



Abstract
The formation of nitrogen hydrides in the interstellar medium is initiated by the nearly thermoneutral reaction of N+ + H2 → NH+ + H. Here, we experimentally determine the enthalpy of this reaction using the principle of detailed balance from a measurement of the rate coefficient of the reverse reaction NH+ + H → N+ + H2. The measurements were carried out in a linear radiofrequency 22-pole trap combined with an effusive beam source of atomic hydrogen at temperatures between 10 and 100 K. The resulting ground-state energy difference (or reaction enthalpy at 0 K) of ΔE0 = (18 ± 4) meV confirms that there are no significant energy barriers on the reaction path.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
This work aims to resolve the longstanding question of the enthalpy of the nearly thermoneutral reaction
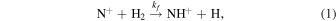
which initiates the formation of nitrogen hydrides in interstellar clouds (Le Gal et al. 2014; Gerin et al. 2016). We will evaluate the reaction enthalpy at 0 K, which is equal to the energy difference between the product and reactant ground states, ΔE0. This quantity is an essential parameter in calculations of state-specific NH+ formation rate coefficients in astrochemical models (Grozdanov et al. 2016; Gómez-Carrasco et al. 2022). At temperatures relevant to the chemistry of molecular clouds (Millar 2015), the temperature dependence of the rate coefficient, kf
, of this forward reaction can be represented by the Arrhenius dependence,  , with activation energy EA ≈ 19 meV (Marquette et al. 1985; Plašil et al. 2022). The presence of activation energy indicates that the reaction cross section has a certain threshold energy ET (Menzinger & Wolfgang 1969). However, neither the activation energies nor the threshold energies of cross sections can be directly identified with the reaction enthalpy, as the data can also be explained by the presence of energy barriers. To elucidate the issue, the variation of the activation energy with the internal excitation and isotopic composition of reactants has been studied in a number of experiments (Marquette et al. 1985, 1988; Gerlich 1993; Zymak et al. 2013; Plašil et al. 2022) and some have also deduced the threshold energy (Ervin & Armentrout 1987; Marquette et al. 1988; Gerlich 1993; Tosi et al. 1994). A notable result was obtained by Tosi et al. (1994), who observed that the change of the energy threshold due to deuteration cannot be explained by the different zero-point energies of the deuterated species, suggesting that the energy threshold is not purely due to reaction enthalpy and an energy barrier is involved. On the other hand, several studies (Adams & Smith 1985; Marquette et al. 1988; Plašil et al. 2022), have observed isotope effects consistent with the hypothesis of no barrier. The recent study by Plašil et al. (2022) shows that the available data are compatible with the hypothesis of no reaction barrier when only low-temperature thermal reaction rate coefficients with known error estimates are taken into account. The isotopic variation of activation energies in accordance with the hypothesis of the absence of energy barriers is a strong indication in favor of this hypothesis.
, with activation energy EA ≈ 19 meV (Marquette et al. 1985; Plašil et al. 2022). The presence of activation energy indicates that the reaction cross section has a certain threshold energy ET (Menzinger & Wolfgang 1969). However, neither the activation energies nor the threshold energies of cross sections can be directly identified with the reaction enthalpy, as the data can also be explained by the presence of energy barriers. To elucidate the issue, the variation of the activation energy with the internal excitation and isotopic composition of reactants has been studied in a number of experiments (Marquette et al. 1985, 1988; Gerlich 1993; Zymak et al. 2013; Plašil et al. 2022) and some have also deduced the threshold energy (Ervin & Armentrout 1987; Marquette et al. 1988; Gerlich 1993; Tosi et al. 1994). A notable result was obtained by Tosi et al. (1994), who observed that the change of the energy threshold due to deuteration cannot be explained by the different zero-point energies of the deuterated species, suggesting that the energy threshold is not purely due to reaction enthalpy and an energy barrier is involved. On the other hand, several studies (Adams & Smith 1985; Marquette et al. 1988; Plašil et al. 2022), have observed isotope effects consistent with the hypothesis of no barrier. The recent study by Plašil et al. (2022) shows that the available data are compatible with the hypothesis of no reaction barrier when only low-temperature thermal reaction rate coefficients with known error estimates are taken into account. The isotopic variation of activation energies in accordance with the hypothesis of the absence of energy barriers is a strong indication in favor of this hypothesis.
The reaction enthalpy can, in principle, be calculated as the difference between the H2 and NH+ dissociation energies. However, despite the recent theoretical and experimental works (Tarroni et al. 1997; Hübers et al. 2009; Shi et al. 2009; Lecointre et al. 2010; Beloy et al. 2011; Zhang et al. 2017; Yang et al. 2019; Ghosh et al. 2022; Gómez-Carrasco et al. 2022), the dissociation energy of NH+ has not been determined with sufficient precision yet and the most recent theoretical reaction studies still approximate the energy difference with the experimental activation energies (Gómez-Carrasco et al. 2022). We have therefore chosen an experimental approach to study the reaction endothermicity by measuring the rate coefficient kr of the reverse reaction
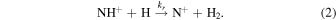
Knowing the forward and reverse thermal rate coefficients and accounting for all the relevant states of the NH2 + system (see Figure 1), we can calculate ΔE0 using the detailed balance principle.
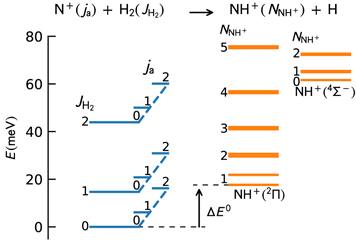
Figure 1. Energy levels of the NH2
+ system. The energies of the reactants as a function of the N+ fine-structure state (ja
) and H2 rotational state ( ) are indicated on the left. The energies of the products as a function of the NH+ rotational state are shown for the two lowest electronic states of NH+. The rotational states of NH+ are labeled by the rotational angular momentum quantum number
) are indicated on the left. The energies of the products as a function of the NH+ rotational state are shown for the two lowest electronic states of NH+. The rotational states of NH+ are labeled by the rotational angular momentum quantum number  . See the text for details of the internal structure calculations. The present value of ΔE0 = 17.6 meV is indicated in the figure.
. See the text for details of the internal structure calculations. The present value of ΔE0 = 17.6 meV is indicated in the figure.
Download figure:
Standard image High-resolution imageThe experiments were carried out in the Atomic Beam with 22-pole Trap instrument (AB-22PT; Gerlich 1992, 2012; Borodi et al. 2009; Plasil et al. 2011; Roučka et al. 2015). The NH+ ions were produced by electron bombardment from two different precursor gas mixtures in the storage ion source (Gerlich 1992)—either N2: H2 or NH3:He mixtures in pressure ratios of 1: 2 or 1: 10, respectively (with identical resulting rate coefficients within the error margin). After mass selection, the ions were injected into a linear 22-pole radiofrequency trap, where they were cooled by collisions with helium buffer gas. To freeze out the N2 or NH3 leaking from the ion source and to avoid parasitic reactions with these species, the trap was kept at temperatures T22PT < 30 K. Based on our previous studies of endothermic reactions (Mulin et al. 2015; Roučka et al. 2018; Plašil et al. 2022) and photodetachment spectroscopy (Plašil et al. 2023), we assume that neither the translational nor the internal temperatures of NH+ exceed the trap temperature by more than 10 K under the present operating conditions. We therefore denote both temperatures as  , which is given as
, which is given as  . The H atoms were produced in a radiofrequency discharge in pure H2 and they were passing through a cold nozzle (accommodator) with temperature Tacc in the range from 7 to 150 K. The H atom temperature TH is close to Tacc (Borodi et al. 2009) and we assume TH = Tacc. The atoms exit the accommodator in an effusive beam, which passes through the trap and can be blocked using a mechanical shutter.
. The H atoms were produced in a radiofrequency discharge in pure H2 and they were passing through a cold nozzle (accommodator) with temperature Tacc in the range from 7 to 150 K. The H atom temperature TH is close to Tacc (Borodi et al. 2009) and we assume TH = Tacc. The atoms exit the accommodator in an effusive beam, which passes through the trap and can be blocked using a mechanical shutter.
The effective translational temperature Tt of the collisions between H atoms with mass MH and NH+ ions with mass  is given as (Light et al. 1969; Gerlich & Horning 1992; Paul et al. 1995)
is given as (Light et al. 1969; Gerlich & Horning 1992; Paul et al. 1995)  . The most relevant parameter for determining the rate coefficients of endothermic reactions is the total available collisional energy—the sum of the translational and internal energies
. The most relevant parameter for determining the rate coefficients of endothermic reactions is the total available collisional energy—the sum of the translational and internal energies  . We therefore define the corresponding collisional temperature Tcoll through the following implicit formula,
. We therefore define the corresponding collisional temperature Tcoll through the following implicit formula,  , as the temperature of hypothetical thermal reactants with total collisional energy Ecoll.
, as the temperature of hypothetical thermal reactants with total collisional energy Ecoll.
The effective number density of H atoms, nH, was determined by chemical probing with CO2 + using the reaction CO2 + + H → HOC+ + O (Borodi et al. 2009). The nH calibration and reaction measurements were always carried out at similar values of the trap temperature, effective trapping potential, and accommodator temperature in order to maintain the same overlap of the H beam with the ion cloud and the level of dissociation (Roučka et al. 2015).
2. Results
The typical experimental data are shown in Figure 2. Every measurement is performed with the beam shutter open (H on) and closed (H off) to resolve the reactions with H atoms from other background reactions. Since our quadrupole mass filter cannot resolve ions with the same number of nucleons, there is also a minor amount of 15N+ isotope mixed in with the studied 14NH+ ions. The dominant loss process for the 14NH+ ions is the fast hydrogen abstraction reaction with the background H2 gas (Rednyk et al. 2019):
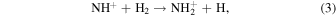
whereas the 15N+ are destroyed by the much slower reaction (1). This is manifested by the bi-exponential decay of the number of ions with m/z = 15. These different loss rates allow us to resolve 14NH+ from 15N+ in the data analysis. The loss of 15N+ on the timescale of our experiment is practically negligible and we assume that its reaction rate coefficient with H2 is equal to that of 14N+ (Zymak et al. 2013).

Figure 2. Measured numbers of trapped ions, Ni
, as a function of trapping time t, showing the loss of the 14NH+/15N+ ions (squares) and formation of the N+ ions (circles) in the trap. The curves represent least-squares fits of the data with a kinetic model. The data were measured with the H beam on (closed symbols and solid lines) and H beam off (open symbols and dashed lines). The fitted numbers of 14NH+ and 15N+ in the measurement with the H beam on are indicated by the dotted lines. The measurement was performed at ion and atom temperatures of  , TH = 75 K, with an H number density of nH = 3.2 × 107 cm−3.
, TH = 75 K, with an H number density of nH = 3.2 × 107 cm−3.
Download figure:
Standard image High-resolution imageBy least-squares fitting (Newville et al. 2014, 2020) of the measured data with a kinetic model that includes reactions (1), (2), and (3), we obtain the rates of N+ production from NH+ with the beam on and off (ron and roff, respectively), where roff is typically close to zero (see Figure 2). The production rates of N+ due to the interaction of NH+ with H atoms are calculated as r = ron − roff. The rate coefficients of the reverse reaction are finally obtained by dividing the rate with the H atom number density calibrated at equivalent conditions using chemical probing.
The measurements were performed at four H atom temperatures—7, 50, 75, and 100 K—and  varied from 15 to 30 K. Since the translational temperature Tt is predominantly determined by TH, we group the results by the accommodator temperature and calculate a weighted average of the measured reaction rate coefficients in each group. The averaged reverse reaction rate coefficients are plotted as a function of Tt and Tcoll in the upper panel of Figure 3. We also plot the data as a function of
varied from 15 to 30 K. Since the translational temperature Tt is predominantly determined by TH, we group the results by the accommodator temperature and calculate a weighted average of the measured reaction rate coefficients in each group. The averaged reverse reaction rate coefficients are plotted as a function of Tt and Tcoll in the upper panel of Figure 3. We also plot the data as a function of  (measurements were done mostly at ion temperatures
(measurements were done mostly at ion temperatures  and 27 K). The data point at the lowest Tt and Tcoll (TH = 7 K) has large statistical uncertainty due to the low number density of H atoms in these conditions. The reaction rate coefficients have no significant dependence on temperature. Hence, the reverse reaction has no significant activation energy.
and 27 K). The data point at the lowest Tt and Tcoll (TH = 7 K) has large statistical uncertainty due to the low number density of H atoms in these conditions. The reaction rate coefficients have no significant dependence on temperature. Hence, the reverse reaction has no significant activation energy.
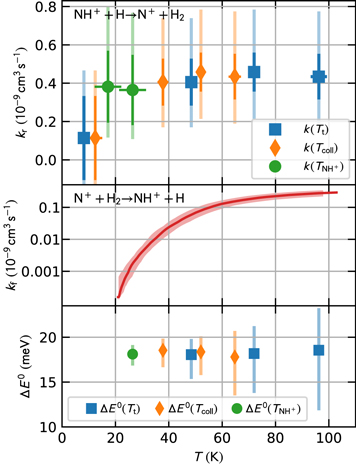
Figure 3. Upper panel: the measured rate coefficients of the reverse reaction as a function of translational, collisional, and NH+ internal temperatures. The inner error bars indicate the statistical errors, whereas the outer error bars also include the systematic uncertainty due to nH calibration. Middle panel: the rate coefficient of the forward reaction extracted from the data of Zymak et al. (2013). The systematic error of the data is indicated by the filled band. Lower panel: values of ΔE0 obtained from the detailed balance principle Equation (5). The overall (systematic + statistical) uncertainties obtained using interval arithmetic from the overall uncertainties of the input data are indicated by the error bars.
Download figure:
Standard image High-resolution image3. Endothermicity Analysis
To obtain quantitative information about the reaction enthalpy, we employ the principle of detailed balance. The full derivation of this principle from quantum mechanics can be found, e.g., in Henriksen & Hansen (2008), Light et al. (1969), and Sakurai & Napolitano (2010). For a general bimolecular reaction  , the forward and reverse reaction rate coefficients are related by the formula
, the forward and reverse reaction rate coefficients are related by the formula
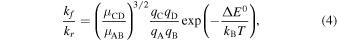
where qX denotes the internal partition sum of species X and μXY is the reduced mass of species X and Y (Light et al. 1969).
For H atoms, we take into account the nuclear spin contribution  (I = 1/2 for 1H) and the electron spin multiplicity
(I = 1/2 for 1H) and the electron spin multiplicity  of the 2S ground state, which leads to
of the 2S ground state, which leads to  . For N+ ions,
. For N+ ions,  , where
, where  (I = 1 for 14N; Fuller 1976) and we sum over the
(I = 1 for 14N; Fuller 1976) and we sum over the  fine-structure (FS) states
fine-structure (FS) states  , with FS energies
, with FS energies  (Kramida et al. 2020). The partition sum of H2 is calculated over the five lowest rotational states of the vibrational ground state, accounting for the different nuclear spin multiplicities gJ
of odd and even (ortho and para) rotational states,
(Kramida et al. 2020). The partition sum of H2 is calculated over the five lowest rotational states of the vibrational ground state, accounting for the different nuclear spin multiplicities gJ
of odd and even (ortho and para) rotational states,  , where gJ
= 1 and 3 for even and odd J, respectively. The H2 rotational energies EJ
are calculated from the molecular constants in Huber & Herzberg (1979). In the case of NH+, we consider the vibrational ground states of the two lowest electronic states, 2Π and 4Σ−, where we account for the rotational states with J ≤ 19/2 according to the effective Hamiltonian of Kawaguchi & Amano (1988). We checked that the neglect of the higher energy levels of NH+ and H2 does not influence the results of our analysis. The nuclear spin partition function of NH+ is the product of the hydrogen and nitrogen nuclear spin partition functions,
, where gJ
= 1 and 3 for even and odd J, respectively. The H2 rotational energies EJ
are calculated from the molecular constants in Huber & Herzberg (1979). In the case of NH+, we consider the vibrational ground states of the two lowest electronic states, 2Π and 4Σ−, where we account for the rotational states with J ≤ 19/2 according to the effective Hamiltonian of Kawaguchi & Amano (1988). We checked that the neglect of the higher energy levels of NH+ and H2 does not influence the results of our analysis. The nuclear spin partition function of NH+ is the product of the hydrogen and nitrogen nuclear spin partition functions,  .
.
When the partition sums are known, we can express ΔE0 from the measured forward and reverse reaction rate coefficients by applying Equation (4) to reaction (1) as
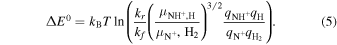
To evaluate kf , we first calculate the rate coefficients of reaction (1) with ortho- and para-H2 from the experimental data with normal and para-enriched H2 (Zymak et al. 2013), and we take kf as a weighted average of the state-specific reaction rate coefficients over the thermal population of H2 nuclear spin states. The systematic uncertainties are not provided by Zymak et al. (2013) and we account for the typical 20% uncertainty of pressure determination and temperature uncertainty defined as Tcoll = T22PT + (5 ± 5) K. The thermal rate coefficient of the forward reaction can be obtained down to approximately 20 K (see Figure 3). At lower temperatures, the rate coefficient for reaction with para-H2 is below the noise level in the measurements with para-enriched H2. Hence, the reaction enthalpy cannot be extracted from the present data obtained at 7 K accommodator temperature.
When calculating ΔE0 from Equation (5), we assumed that the temperature in the present experiment is equal to either Tt or Tcoll. This corresponds to two extreme cases: if we take T = Tt, we assume that the internal energy of NH+ is not relevant for promoting the (possibly endothermic) reverse reaction. Conversely, assuming T = Tcoll means that the internal energy is equivalent to translational energy in promoting the reverse reaction. For completeness, we also consider the case  , i.e., that only the internal energy is relevant.
, i.e., that only the internal energy is relevant.
As expected, the detailed balance analysis leads to positive ΔE0, i.e., the reverse reaction is exothermic. The obtained data shown in the lower panel of Figure 3 as a function of Tt, Tcoll, or  do not depend on temperature, in accordance with the fact that ΔE0 is a constant. Averaging the data obtained at different accommodator temperatures leads to
do not depend on temperature, in accordance with the fact that ΔE0 is a constant. Averaging the data obtained at different accommodator temperatures leads to  ,
,  , and
, and  when assuming T = Tt, T = Tcoll, and
when assuming T = Tt, T = Tcoll, and  , respectively. The averages are obtained as arithmetic means of the data and the stated error estimates represent the mean upper/lower systematic error bounds due to the systematic uncertainties (40% of nH and
, respectively. The averages are obtained as arithmetic means of the data and the stated error estimates represent the mean upper/lower systematic error bounds due to the systematic uncertainties (40% of nH and  ). Regardless of our assumptions on temperature, ΔE0 is contained between 13.8 and 21.4 meV, i.e., our experimental value of ΔE0 can be represented as ΔE0 = (17.6 ± 3.8) meV or, after rounding, as
). Regardless of our assumptions on temperature, ΔE0 is contained between 13.8 and 21.4 meV, i.e., our experimental value of ΔE0 can be represented as ΔE0 = (17.6 ± 3.8) meV or, after rounding, as
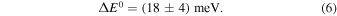
The vibrationless energy change ΔEe (Grozdanov et al. 2016; Plašil et al. 2022) can be calculated by correcting for the zero-point vibrational energies (ZPEs) of reactants and products. Subtracting the ZPE of NH+ (186.9 meV Colin 1989) and adding the ZPE of H2 (270.2 meV Huber & Herzberg 1979) leads to
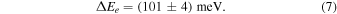
The resulting vibrationless energy change is consistent with ΔEe = (103 ± 3) meV obtained by Plašil et al. (2022) in the ion trap study of N+ + HD/D2 reactions under the assumption of no reaction barriers.
The measured value of the reaction enthalpy ΔE0 can be utilized to calculate the dissociation energy of NH+ in the 2Π ground state toward the N+ + H asymptote as
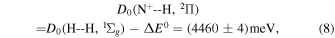
where the H2 dissociation energy is D0(H − H, 1Σg ) = 4478.1 meV (Liu et al. 2009). This value is in agreement with the theoretically calculated dissociation energy D0(N+–H, 2Π) = 4462 meV (Tarroni et al. 1997). The dissociation energy toward the N + H+ asymptote can be calculated from the known ionization potentials (IPs) of H and N (Kramida et al. 2020), resulting in D0(N–H+, 2Π) = D0(N+–H, 2Π) + IP(H) − IP(N) = (3525 ± 4) meV, in agreement with the experimental values (3531 ± 3) meV of Adams & Smith (1985) and (3524 ± 3) meV of Marquette et al. (1988), which were obtained from measurements of kf with para-enriched and normal H2, HD, and D2 with the assumption of no energy barrier. Since the process NH+ (2Π) → N (4S) + H+(1S) involves an electronic transition, some works report instead the adiabatic energy of dissociation from the NH+ (4Σ−) state (Tarroni et al. 1997; Amero & Vázquez 2005). This can be calculated as D0(N–H+, 4Σ−) = D0(N–H+, 2Π) − ΔEΣΠ = (3481 ± 4) meV, where ΔEΣΠ = 44.0 meV is the energy difference between the lowest levels of the 4Σ− and 2Π electronic states (Kawaguchi & Amano 1988). The theoretically determined value D0(N–H+, 4Σ−) = 3496 meV (Tarroni et al. 1997) is in satisfactory agreement with our data, considering that the theory neglects the spin–orbit interaction. More information concerning NH+ dissociation energies can be found in Amero & Vázquez (2005) and Tarroni et al. (1997).
4. Discussion and Conclusion
We have used the principle of detailed balance to derive the enthalpy of reaction (1) from the measurements of forward and reverse reaction rate coefficients. This method is accurate if the reactants both in the forward and reverse reaction measurements are in thermal equilibrium. In our study, we had to deal with a few deviations from thermal equilibrium:
(1) The nuclear spin states of H2 in the kf measurement were not in equilibrium. However, measurements with two different nuclear spin state populations (Zymak et al. 2013) allowed us to derive the thermal reaction rate coefficients.
(2) The FS-level population of N+ in the kf measurement was not known. However, our experimental results (Zymak et al. 2013; Plašil et al. 2022) are compatible with the assumption that the FS of N+ ions is relaxed efficiently in collisions with He, and this conclusion is also supported by a recent theoretical prediction of relatively fast FS relaxation in collisions of N+ with H2 and He (Gueguen & Lique 2023).
(3) The internal and translational temperatures,  and Tt, are not in equilibrium, but we show in Figure 3 that the obtained reaction enthalpy is independent of the assumptions concerning the role of the translational and internal temperature of the reactants, owing to the fact that the reverse reaction rate coefficients are nearly constant throughout the studied temperature range.
and Tt, are not in equilibrium, but we show in Figure 3 that the obtained reaction enthalpy is independent of the assumptions concerning the role of the translational and internal temperature of the reactants, owing to the fact that the reverse reaction rate coefficients are nearly constant throughout the studied temperature range.
The present value of ΔE0 is compared to the previously measured activation energies, EA, and energy thresholds, ET, of the forward reaction in Figure 4. If there are no barriers on the reaction paths, ET should be equal to ΔE0 and EA should converge to ΔE0 at low temperatures (kB T ≪ EA; Menzinger & Wolfgang 1969). The good agreement of the previously determined EA and ET with the measured value of ΔE0 confirms that the assumption of no barriers is valid (the agreement with the data of Marquette et al. 1985; Gerlich 1993 with unknown uncertainties cannot be statistically evaluated). The height of the hypothetical energy barrier can be calculated as Eb = EA − ΔE0 and using the most recent value of EA = (19.3 ± 2.7) meV for H2 in thermodynamic equilibrium (TDE; Plašil et al. 2022) leads to Eb = (2 ± 5) meV. Therefore, the height of the possible barrier is, within the accuracy of our measurement, equal to zero. This result confirms that the interpretation of the activation energy as reaction endothermicity in the previous theoretical (Grozdanov & McCarroll 2015; Grozdanov et al. 2016; Yang et al. 2019; Gómez-Carrasco et al. 2022) and astrochemical (Dislaire et al. 2012; Roueff et al. 2015) studies was justified, providing solid ground for further research of the N+ + H2 collision system.

{kind=link}
{kind=link}
{kind=link}
Figure 4. Comparison of the reaction enthalpy determined in the present study with the upper estimates obtained in previous studies of the forward reaction: Pla22 (Plašil et al. 2022), Ger93 (Gerlich 1993), Mar88 (Marquette et al. 1988), Erv87 (Ervin & Armentrout 1987), Mar85 (Marquette et al. 1985), and Ada85 (Adams & Smith 1985). The circles indicate activation energies and the diamonds indicate evaluated reaction enthalpies with the assumption of no barrier. The values labeled as "global fit" were evaluated by combining the experimental data for multiple isotopic variants of the forward reaction.
Download figure:
Standard image High-resolution image{kind=link}