; Napoleoni et al. 2023a, 2023b), and molecular biosignatures (Klenner et al. 2020a, 2020b; Dannenmann et al. 2022; Klenner et al. 2023).
Here we measure the cation and anion LILBID mass spectra of iron (II) and iron (III) chlorides and sulfates, to investigate the spectral signatures of iron in different oxidation states as detectable by impact ionization mass spectrometers on board space missions to icy moons. The recorded LILBID spectra are analog for the SUDA instrument and complement a database (Klenner et al. 2022) for Europa Clipper and future Enceladus missions that might allow the detection and characterization of iron on these icy moons, providing valuable information on their geochemistry and habitability.
2. Methods
Four types of aqueous solutions were prepared at concentrations of 0.1 wt%: (a) iron (II) chloride, (b) iron (III) chloride, (c) iron (II) sulfate, and (d) iron (III) sulfate. Molar concentrations are [FeCl2] = 7.9 × 10−2 mol L−1, [FeCl3] = 6.2 × 10−2 mol L−1, [FeSO4] = 6.6 × 10−2 mol L−1, and [Fe2(SO4)3] = 2.5 × 10−2 mol L−1. Additionally, iron (III) sulfate was measured at additional concentrations (1, 0.01, 0.005, and 0.0001 wt%), and its detection limit in cation mode was determined by decreasing its concentration until the signal was not detectable anymore. The solutions were prepared only a few minutes before their measurement, to minimize oxidation reactions of Fe2+ to Fe3+. The salts were measured in solutions of both argon-sparged deionized water and nonsparged deionized water, and no major differences were observed in the mass spectra (nor in the solutions' colors). The spectra presented here are therefore those of salts dissolved in nonsparged deionized water. The pH values of the solutions (measured at 20°C) are pH = 3.9 for iron (II) chloride, pH = 2.2 for iron (III) chloride, pH = 4.9 for iron (II) sulfate, and pH = 2.5 for iron (III) sulfate.
The solutions were measured with LILBID coupled to ToF MS (Figure 1) in a vacuum chamber (5 × 10−5 mbar). The aqueous sample solutions were injected in a micron-sized liquid water beam irradiated by a pulsed infrared laser (λ = 2840 nm, energy up to 4mJ), which desorbs analyte molecules in a thermal explosion. This process is analogous to the impact ionization of dust grains in space, when those impact the metal targets of space-borne mass spectrometers at hypervelocities (i.e., >1 km s−1; Postberg et al. 2009; Klenner et al. 2019). In both cases (i.e., LILBID and impact ionization in space), a cloud of neutral and ionized species is created from the sample/dust grain, and the fraction of ionized species is measured by ToF MS. The LILBID ionization method tends to produce singly charged ions. Different impact speeds of the ice grains onto space-borne mass spectrometers are simulated in LILBID by a delayed extraction of the ions: ions of specific velocities are selected thanks to a repeller electrode switched on after a defined delay time, preventing ions arriving later than the delay time from entering the mass spectrometer detector. Different impact speeds of the dust grains, ranging from 3 to >20 km s−1, can thus be simulated by adjusting the extraction delay time and the laser's power intensity (Klenner et al. 2019).
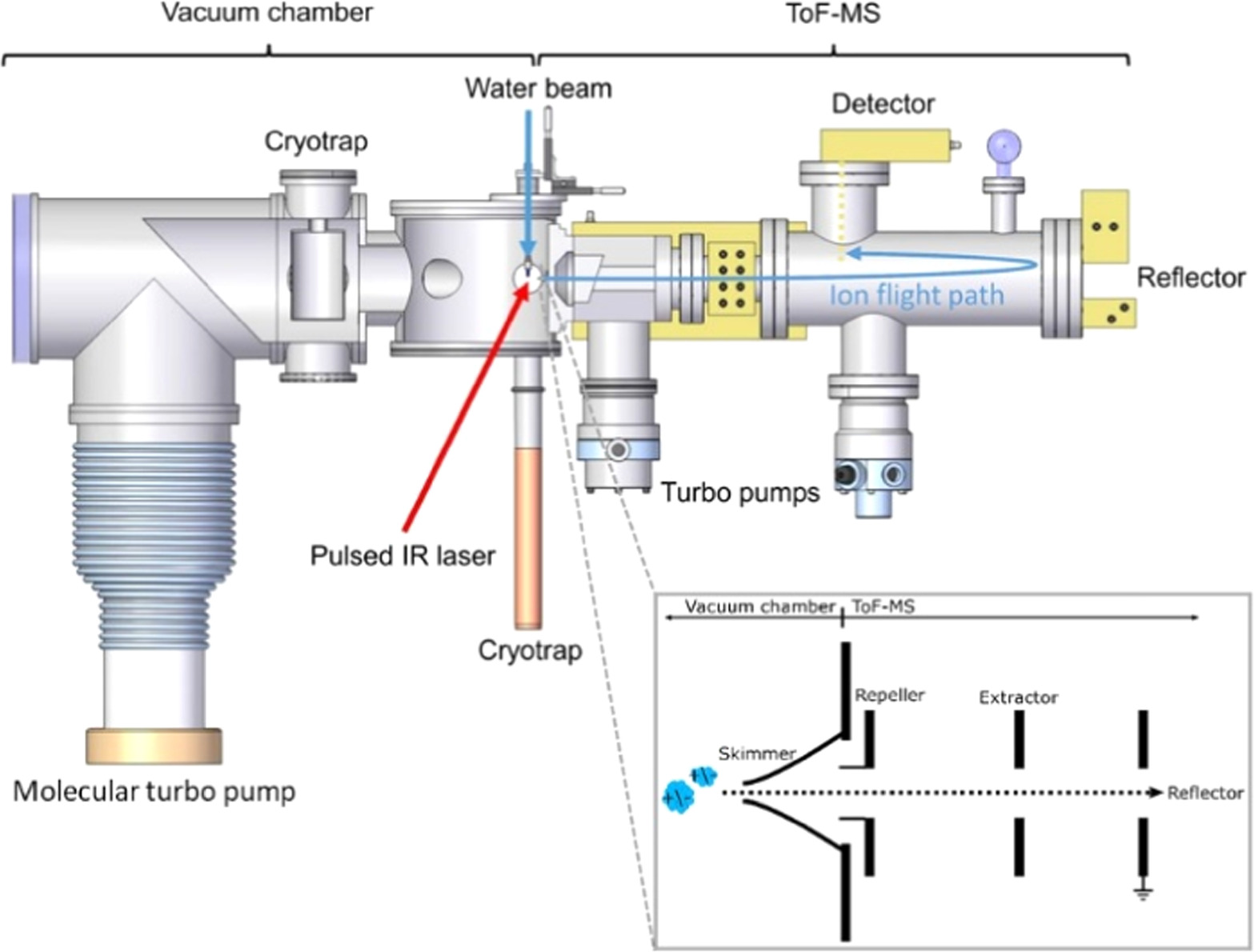
Figure 1. The LILBID laboratory setup coupled to a ToF mass spectrometer, reproducing the impact ionization mass spectra of ice grains recorded in space (figure reproduced from Klenner et al. 2019). The principle of delayed extraction of the ions as a function of their initial velocities is shown in the lower right inset.
Download figure:
Standard image High-resolution imageHere the samples were measured with LILBID in both cation and anion mode, rendering complementary mass spectra, with a mass resolution of 600–800 m/Δm. Each recorded spectrum was an average of 300 individual spectra, co-added to improve the signal-to-noise ratio, and was both baseline corrected and recalibrated. The LILBID setup was calibrated before each measurement session. The recorded mass spectra are stored in a database 5 (Klenner et al. 2022) in which LILBID analog data for impact ionization mass spectrometers are available.
3. Results
Both cations and anions bearing Fe2+ and/or Fe3+ were identified in the recorded LILBID mass spectra of iron (II) and iron (III) chlorides and sulfates (Figures 2–4 and A1–A4). Distinctive spectral characteristics are highlighted in Table 1 for each type of sample in both ion modes and discussed in detail in the following subsections. Peaks corresponding to Fe-bearing and Cl-bearing cations show respectively characteristic Fe isotope patterns (54Fe, 56Fe, and 57Fe with intensities of 5.82%, 91.66%, and 2.19%) and Cl isotope patterns (35Cl and 37Cl with intensities of 75.87% and 24.22%). Isotopic patterns were used to resolve ambiguities in identifying peaks (e.g., Figure 2(a)).

Figure 2. LILBID cation mass spectra recorded with a delay time of 5.1 μs for (a) iron (II) chloride (FeCl2), with an example of the iron isotope pattern for [Fe2+ (OH)]+, and (b) iron (III) chloride (FeCl3). In the spectrum in panel (b), two distinct peaks are observed at m/z 73 at a similar intensity, corresponding to a pure water cluster [H3O(H2O)3]+ and to [Fe2+(OH)]+ (Figure A2).
Download figure:
Standard image High-resolution imageTable 1. Characteristic Peaks for Fe2+ and Fe3+ Chlorides and Sulfates, Highlighting Distinctive Spectral Appearance between the Fe2+ and Fe3+ Salt Solutions
| Chlorides | Sulfates | |||
|---|---|---|---|---|
| Fe2+ | Fe3+ | Fe2+ | Fe3+ | |
| (FeCl2) | (FeCl3) | (FeSO4) | (Fe2[SO4]3) | |
| Characteristic peak(s) in cation mode | [aFe2+ (OH)b ]+ e.g., m/z 73 [Fe2+ (OH)]+, m/z 163 [2Fe2+ (OH)3]+ | [aFe2+ (OH)b ]+ e.g., m/z 73 [Fe2+ (OH)]+, m/z 163 [2Fe2+ (OH)3]+ [aFe3+ (OH)b ]+ e.g., m/z 90 [Fe3+ (OH)2]+, m/z 197 [2Fe3+ (OH)5]+ [Fe2+ Fe3+ (OH)4]+ m/z 180 | [aFe2+ (OH)b ]+ e.g., m/z 73 [Fe2+ (OH)]+, m/z 163 [2Fe2+ (OH)3]+ | [aFe2+ (OH)b ]+ e.g., m/z 73 [Fe2+ (OH)]+, m/z 163 [2Fe2+ (OH)3]+ [aFe3+ (OH)b ]+ e.g., m/z 90 [Fe3+ (OH)2]+, m/z 197 [2Fe3+ (OH)5]+ [Fe2+ Fe3+ (OH)4]+ m/z 180 |
| Characteristic peak(s) in anion mode | [aFe2+ (OH)b Clc ]− e.g., m/z 107 [Fe2+ (OH)3]−, m/z 125 [Fe2+ (OH)2 Cl]−, m/z 143 [Fe2+ (OH) Cl2]−, m/z 161 [Fe2+ Cl3]−, m/z 197 [2Fe2+ (OH)5]− | [aFe3+ (OH)b Clc ]− e.g., m/z 124 [Fe3+ (OH)4]−, m/z 142 [Fe3+ (OH)3 Cl]−, m/z 160 [Fe3+ (OH)2 Cl2]−, m/z 231 [2Fe3+ (OH)7]− | [aFe2+ (SO4)b (OH)c ]− e.g., m/z 169 [Fe2+ (SO4) (OH)]−, m/z 321 [2Fe2+ (SO4)2 (OH)]− | [aFe3+ (SO4)b (OH)c ]− e.g., m/z 186 [Fe3+ (SO4) (OH)2]−, m/z 248 [Fe3+ (SO4)2]− [aFe3+ (SO4)b (OH)c -H]− e.g., m/z 168 [Fe3+ (SO4) (OH) -H]−, m/z 275 [2Fe3+ (SO4) (OH)4 -H]−, m/z 337 [2Fe3+ (SO4)2 (OH)2 -H]− |
Note. Interestingly, in cation mode Fe (iii) salts show characteristic molecular cations related to both Fe2+ and Fe3+. Bold entries are the general formulas of the ions.
Download table as: ASCIITypeset image
3.1. Cation Mode Spectra
3.1.1. Iron (II) Chloride (Figures 2(a) and A1)
In the cation mass spectrum of Fe (ii) chloride solution (Figure 2), [Fe2+ (OH)]+, [2Fe2+ (OH)3]+ cations, and water cluster peaks corresponding to [aFe2+ (OH)b (H2O)c ]+ cations are highly prominent (with a > 0, b > 0, and c ≥ 0). Minor peaks at m/z 90 and 108 are attributed to [Fe3+ (OH)2]+ and [Fe3+ (OH)2(H2O)]+ cations. These peaks vanish completely in the cation mass spectra of the same solution at lower delay time (Figure A1). The low intensities of these characteristic peaks indicate very low concentrations of Fe3+ in this sample, as compared to Fe2+.
3.1.2. Iron (III) Chloride (Figure 2(b))
In the cation mass spectra of Fe (iii) chloride solution (Figure 2), ions bearing Fe2+ and/or Fe3+ were detected. Both [Fe2+(OH)]+ (interfering with a pure water cluster peak, as seen in Figure A2) and [Fe3+(OH)2]+ cations are highly prominent. Cations identified as [2Fe2+(OH)3]+, [2Fe3+(OH)5]+, and water cluster peaks [aFe2+(OH)b (H2O)c ]+ and [aFe3+(OH)b (H2O)c ]+ were detected. [aFe3+(OH)b (H2O)c ]+ cations are drastically increased compared to the Fe (ii) solution and—in contrast to the Fe (ii) solution—are still present at a lower delay time, indicating a much higher concentration of Fe3+ in this sample. Cations including both Fe2+ and Fe3+, i.e., [Fe2+Fe3+(OH)4 (H2O)a ]+, were detected at lower intensity.
3.1.3. Iron (II) Sulfate (Figure A3)
The cation mass spectrum of iron (II) sulfate (Figure A3) is quite similar to those of iron (II) chloride (Figure 2), in the sense that in both spectra [Fe2+ (OH)]+ cations show high intensities, together with [2Fe2+ (OH)3]+ and [aFe2+ (OH)b (H2O)c ]+ water clusters peaks and small [Fe3+ (OH)2 (H2O)a ]+ peaks. In the cation mass spectra of Fe (ii) sulfate (Figure A3), a small number of additional cations were detected at lower intensities: [Fe2+ Fe3+ (OH)4]+ (m/z 180) and unidentified peaks at m/z 269 and m/z 307.
3.1.4. Iron (III) Sulfate (Figure A4)
Similarly to the Fe (ii) sulfate, the cation mass spectrum of Fe (iii) sulfate (Figure A4) shares many similarities with the respective chloride (Figure 2): in both spectra, (1) [Fe2+(OH)]+ and [Fe3+(OH)2]+ cations are highly prominent, as well as their water cluster peaks [Fe2+(OH) (H2O)a ]+ and [Fe3+(OH)2(H2O)a ]+; (2) [2Fe2+(OH)3]+, [2Fe3+(OH)5]+, and [Fe2+Fe3+(OH)4]+ cations are detected at lower intensities; (3) water clusters are detected for most species (i.e., [aFe2+(OH)b (H2O)c ]+, [aFe3+(OH)b (H2O)c ]+, [aFe2+ bFe3+ (OH)c (H2O)d ]+); and (4) a peak at m/z 286 was identified as [3Fe3+(OH)8-H2O]+ cations. In the cation mass spectra of Fe (iii) sulfate (Figure A4), a few additional cations are detected: [2Fe3+(OH) (SO4)2]+ at m/z 304, [2Fe3+(OH) (SO4)2]+ at m/z 321, and unidentified peaks at m/z 268 and 269. Additionally, the detection limit of Fe (iii) sulfate in cation mode was determined to be 0.0001 wt%, i.e., 2.5 × 10−6 mol L−1.
3.2. Anion Mode Spectra
In contrast to positive mode, the negative-mode spectra show fundamental differences between sulfate and chloride solutions.
3.2.1. Iron (II) Chloride (Figures 3(a) and A5)
In the anion mass spectra of Fe (ii) chloride (Figure 3), [Fe2+(OH)a Clb ]− anions are detected at very prominent intensities, and [2Fe2+ (OH)a Clb ]− and [3Fe2+ (OH)a Clb ]− anions at lower intensity. Peaks at m/z 125, 143, and 161 could also be a water cluster series of [Fe2+ (OH)3]− (m/z 107), but the presence of smaller peaks at m/z 127, 145, 163, and 165 confirms the presence of chlorine atoms in those anions. A small peak corresponding to [Fe3+ (OH)4]− was detected at m/z 124 (Figure 3), but it was not detected at lower delay time (Figure A5), indicating a very low concentration of Fe3+ in this sample.

Figure 3. LILBID anion mass spectra recorded with a delay time of 6.0 μs for (a) iron (II) chloride (FeCl2) and (b) iron (III) chloride (FeCl3).
Download figure:
Standard image High-resolution image3.2.2. Iron (III) Chloride (Figure 3(b))
In the anion mass spectra of Fe (iii) chloride (Figure 3), [Fe3+(OH)a Clb ]− anions are detected at prominent intensities, and [2Fe3+(OH)a Clb ]− and [3Fe3+ (OH)10]− anions at lower intensities. [Fe2+ (OH)a Clb ]− anions were also detected, but at lower intensities than [Fe3+ (OH)a Clb ]− anions.
3.2.3. Iron (II) Sulfate (Figure 4(a))
The anion mass spectra of iron (II) sulfate (Figure 4) show prominent sulfate peaks [HSO4 (H2O)a ]− and Fe2+-bearing anions: highly prominent [Fe2+(SO4) (OH)]− anions, as well as smaller [2Fe3+(SO4)2 (OH)]− peaks. Some smaller peaks were identified as Fe3+-bearing species: [aFe3+(SO4) (OH)b -cH]− anions.

Figure 4. LILBID anion mass spectra recorded with a delay time of 5.4 μs for (a) iron (II) sulfate (FeSO4) and (b) iron (III) sulfate (Fe2[SO4]3).
Download figure:
Standard image High-resolution image3.2.4. Iron (III) Sulfate (Figure 4(b))
In the anion mass spectra of iron (III) sulfate (Figure 4), [aFe3+(SO4)b (OH)c -dH]− anions are detected at high intensity, as well as [Fe2+(SO4) (OH)]− anions at m/z 169.
4. Discussion
4.1. Spectral Analysis
While our work focuses on applications for space-borne instruments, previous laboratory work by Charvat & Abel (2007) and Mayer et al. (2022) performed comparable experiments and showed different applications and mechanistic details of laser-assisted liquid-phase dispersion MS when analyzing iron at different oxidation states. Both studies have highlighted the possibility of measuring oxidation states of ions in complexes with IR laser desorption MS. In these two papers, aqueous solutions containing Fe in different oxidation states (in both organic and inorganic complexes) were measured. It was shown that IR laser desorption of ions combined with ToF MS detects the "situation" (concentrations of species) in solution, i.e., the solution concentration with a linear response over several orders of magnitude. This result is of prime importance, as it shows that the method is suitable to measure Fe ion concentrations in solutions and/or in the condensed phase (e.g., ice), making it a powerful technique to work in analog experiments also for ions in different oxidation states. Other techniques such as electrospray ionization (ESI) and UV-matrix-assisted laser desorption ionization (MALDI) change oxidation states through electrochemistry at the ESI tip or through electron transfer and plasma chemistry during ablation. Still other MS methods obtained some results comparable to ours, which must be treated with caution in the context of analog experiments for space missions. Hellman et al. (2006) measured FeCl3 (0.1mM) in aqueous solution with ESI MS and identified a range of [Fea Ob (OH)c Cld ] ions, including species containing both Fe2+ and Fe3+. In cation mode, they detected [Fe3+Cl2]+ (m/z 126) and [Fe2+ 2(OH) Cl2]+ (m/z 160) cations. We have not detected these two species in every cation mass spectrum of Fe2+ or Fe3+ chlorides, despite the presence of a peak at m/z 126 (Figure 2), which has been attributed to [Fe3+(OH)2(H2O)2]+ owing to the lack of a characteristic 37Cl isotopologue peak at m/z 128. In anion mode, Hellmann et al. (2006) detected [Fe2+ Cl3]− (m/z 161), [2Fe2+ Cl5]− (m/z 287), [Fe3+ (OH) Cl3]− (m/z 178), and [Fe3+ Cl4]− (m/z 196) anions, species that we also identified (Figure 3). Radisavljević et al. (2013) studied FeCl3 with MALDI and laser desorption ionization ToF MS and also identified [Fe2+ Cl3]− and [Fe3+ Cl4]− anions. The hydrolysis of Fe (ii) in water leads to an acid solution where [Fe(H2O)6]2+ is the predominant species, and another hydrolysis product is [Fe(OH)3]− (Guimarães et al. 2007), which we observe at high intensity in the anion mass spectra of Fe (ii) and Fe (iii) chlorides (Figure 3).
Moreover, our present results show that it is possible to detect and discriminate Fe2+ from Fe3+ with impact ionization mass spectrometers such as SUDA. The determination of the oxidation state (II or III) of iron is done by the identification of Fe2+- and/or Fe3+-bearing ions at different relative intensities. While Fe2+ and Fe3+ have practically the same mass, they form different characteristic clusters owing to their different charges. While peaks from Fe2+ species appear in both Fe (ii) and Fe (iii) samples, major peaks bearing Fe3+, such as [Fe3+ (OH)2]+, [Fe3+ (OH)a Clb ]−, and [aFe3+(SO4)b (OH)c -dH]−, only appear in Fe (iii) samples and thus are particularly important to discriminate between the two oxidation states of iron (Figures 2, 3, 4). Our results, combined with those of Mayer et al. (2022) showing the complexation of iron ions in different organic complexes, suggest that organic clusters with Fe2+ or with Fe3+ might also display characteristic spectral differences in SUDA-type mass spectra.
In the mass spectra of Fe (iii) chlorides and Fe (iii) sulfates, we observe iron in two oxidation states (Fe2+ and Fe3+), although we used only Fe3+ salts. This was also previously observed by Mayer et al. (2022), who observed Fe(I), Fe (ii), and Fe (iii) in solutions of Fe (iii) with organic complex partners (e.g., oxalate). This can be explained by the fact that Fe (iii) is not very stable in solution (it is a strong oxidation agent). A reduction of Fe3+ to Fe2+ upon laser-induced desorption and ionization has been previously observed in MALDI-MS with iron (III) (Radisavljević et al. 2013) and with other metal ions such as Cu (Zhang et al. 2003); however, contrary to our ionization method, in MALDI the oxidation state can change through electrochemistry at the ESI tip or through electron transfer and plasma chemistry during ablation.
We find that cation mass spectra of both Fe (ii) and Fe (iii) chloride are quite similar to those from respective sulfates (Figures 2, A3, A4). This result was expected since similar cations are present in those samples. An important lesson for impact ionization mass spectrometers is that the anion mode is much better at discriminating sulfates from chlorides, even in small quantities (Figures 2–4 and A1–A5); therefore, SUDA's ability to use both ion modes provides great improvement over CDA, which only was able to detect cations. Indeed, in contrast to positive mode, the negative-mode spectra show fundamentally different spectra between sulfate and chloride solutions. However, the oxidation state of iron can be detected in both ion modes.
Several ionic species detected in this work have also been described in previous MS experiments. In Napoleoni et al. (2023b), sulfate anions were identified with the LILBID technique, in the anion mass spectra of both magnesium sulfates (MgSO4) and sulfuric acid (H2SO4). We here observe similar sulfate anions, such as [SO3]− and [HSO4]− (Figures 3 and 4). We conclude that anion spectra always show characteristic sulfate peaks, regardless of the cation these were previously bound to, together with unique molecular peaks that allow the identification of such cations.
4.2. Implications for Europa and Enceladus
Iron could be present as soluble ions in the oceans of Europa and Enceladus over a wide range of pH (0–10) and oxidation states (Figure 5). Soluble iron-bearing species could be transported from depths to the surface of the ocean, helped by the buoyancy of hydrothermal plumes of hot fluids (Schoenfeld et al. 2023) containing metal species and/or upwelling bubbles of volatile gases (Postberg et al. 2018b). The detection of Fe2+ and/or Fe3+on Enceladus's and Europa's surfaces would therefore have implications for the characterization of their subsurface oceans, potentially including hydrothermal systems, but surface processes could also play an important role. Both Fe2+ and Fe3+ are expected to be present on hydrothermal vent minerals and released as ions into the ocean by hydrolysis of these minerals. While (some) Fe3+ may cross the ocean and reach the surface in the same oxidation state, Fe2+ would likely be transformed to Fe3+ during its journey to the exterior of the moon because the redox gradients created between the reduced core's interior and the more oxidized subsurface ocean entail that all ionic ferrous compounds are subject to mildly oxidizing conditions. Thus, the detection of Fe2+ ions (or simultaneous presence of both ferrous and ferric ions) in freshly ejected material might hint at the presence of protective mechanisms taking place at the hydrothermal vents and hydrothermal plumes, preventing the oxidation of Fe2+ ions. These protective mechanisms may be achieved by complexation of Fe2+ by organic compounds, as observed in terrestrial hydrothermal systems (Toner et al. 2009; Sander & Koschinsky 2011). Besides, a lack of detection of Fe2+ in the surface of these moons does not imply the absence of Fe2+ in the subsurface ocean of the moon.

Figure 5. Example of a Pourbaix diagram of the Fe–O–H system. The driving variables are the pH and reduction potential Eh; the latter values are expressed relative to the standard hydrogen electrode. Eh is a measure of the oxidation state of the system. Within a region demarcated by black lines, the indicated species/mineral is the most stable form of iron. The aqueous species regions are shaded in color to highlight that they are of greatest interest in the present application of measuring soluble salts from samples of ocean water. The sizes of their regions depend on the activity of the species. Here they are assumed to have activities of 10−6—close to our experimentally determined detection limit for ferric sulfate. Dashed lines show cases of strongly oxidized and reduced conditions as points of reference. This diagram was constructed using thermodynamic data from the SUPCRT database (Helgeson 1978; Shock et al. 1997), together with the Nernst equation. Note: Fe2O3 = hematite, Fe3O4 = magnetite, Fe0 = native iron.
Download figure:
Standard image High-resolution imageGenerally, it is important to remember that the oxidation state of Fe in solution, including in the subsurface oceans of icy moons, is highly dependent on the surrounding conditions, such as the dissolved O2 or H2 and pH (Figure 5). Fe (iii) in solution can act as an oxidizing agent, which makes both Fe (ii) and Fe (iii) ions available in aqueous solution. A detection of the Fe (ii)/Fe (iii) ratio, combined with related modeling work, might be an effective tool to provide information about the geochemistry of subsurface oceans. It may also bring information about the timescales of exposition of iron-bearing material to surface irradiation. In the case of plumes ejecting frozen material to high altitudes, SUDA could sample material that has been exposed to radiation for less than a few minutes, and thus it may be possible to sample Fe2+ in concentrations accurately reflecting those present in Europa's subsurface liquid reservoirs. For Enceladus specifically, fresh plume material—as sampled by the Cassini spacecraft in the vicinity of Enceladus—should not undergo considerable modifications of the oxidation state due to radiation. On the contrary, the E-ring of Saturn is mainly fed by the Enceladus plume (Postberg et al. 2008), so its material might have been exposed to space weathering (Hendrix et al. 2018) for much longer timescales, up to hundreds of years.
On Europa specifically, the surface is highly oxidizing owing to radiation; thus, if Fe2+ reaches the surface, it would be quickly oxidized—forming Fe3+, or reacting with water or other compounds in its environment. Therefore, if Fe2+ is detected in surface ejecta by spacecraft measurement, it is an indicator that the ejecta is coming from fresh surface deposits that have not been exposed to radiation for a long time. To our knowledge, the timescales of Fe (ii) oxidation have not been accurately estimated for Fe (ii) in water ice at Europa's surface conditions. Hibbitts et al. (2019) irradiated ferrous sulfate (FeSO4, without water-ice matrix) with doses of 40 keV electrons equivalent to exposure of months to years on Europa's surface. They observed a darkening of the sulfate, which was possibly attributed to the oxidation of the ferrous iron to ferric iron. As compared to Fe (ii) without water-ice matrix, Fe (ii) irradiated in water-ice matrix might undergo much faster oxidation owing to possible interactions with the abundant strong oxidants formed by irradiation of water ice at Europa surface conditions (e.g., H2O2; Hand & Carlson 2011). However, in an icy medium, oxidation of iron by oxidants such as H2O2 is limited by both low diffusion rates and low reactivity at cold temperatures; this reaction therefore requires a liquid medium to be efficient and occurs at a timescale of minutes at 4°C (Croot & Laan 2002), but this probably takes much longer in cold ice.
The detection and quantification of Fe2+ and/or Fe3+ in ejected ice grains could constrain the geochemistry of the subsurface oceans of Enceladus and Europa. The pH of oceanic water plays a decisive role in the solubility of ferric and ferrous ions (Figure 5). Ferrous ions would have a substantial dissolved concentration (∼1 μM) if the ocean water is relatively reduced and the pH is neutral to low. Ferric iron has a much lower solubility unless the pH is quite low. The pH of Enceladus, currently thought to be mildly alkaline (Glein et al. 2018), may therefore not allow for a high solubility of any form of iron. As shown in Figure 6, the characterization (or a lack of detection) of iron would have implications for the geochemistry of subsurface oceans, depending on the origin (plume or surface) of the sampled ice grains. For example, a detection of Fe (iii)-dominated plume ice grains (i.e., with a direct origin from subsurface fluids) would indicate an oxidized, acidic ocean (i.e., lower left of Figure 5). Besides iron, a detection of aluminum ions (Al3+) would hint at an acidic ocean composition, as this ion only occurs under acidic conditions. Other ions, such as Mg ions, could also be used similarly to get indications about the pH range of the original liquid source.

Figure 6. Simplified interpretations of the (non)detection and characterization of iron in ice grains from ocean worlds with SUDA-type instruments. Three scenarios are considered: (a) detection of Fe (ii)-dominated grains, (b) detection of significant Fe (iii) in ice grains, and (c) no Fe detection. In addition to iron, the detection of aluminum ions (Al3+) in ice grains would support a salt source from an acidic ocean composition. In both scenarios (a) and (b), the case of ice grains originating from a plume (i.e., fresh surface material) is distinct from ice grains originating from the surface, where older material has likely undergone oxidation. In the case of surface material, the age of this material may be constrained by investigating its surface appearance, including geological features. In case (c), the characterization of the pH (e.g., by detection in the grains of other ions, like hydroxide) could put constraints on the oxidation state in the case of a circumneutral pH, while the case of a low pH would hint at a nonoceanic fluid origin (e.g., a perched melt of water and sulfuric acid) of the ice grains (as seen in Figure 5, iron is soluble at low pH, whatever the oxidation state of the ocean). In the case of a high-pH ocean, iron would not be present as ions (but incorporated in very poorly soluble minerals; Figure 5) and therefore not detectable in ice grains.
Download figure:
Standard image High-resolution imageIn Figure 6, we assumed that the iron in the detected ice grains once resided in liquid water (i.e., the sampled source material—either plume or surface ice—developed from a liquid). However, another possible scenario would be that Fe ions have never been dissolved in a liquid and occur from the depositions of meteoritic material such as interplanetary dust particles. In this putative case, Fe might be associated with Ni.
Currently, Fe-bearing species have not been detected in ice grains from Enceladus with the CDA instrument. However, due to the large amount of CDA data yet to analyze, it may be that Fe-bearing species are only present in a small number of ice grains sampled by Cassini (similarly to phosphates; Postberg et al. 2023) and that the spectra showing Fe-bearing species have just not been characterized yet. It may otherwise hint that iron is preferentially present as insoluble hydroxide compounds, thus suggesting that the pH tends to more alkaline values (Figure 5), in accordance with our current understanding of the pH of Enceladus's ocean. On the contrary, a finding of Fe-bearing species in Enceladan ice grains would be an indication of lower pH values. Even if not detected in Enceladean ice grains, establishing the detection limit of Fe2+- and Fe3+-bearing salts in Enceladus ice grains would yield a lower limit for their concentrations and therefore constrain the solubility of these ions and thus the pH. Some complementary measurements of other elements could bring additional constraints, such as Al3+ in chloride or sulfate salts (Figure 6).
While salts were thoroughly studied in previous LILBID experiments (Charvat & Abel 2007; Postberg et al. 2009; Mayer et al. 2022; Napoleoni et al. 2023a, 2023b; Postberg et al. 2023), this is the first time that a metallic element with multiple oxidation states was analyzed with LILBID serving as an analog experiment relevant for Europa and Enceladus missions. We showed that Fe2+ and Fe3+ display characteristic mass patterns, due to the formation of different charged clusters and molecular aggregates, and expect similar results for other transition metals with oxidation states comparable to those of iron, e.g., nickel (oxidation state of +2 is most common, but +3 occurs as well), which could also be relevant for the origin of life on icy moons (Camprubi et al. 2017). This work is also a first step toward further characterization of different transition metals with a greater number of possible oxidation states, such as manganese (oxidation states from +2 to +7). Besides salts, our results are thought to be applicable to other Fe-bearing phases such as mineral phases, because the formation of Fe(OH)n species from such minerals in a water matrix on the instrument post-impact should be common.
5. Conclusion
This study indicates that impact ionization mass spectrometers—like SUDA—could be useful tools for the characterization of the oxidation state of subsurface oceans by quantification of iron-bearing salts in Enceladus's and Europa's ice grains. In both our analog experiment and future flight data (e.g., SUDA mass spectra), the intensities of Fe-bearing ions of different oxidation states and the isotope distribution patterns are informative features that can be used to determine the presence and oxidation state of iron-bearing compounds.
While Fe-rich samples have been measured with the laser dispersion MS before (Charvat & Abel 2007; Mayer et al. 2022), our experiments complement these previous results, showing that LILBID appears as a reliable technique to characterize the oxidation state of the analyte (contrary to ESI and MALDI, where the oxidation state is possibly changed during the ionization process). This is an especially important result, as detecting oxidation states via MS is very challenging. Moreover, the consistency of our results with those of Charvat & Abel (2007) and Mayer et al. (2022) shows that our experiments can specifically serve as relevant analogs for ice grain analysis emitted by icy moons in future and past space missions.
The LILBID spectra recorded in this work could allow the detection of iron and the characterization of its oxidation state on the surfaces of icy moons and potentially from their subsurface oceans. The recorded LILBID spectra are stored in a database (Klenner et al. 2022) providing analog data for space missions targeting dust grains from icy moons, such as Europa Clipper and potential future Enceladus missions. Future laboratory work will investigate whether characteristic spectral differences between organic clusters with Fe2+ or with Fe3+ could also be detected (Mayer et al. 2022).
The characterization of the oxidation states of Fe in icy moons' ice grains with SUDA-type instruments may provide major insight into the geochemistry of subsurface oceans. A quantification of the oxidation states of iron for Enceladus or Europa may be used to constrain the concentrations of reduced iron in the ocean and thus the likelihood of different models of redox chemistry in the ocean (Ray et al. 2021). In perspective, such results would also constrain potential metabolic pathways, such as iron reduction metabolisms (Roche et al. 2023), that may be used by possible extant life in icy moons' oceans, where the necessary ingredients and environmental conditions favorable for sustaining life seem to be present. Future space missions to icy moons, such as the upcoming Europa Clipper, may detect the chemical signatures of a biogeochemical iron cycling, providing information about extant life that may have started in extraterrestrial hydrothermal systems.
Acknowledgments
The experiments presented in this work were conducted at Freie Universität Berlin and supported by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program by the Consolidator Grant 724908 Habitat-OASIS. B.A. thanks the German Science Foundation (DFG) for funding through grant AB 63/25-1. We would like to acknowledge and thank Rene Reviol for his technical support and aiding with the acquisition of materials required for the experiments, as well as Taylor Price (University of Minnesota) for valuable discussion about iron on ocean worlds.
Appendix:

Figure A1. LILBID cation mass spectra of iron (II) chloride (FeCl2), recorded with a delay time of 4.5 μs.
Download figure:
Standard image High-resolution image
Figure A2. Zoomed-in section of the LILBID cation mass spectra of iron (III) chloride (FeCl3), recorded with a delay time of 5.1 μs. Two distinct peaks are observed at m/z 73 at a similar intensity, corresponding to a pure water cluster [H3O(H2O)3]+ (m/z 73.05) and to [Fe2+(OH)]+ (m/z 72.94). To resolve the two peaks, a mass resolution of about 500 is required.
Download figure:
Standard image High-resolution image
Figure A3. LILBID cation mass spectra of iron (II) sulfate (FeSO4), recorded with a delay time of 5.0 μs.
Download figure:
Standard image High-resolution image
Figure A4. LILBID cation mass spectra of iron (III) sulfate (Fe3+ 2[SO4]3), recorded with a delay time of 5.0 μs.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure A5. LILBID anion mass spectra of iron (II) chloride (FeCl2), recorded with a delay time of 5.3 μs.
Download figure:
Standard image High-resolution image{kind=link}