



Abstract
A newly calculated ab initio potential energy surface is used to compute collision-driven state-changing cross sections and rate coefficients over a range from 5 to 100 K for CN−(1Σ), the smallest anion detected in the interstellar medium, interacting with He, an abundant species in this environment. We compare our presently computed rate coefficients with those previously published for the similar and important systems CN–He, CN-H2, and CN−–H2 to illustrate the broader network of inelastic, state-changing processes for these four systems. We also discuss the size-scaling effects that occur when changing partners from He to H2. We further analyze the differences in size between collision-driven rate coefficients when going from neutral CN to its anion. All the present results are discussed in detail, to provide accurate and realistic data for chemical networks that wish to include the CN− anion in their modeling of astrochemical environments.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Negatively charged molecules have always been considered important precursors for neutral prebiotic systems and are expected to play important roles as reactants in interstellar media and planetary atmospheres. The presence of anions in the interstellar medium (ISM) and circumstellar envelopes (CSE) was first predicted theoretically on the basis of linear chain molecular stabilities and electron affinities. An extensive review of astrochemical anion formation and evolutionary processes, as well as observational data, has been presented recently up to 2017 (Millar et al. 2017). Very early on, in fact, they were considered to be present in the ISM by astrochemical models that assumed the relevance of ion–molecule collision processes at low temperatures (Dalgarno & Cray 1973; Herbst 1981). However, detections have been hard to come by and have occurred only very recently due to a general lack of experimental data and because at first, estimated abundances seemed to be very low. Given the capabilities of powerful new telescopes and instruments, however, new findings are increasingly expected to provide additional evidence on the relevance of ion-driven processes involving anions.
The first discovery of a molecular anion in the ISM occurred relatively recently in 2006 (McCarthy et al. 2006), leading to renewed interest from chemists, physicists, and astrophysicists in molecular anions in general and motivated new surveys as well as new theoretical and laboratory studies of species now expected to survive under CSE conditions. The aim is to determine their role and influence on the composition of extraterrestrial sources and in environments where gas-phase ion–molecule reactions are expected to take place (Bierbaum 2011; Cernicharo et al. 2011). On the other hand, among the multitude of anions already predicted by astrochemical models, only five carbon chains of type C2nH− and C2n+1N− have been detected thus far (Cernicharo et al. 2011). Although no additional negative species have been observed since 2010, the search for anions continues (Cernicharo et al. 2011) and has recently provided evidence for the existence of the smallest molecular negative ion detected to date, the CN− species (Agúndez et al. 2010). Astronomers are therefore hopeful for yet more accomplishments through analysis of results from, for example, ALMA, one of the most recent astronomical observatories.
In the laboratory, ion–molecule reactions have also been the subject of many recent inquires (Bierbaum 2011) to elucidate the possible chemical mechanisms for anion formation in the ISM (Satta et al. 2015). The question of the formation mechanisms is, in fact, still open since the conventional suggestion of the radiative electron attachment paths (REA) has been found ineffective for the smaller anionic species observed so far (Douguet et al. 2015) while specific chemical paths suggested earlier for temperatures well below room values (Satta et al. 2015) are still awaiting laboratory confirmations.
Experimental work on the detection of CN− anions (Agúndez et al. 2010) has confirmed observation of the j = 2–1 and j = 3–2 rotational lines in the IRC +10216 spectral emission, as well as the line from the j = 1-0 emission. In those observations a column density of 5 × 1012 cm−2 and a rotational temperature of 16 K were derived from a rotational diagram constructed with the velocity-integrated intensities of the j = 2–1 and j = 3–2 lines, where both data were found to be consistent with CN− line emissions from the cooler outer envelope of the interstellar source. In the modeling employed in that work the computed collisional, inelastic rotational state-changing rates for CN− with H2 molecules were employed and scaled in order to also model the same, but still unavailable, collisional rate coefficients for He as an atomic partner of CN−. Direct knowledge of these collisional cross sections and rate constants would therefore be an important ingredient for a realistic modeling of the correct collision kinetics for both He and H2 partners, albeit considering their estimated differences in column densities in CSE environments (Cordiner & Millar 2009), where the relative ratio of He to H2 in the models is taken to be around 0.2. Our new data would also allow us to question the possible validity of local thermal equilibrium (LTE) assumptions in the observed environments (Kłos & Lique 2011; Wakelam et al. 2015). A fairly comprehensive review of collisional processes involving interstellar molecules interacting with He, H, and H2 up to 2013 has also been published recently (Roueff & Lique 2013). Detailed computational work on the rotational processes of neutral CN interacting with H2 has been recently presented in the literature (Kalugina & Lique 2015; Yang et al. 2016; Burton et al. 2018), their accurate results complementing our present study. The latter will be focused instead on the comparative analysis of rotational excitation/de-excitation of the CN− anion and the CN molecule with different partners.
In this work we obtain, for the first time to the best of our knowledge, accurate rotationally inelastic cross sections for the CN−–He system over a broad range of collision energies and employ them for the calculation of the corresponding state-changing (i.e., rotationally inelastic) rates over a range of temperatures of direct interest to their ISM environments. Furthermore, by making use of previously published inelastic rates for CN−–H2 (Kłos & Lique 2011) we verify whether one can reliably scale one set of rates to obtain the other, as often suggested and done in the current literature (e.g., see Agúndez et al. 2010). We shall also discuss below what has already been discovered for the same problem involving the corresponding neutral molecule CN (Kalugina & Lique 2015) and also CO (Walker et al. 2014).
We compute from first principles the quantum potential energy surface (PES) for the CN− interaction with He and compare our present findings for it with those for both CN−–H2, in which the hydrogen molecule is considered to be the para-H2 partner in its j = 0 state, and the PES for its neutral molecular counterpart CN interacting with He and H2, again employing previously published PESs (Lique et al. 2010; Kłos & Lique 2011; Kalugina et al. 2012).
The analysis of the relevant interaction forces will be briefly outlined in Section 2, while Section 3 will describe the calculations of the inelastic cross sections and of the corresponding state-to-state collisional rates. We then go on to compare rates for all four systems mentioned above in Section 4. Section 5 will briefly discuss the Einstein A coefficients for spontaneous radiative emission from the relevant rotational levels of CN− and CN to provide more quantitative estimates on the relative competitions between radiative and collisional channels of decay via the presentation of critical density values. Finally, Section 6 will present our conclusions.
2. Analysis of Potential Energy Surfaces
2.1. The Interaction Potential between CN−(1Σ) and He(1S)
The interaction potential between closed-shell CN−, treated as a rigid rotor with a bond length fixed at req = 1.181431 Å, and He has been calculated here, using a large basis set expansion at the post-HF level, through the suite of codes provided by MOLPRO (Werner et al. 2012, 2019). The calculations used the Couple-Cluster-Singles-Doubles and perturbative treatment of Triples (CCSD(T)) (Purvis & Bartlett 1982; Pople et al. 1987) method with an aug-cc-pV5Z basis set (Kendall et al. 1992), thereby providing an initial evaluation of the charge-distribution in the system. We then extended the quality of the correlation effects by employing Complete-Active-Space Self-consistent-Field (CASSCF) calculations (Werner & Knowles 1985; Knowles & Werner 1985) followed by a Multi-Reference-Configuration-Interaction (MRCI; Shamasundar et al. 2011) treatment employing the same aug-cc-pV5Z basis set. Both treatments confirm the importance of including the basis-set-superposition-error (BSSE) treated via the counterpoise method (Boys & Bernardi 1970). More specifically, we have employed the Cs point-group symmetry within several choices for the CAS(X,Y) approach followed by the MRCI treatment with an aug-cc-pV5Z basis, as defined in MOLPRO and have varied the number of the active orbitals within the CAS approach up to the convergence of the calculated quantities, reached when using the CAS(11,2) expansion. The PES employed for the dynamics in the following sections was kept at the CAS(8,4) level of the correlation treatment as it was found not to change when the next CAS(11,2) level was employed.
The data collected in Table 1 show the variation of several properties of CN− as the quality of the post-Hartree–Fock treatment of correlation effects is improved by going from the MCSCF level of the first two rows in the table to the further inclusion of MRCI treatments with different basis set expansions as indicated in the next eight rows. The following comments can be made:
Table 1. Convergence of Molecular Properties for the Isolated Molecular Anion as Basis Sets Are Increased for Different Ab Initio Methods
| Method | Basis | μ/Debye | C Charge | N Charge | R/Å | Energy/Eh |
|---|---|---|---|---|---|---|
| MCSCF | aug-cc-pV5Z | 0.8468 | −0.4937 | −0.5063 | 1.1870 | −92.4673 |
| cc-pVTZ-F12 | 0.8432 | −0.4491 | −0.5509 | 1.1872 | −92.4661 | |
| MCSCF+MRCI | aug-cc-pVQZ | 0.7069 | −0.5388 | −0.4612 | 1.1827 | −92.7144 |
| aug-cc-pV5Z | 0.7096 | −0.5025 | −0.4975 | 1.1818 | −92.7207 | |
| cc-pVTZ-F12 | 0.7118 | −0.4443 | −0.5557 | 1.1834 | −92.7061 | |
| cc-pVQZ-F12 | 0.7106 | −0.5909 | −0.4091 | 1.1822 | −92.7175 | |
| aug-cc-pVQZ-F12 | 0.7097 | −0.6508 | −0.3492 | 1.1823 | −92.7178 | |
| aug-cc-pV5Z-F12 | 0.7091 | −0.6166 | −0.3834 | 1.1820 | −92.7205 | |
Download table as: ASCIITypeset image
- (i)The values of the dipole moment in the first column of the table seem to be converging to a stable value of about 0.71 Debye, which is now the best available estimate of this quantity with the largest basis set employed.
- (ii)The residual partial charge on the C-end of the molecule is also converging to a value of about −0.61 excess negative charge, while the corresponding N-end of the molecule is seen to converge to a smaller value of about −0.38, thus indicating that the negative end of the dipole charge is on the C-atom as suggested also in earlier studies in our group (Satta et al. 2015), where a positive dipole moment was also suggested.
- (iii)At the lowest value of the total electronic energy (bottom of table) the molecular geometry is also seen to converge to about 1.18 Å, which is also therefore the best available estimate of this bond length, using the largest basis set expansion, which we have found for this system.
The data in Table 1 indicate how delicate the relative balance of the partial distributions of the excess negative charge is within the atoms of the anion and how much such values are influenced by the level of treatment of correlation forces. This also means that to describe correctly the partial charge redistributions as He approaches the anion requires an extensive inclusion of correlation effects in order to ensure the correct spatial polarization of the additional electronic charge of that system, as we shall further discuss below. The largest active space, obtained using CAS(11,2) followed by the MRCI treatment, indicates that the C-end of the anion remains the part with the slightly larger residual negative charge, although the N atom is also very close in value. The last effect explains why the angular extent of the attractive well region is fairly wide, although shifted to the N-side of the target molecule, as shown by Figure 1.
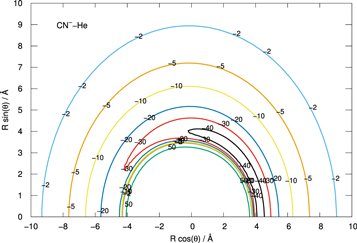
Figure 1. In-plane presentation of the CN−–He PES, showing the angular extension of the collinear region with an attractive well mainly located at the N-end of the molecule. See the main text for further details.
Download figure:
Standard image High-resolution imageThe data in Figure 1 present an in-plane view of the CN−–He PES, showing a rather marked anisotropy of the spatial interaction potential and therefore that collisional encounters between the partners would lead to an efficient dynamical torque applied to the molecular anion, as we have often discussed in our earlier studies with similar systems (e.g., see González-Sánchez et al. 2015) and which we shall further elaborate on in Section 4. We can also see that the information given by Figure 1 indicates, at the present level of correlation calculations, that the deeper region of the angularly extended attractive well is located at the N-end of the molecular anion.
The interaction forces can be represented by a multipolar expansion in Legendre polynomials, which provides a transparent assessment of the spatial anisotropic features, given as Equation (1)
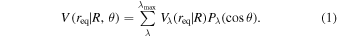
The corresponding pictorial view of the radial coefficients is shown in Figure 2. The results are from the numerical quadrature of the points from the original computed grid of radial and angular values of the PES and the calculation has been carried out up to λmax = 20 to ensure convergence of the quadrature. Only the lower coefficients are shown as they will be the ones dominating the dynamical coupling between rotational states via the scattering equations discussed below. One sees the dominant strength of the isotropic component, given by the λ = 0 coefficient, in driving the interaction dynamics and asymptotically linked to the spherical polarizability of the He atom. By the same token, we also see that the anisotropic terms given by λ = 1 and λ = 2 are the coefficients with the next stronger interactions, an indication that dynamical torques caused by their strength will favor rotational transition processes with Δ j = 1 and Δ j = 2 direct excitations, as we shall further discuss below. In the .tar.gz package of supplementary information we provide the  coefficients for the CN−–He PES fit.
coefficients for the CN−–He PES fit.

Figure 2. Computed multipolar radial coefficients from the expansion of Equation (1). Only the lower coefficients are shown in the figure.
Download figure:
Standard image High-resolution image2.2. Comparison of Interaction Forces for CN− and CN with He and H2
Earlier computational studies have been carried out for the interaction of the neutral CN molecule with He atoms and to further compute rotational state-changing collisions including spin-rotation effects (Lique et al. 2010; Lique & Kłos 2011). It is therefore of interest to compare that PES with the present one since changes in the strength and anisotropy between the two PES will help us to study differences in the efficiency of rotationally inelastic processes from the neutral to the anionic partners. The data in Figure 3 compare the lowest five multipolar coefficients for CN−–He with those for neutral CN–He (Lique et al. 2010).
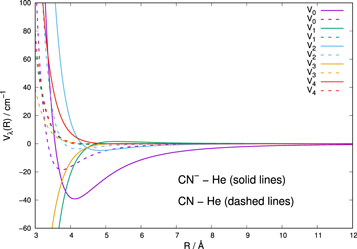
Figure 3. Comparison between the first five multipolar radial coefficients computed for the CN−–He interaction in the present study and that for the CN–He potential from earlier work (Lique et al. 2010). See the main text for further details.
Download figure:
Standard image High-resolution imageThe weaker interaction pertaining to the neutral molecule is clearly visible: the attractive wells are all located further out for the negative ion and their relative depths are all much larger than for the neutral. We should therefore expect that both elastic processes and state-changing collisions with He should be markedly larger for CN−. As a consequence, one should envisage that a larger number of ionic molecules would be rotationally excited in CSE environments and could therefore become more visible during their radiative decay. Such aspects of the rotational dynamics will be analyzed in more detail in the following sections.
Molecular hydrogen is one of the most abundant species in the ISM, together with He and H. It is therefore of direct interest to know the specific features of its interaction with the CN− molecular anion and compare them with those for He presented in the previous subsection. It is also interesting to note that the interaction of CN− with hydrogen atoms is known to give rise to a reaction with formation of metastable HCN− at short distances, a step leading to the destruction of the CN− partner by stabilizing the neutral molecule and emitting the extra electron: the processes is called associative detachment (AD) and has been studied many times before, most recently also by our group (Jerosimić et al. 2018). Because of the presence of this destruction channel, and because of the lower abundances detected for H in comparison with He, inelastic collision processes are expected be less significant and therefore the formation of rotationally excited states of CN− by collision with H should play a minor role with respect to that occurring with either He or H2. In the present work we shall therefore omit discussing collisional rotational state-changing effects coming from H atoms but deal instead with those linked to the interactions of CN− with He and H2.
The CN−–H2 PES has already been obtained with ab initio methods and employed for studying rotationally inelastic collisions (Kłos & Lique 2011) using the full dimensionality of the problem. The geometry of the CN−–H2 system with H2 and the CN− treated as rigid rotors, is characterized by three angles θ, θ, and ϕ and the distance R between the centers of masses of H2 and CN−. The polar angles of the CN− and H2 molecules with respect to R were denoted, respectively, θ and θ', while ϕ was the dihedral angle, i.e., the relative polar angle between the anions and H2 bonds (Kłos & Lique 2011). The final  potential is therefore a 4D PES between the two rigid-rotor partners. In the calculations reported by those authors, they found that there were not significant differences between the inelastic cross sections for the two species of H2 and therefore the collisional rates for the p-H2(j = 0) and for the o-H2 (j = 1) were found to be very similar (Kłos & Lique 2011).
potential is therefore a 4D PES between the two rigid-rotor partners. In the calculations reported by those authors, they found that there were not significant differences between the inelastic cross sections for the two species of H2 and therefore the collisional rates for the p-H2(j = 0) and for the o-H2 (j = 1) were found to be very similar (Kłos & Lique 2011).
In the present work we shall only employ the rates for the collisional excitation of CN− by para-H2, to compare them with our new findings for the anion interacting with He atoms. For collisions at low temperatures (T ≤ 100 K), in fact, the rotational excitation probability of H2 itself is fairly low (Kłos & Lique 2008; Dumouchel et al. 2011) since the energy spacing between the j = 0 and j = 2 levels in p-H2 is around 510 K. In this case, only the leading terms of the 4D multipolar expansion need to be retained in the expansion, thereby allowing the resulting PES to be simplified to the lower dimensionality of that for an atomic partner, as reported by Equation (1).
The data in Figure 4 show a comparison between the leading multipolar coefficients (the first five) of the present CN−–He PES and the same coefficients from the averaged CN−-p-H2 as given by the earlier work on this system (Kłos & Lique 2011). We see that the averaged potential for CN−-p-H2 exhibits stronger coupling coefficients that those we have found for He. The quantum calculations reported below will therefore be able to tell us what effects such differences will have on the relative size of the state-to-state rates between the two systems.
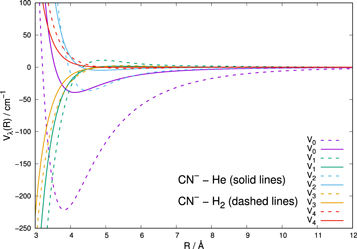
Figure 4. Comparison of the multipolar radial coefficients for the interactions of CN− with He (present results) and with p-H2 (j = 0) (from Kłos & Lique 2011). See the main text for further details.
Download figure:
Standard image High-resolution imageAnother comparison of interest for the present discussion is that between the averaged interaction of p-H2 (j = 0) with either CN− or the neutral CN molecule. The latter PES has also been obtained in earlier work and already reported in the literature (Kalugina et al. 2012, 2013). The comparison is shown in Figure 5, where the multipolar coefficients from both averaged potentials are compared via their first five radial components. The overall strength of the interaction with the anionic partner is clearly larger than in the case of the neutral partner, an effect that is in line with what we have found when comparing neutral and anionic CN with He atoms. As a consequence, we therefore expect that the excitation probabilities for internal rotations of the anion will be markedly larger and more significant than in the case of neutral CN. We shall report below a comparison between state-to-state rotational rates that will bear us out on this point.
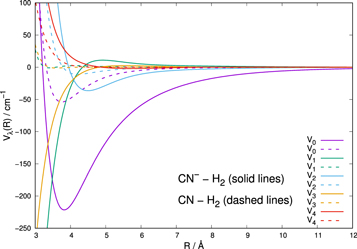
Figure 5. Comparison of the computed averaged potentials between CN− and CN molecules with p-H2 (j = 0). The results given by solid lines are from Kłos & Lique (2011) while the dashed lines are from Kalugina et al. (2012). See the main text for further details.
Download figure:
Standard image High-resolution image3. Rotational Features of CN− and Quantum Collision Dynamics
The present work is directed to study the state-changing of rotations in CN− ISM environments by collision with He. The actual energy ladder involving the lower-lying rotational levels of this molecular anion are reported by Figure 6, showing levels up to about 100 cm−1 of relative collision energies. The chosen rotational constant was the experimental value, found by Gottlieb et al. (2007) to be 1.87239 cm−1. The corresponding distribution of relative population probabilities, obtained by assuming the LTE distribution as the initial condition in the observational regions (e.g., see Wakelam et al. 2015), indicates that up to 50 K, rotational states of the anion up to j = 8 are significantly populated. This means that the collisional de-excitation rates will need to be calculated from a larger range of initial levels of the molecule, as we shall further show in the next section. We can also say that at the lower temperatures in the inner region of the CSE where the anion has been observed (e.g., see Millar et al. 2017), i.e., between 10 to 16 K, the excited rotational levels for j = 1 and j = 2 have a larger relative population than the j = 0 level. This feature will be significant in indicating which excited rotational levels from the collisions would contribute more to radiative emissions from such states.
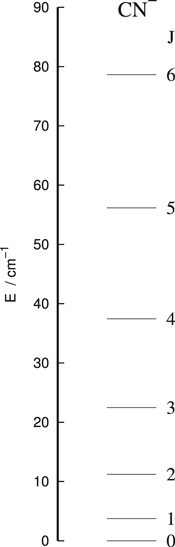
Figure 6. Computed energy spacing for the lower-lying rotational levels of CN− as a rigid rotor with the experimental rotational constant of 1.87239 cm−1 taken from Gottlieb et al. (2007). See the main text for further details.
Download figure:
Standard image High-resolution imageThe usual time-independent formulation of the coupled-channel (CC) approach to quantum inelastic scattering will not be repeated in detail (see, for example, Taylor 2006 for a general treatment) since we have already discussed it in many of our earlier works (López-Durán et al. 2008). Hence, only a short outline will be given below.
When no chemical modifications are induced in the target by the impinging atom (as in our case), the total scattering wave function can be expanded in terms of asymptotic target rotational eigenfunctions (within the rigid-rotor approximation), which are taken to be spherical harmonics and whose eigenvalues are given by Bj(j + 1), where B is the rotational constant. The channel components for the CC formulation are therefore expanded into products of total angular momentum eigenfunctions and of radial functions. These functions are in turn the elements of the solutions matrix that appear in the familiar set of coupled, second order homogeneous differential equations:
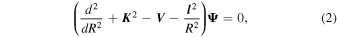
where ![${[{\boldsymbol{K}}]}_{{ij}}={\delta }_{i,j}2\mu (E-{\epsilon }_{i})$](https://content.cld.iop.org/journals/0004-637X/897/1/75/revision1/apjab94a0ieqn3.gif) is the diagonal matrix of the asymptotic (squared) wavevectors and
is the diagonal matrix of the asymptotic (squared) wavevectors and ![${[{\boldsymbol{l}}]}_{{ij}}={\delta }_{i,j}{l}_{i}({l}_{i}+1)$](https://content.cld.iop.org/journals/0004-637X/897/1/75/revision1/apjab94a0ieqn4.gif) is the matrix representation of the square of the orbital angular momentum operator. This matrix is block-diagonal with two sub-blocks that contain only even values of (l + j) or only odd values of (l + j).
is the matrix representation of the square of the orbital angular momentum operator. This matrix is block-diagonal with two sub-blocks that contain only even values of (l + j) or only odd values of (l + j).
The scattering observables are obtained in the asymptotic region where the log-derivative matrix has a known form in terms of free-particle solutions and unknown mixing coefficients. For example, in the asymptotic region the solution matrix can be written in the form:

where J(R) and N(R) are matrices of Riccati–Bessel and Riccati–Neumann functions. Therefore, at the end of the propagation one uses the Log-Derivative matrix to obtain the K matrix by solving the following linear system:
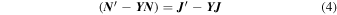
and from the  matrix the S-matrix is easily obtained and from it the state-to-state cross sections. We have already published an algorithm that modifies the variable phase approach to solve that problem, specifically addressing the latter point and we defer the interested reader to these references for further details (Martinazzo et al. 2003; López-Durán et al. 2008). In the present calculations we have followed the above method and generated a broad range of state-to-state rotationally inelastic cross sections from the collisions of CN− with He.
matrix the S-matrix is easily obtained and from it the state-to-state cross sections. We have already published an algorithm that modifies the variable phase approach to solve that problem, specifically addressing the latter point and we defer the interested reader to these references for further details (Martinazzo et al. 2003; López-Durán et al. 2008). In the present calculations we have followed the above method and generated a broad range of state-to-state rotationally inelastic cross sections from the collisions of CN− with He.
Once the state-to-state inelastic integral cross sections are known, the rotationally inelastic rate constants kj→j'(T) are evaluated as the convolution of the cross sections over a Boltzmann distribution of the relative collision energies:
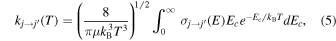
where Ec = μ v2/2 is the kinetic energy.
The above quantities have been obtained over a broad range of initial rotational states of the CN− molecular anion. The actual numerical details will be further discussed in the following section.
4. Rotationally Inelastic Cross Sections and State-to-state Rate Coefficients
To establish the importance of the state-changing collision processes for the CN− anion under ISM conditions, we need to examine the energy/temperature dependence of the inelastic cross sections and rate constants involving several of its rotational levels. We have computed such quantities covering the first 10 rotational levels of the target anion, some examples of which are shown and discussed in the following figures.
Scattering calculations were carried out for collision energies between 1 and 1000 cm−1 using steps of 0.1 cm−1 for energies up to 100 cm−1, 0.2 cm−1 for 100–200 cm−1, 1.0 cm−1 for 200–500 cm−1, and 2 cm−1 for 500–1000 cm−1. This fine energy grid was used to ensure that important features such as resonances appearing in the cross sections were accurately accounted for and their contributions correctly included when the corresponding rate coefficients were calculated. The number of partial waves was increased with increasing energy and went up to J = 100 for the highest energies considered. Inelastic cross sections were computed for all transitions between j = 0 to j = 10, which should be sufficient to model gas dynamics processes up to about 100 K. The number of rotational levels that were coupled during the multichannel treatments went up to j = 20, the latter channel becoming open only at the higher energies sampled in our calculations since j = 20 corresponds to 784 cm−1. The rotational levels considered (up to j = 10) correspond to the levels expected to be most significantly populated during collisions in ISM environments.
A set of examples is given in the four panels of Figure 7 for the excitation processes from some of the initial levels expected to be populated in the CSE environments. For completeness, we also give more examples of excitation cross sections starting from other initial rotational levels. The data in the upper left panel present excitation processes from j = 1 and j = 3 involving transitions with increasing values of the Δ j - changing index: those with Δ j = 1, Δ j = 2, and Δ j = 3 . The same types of processes are also shown in all the other panels, but each time starting from a different set of initial rotational levels: from j = 2, 4 in the upper right panel, from j = 5, 7 in the lower left panel and from j = 6, 8 in the lower right panel. The following considerations can be made by perusing the data in this figure:
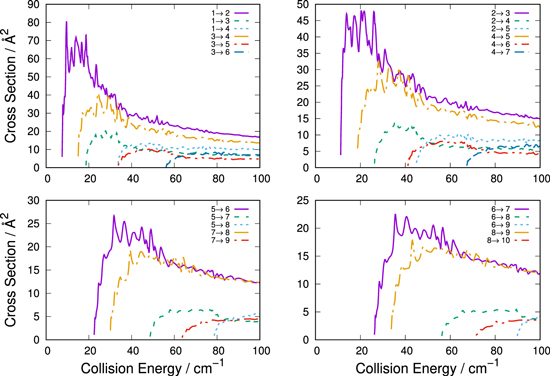
Figure 7. Rotationally inelastic cross sections for CN−–He at low energies. The processes shown in each panel involve excitation collisions from specific initial levels and with increasing Δj values. See the main text for further details.
Download figure:
Standard image High-resolution image(i) As the transitions in each panel involve increasingly higher final levels we see that the cross sections become relatively smaller, indicating that the dominant excitation processes are those from the lower rotational levels with initial j = 1, 2, 3 and with Δ j = 1 transitions, while as the Δ j value increases the size of the cross sections invariably decreases.
(ii) As the initial rotational levels become increasingly higher, i.e., when we are considering initially more excited molecular species, the excitation cross sections become smaller and are seen to vary by nearly an order of magnitude from the upper left to the lower right panels.
These rotational inelastic cross sections already indicate that the excitation processes from the lower rotational levels, those mostly populated at the temperatures of interest, and with Δ j = 1, have the greatest role in the state-changing collisional network we are examining. Such features will naturally affect the corresponding rates that we analyze in Figure 8.
As discussed for the calculations of the cross sections before, we see in all four panels of Figure 8 that the largest rate coefficients pertain to processes involving rotational levels undergoing excitation to their contiguously next higher levels. Furthermore, they indicate that exciting the lower levels, e.g., molecules in their j = 1, 2, 3 states, produces the largest excitation rate constants. Since these would be the levels with the higher population at the temperatures of interest, we can say that the network of rotationally excited molecular anions most likely to be produced by collisions with the He partner would be those discussed in our present data.

Figure 8. Rotationally inelastic excitation rates for CN−–He. The four panels follow the same notation of the previous figure for the cross sections. They span a temperature range of about 100 K.
Download figure:
Standard image High-resolution imageA further comparison between different inelastic rate coefficients is shown by the four panels of Figure 9, which report the de-excitation rate coefficients from several of the lower states discussed earlier. These calculations show again that the largest de-excitation rate constants involve transitions between states with the relatively smaller energy gaps and corresponding to the Δ j = −1 transitions.
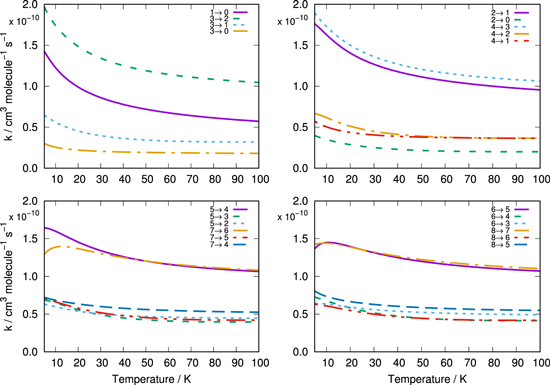
Figure 9. Rotationally inelastic de-excitation rate coefficients for CN−–He. The four panels follow the same notation of the previous figures and involve the same set of rotational levels. They also span a temperature range of about 100 K.
Download figure:
Standard image High-resolution imageThe depopulation rate coefficients from the higher levels, on the other hand, show that their relative values increase with temperature, thereby indicating a larger probability to undergo de-excitation as one moves to the upper rotational states, thereby reducing the energy gap between levels involved in the transitions.
In the .tar.gz package of supporting information we give the rate coefficients for all transitions between j = 0 to j = 10 for temperatures between 5 and 100 K. We also provide the three parameters and rms error for the function often employed to represent the state-changing rates in the ISM environment (Satta et al. 2015; Wakelam et al. 2015)
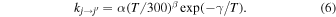
4.1. State-changing Efficiency for CN and CN− in Collision with He
It is well known that the observed abundances of the neutral counterpart of the present anion, the CN(2Σ) molecule, are also significant within the same ISM environments where the anion has been observed. It is therefore interesting to compare the relative excitation efficiency of their rotational states by collision with He atoms. Rate coefficients for rotationally inelastic CN–He collisions have already been obtained from ab initio calculations (Lique et al. 2010; Lique & Kłos 2011).
Figure 10 compares the rotationally inelastic rate coefficients for CN−–He computed here and the previously published rates for CN–He. As the neutral molecule's electronic ground state is (2Σ), spin-rotation coupling effects split each rotational state by j = N ± 0.5 to give fine structure. To provide a more straightforward comparison, the rate constants for relevant CN rotational transitions were summed over their fine structure components, a procedure that results in the molecule being effectively treated as pseudo-singlet (Hernández Vera et al. 2017) but using the exact coupling in the calculations. Excitation rates for CN are consistently one order of magnitude smaller than those for the anion for Δj = 1 transitions, confirming once more that the increased interaction strength of the ionic interaction as shown in Figure 3, invariably lead to more probable collisional excitation processes in encounters with He atoms in the same CSE environments.

Figure 10. Comparison of rotationally inelastic rate coefficients for He scattering from CN− (solid lines) calculated here and CN (dotted lines) obtained from Lique et al. (2010). The color coding is the same for both processes.
Download figure:
Standard image High-resolution image4.2. Comparing State-changing Rate Constants for CN− in Collision with He and H2
The most abundant collision partner for CN− in the ISM environment is the H2 molecule, with next in abundance being the He atom. It is therefore interesting to compare the relative collisional efficiencies of these two partners under the ISM conditions of the present work. We have already compared the two PESs in Section 2. The rates for p-H2 were calculated by Kłos & Lique (2011) and obtained from the Basecol database (Dubernet et al. 2013).
The present comparisons between inelastic rate constants are reported by the four panels of Figure 11 and quantitatively show the size-reduction of these rate coefficients when going from H2 to He as collision partners: the change is consistently of one order of magnitude or more over the range of temperatures of interest. This is an important result confirming general expectations from the findings in Figure 4, but also allowing us to test popular approximate scaling schemes often employed to transfer one set of rates from one system to the other.
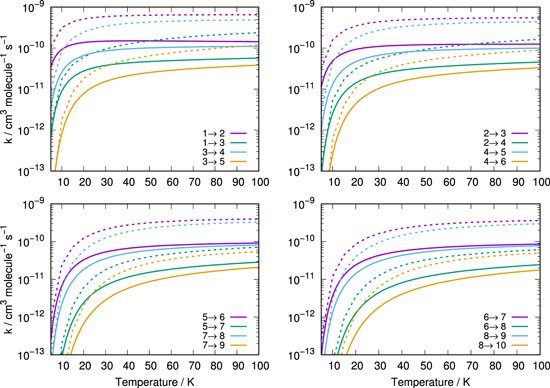
Figure 11. Comparison of rotationally inelastic rate coefficients for scattering of CN− from He (solid lines) and from p-H2 (dotted lines) computed by Kłos & Lique (2011).
Download figure:
Standard image High-resolution imageFor neutral molecules it is a rather common procedure to use the He collisional coefficients to approximate those with H2 by assuming the same cross sections. The ensuing necessary rate coefficients are then obtained as scaled by the ratio of the reduced masses as
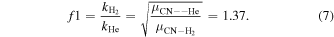
For scaling collisions of He and H2 with molecular ions, a slightly more sophisticated scale factor can be used to additionally account for the differences between the ionic interactions:
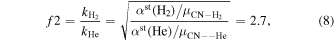
where αst(He) = 1.384 a03 and αst(H2) = 5.314  are the polarizabilities of He and H2 respectively.
are the polarizabilities of He and H2 respectively.
The specific validity of such scaling has been discussed in the literature rather extensively via accurate calculations involving the CO neutral target with different collision partners (Walker et al. 2014) and found to be unreliable. The authors also suggested different scaling procedures that were found to fare better, but underline the need to have the actual rate coefficients correctly computed for the compared systems. This point is what we briefly illustrate below with our calculations.
To verify the differences between both prescriptions presented above, we compare the rate constants previously published for CN−-p-H2 (Kłos & Lique 2011) with those calculated here for CN−–He but scaling them with either of the above scale factors. Our results are given in Figure 12, which shows in its four panels the same set of excitation rate coefficients, giving them for four different temperatures of interest. The values computed for the H2 partner are given by the bars colored in purple, while the scaled values obtained for the CN−–He calculations are given by the green color when using the f1 prescription, and by the blue color for the f2 scaling choice. The following can be gleaned from the four panels in that figure:

Figure 12. Comparison of rotationally inelastic rate coefficients for CN−–H2 (Kłos & Lique 2011) with those computed for the CN−–He system but scaled by the f1 and f2 factors discussed in the main text.
Download figure:
Standard image High-resolution image
- (i)The scaling factor suggested for ionic partners, and therefore explicitly involving the role of long-range polarizability changes, turns out to be more realistic than the simpler choice of mass scaling the rates (green bars), the differences however being increasingly larger as the temperature is increased.
- (ii)Even the more realistic scaling for ionic systems is still not as accurate as using explicitly the correct PES to treat the H2 collisions with CN−.
Such comparisons therefore suggest that any scaling procedure should be used with caution when both sets of rate coefficients are needed to realistically model inelastic processes within reaction networks. Kalugina & Lique (2015) have also shown that for the corresponding neutral systems CN–He and CN-H2, the scaling procedure of Equation (7) is not reliable with up to an order of magnitude difference between scaled and calculated rate constants.
4.3. State-changing Efficiency for CN and CN− in Collision with H2
For the sake of completeness we conclude this section by comparing the rotationally inelastic rate coefficients for both CN and CN− in collisions with p-H2. Figure 13 directly compares the state-changing inelastic data for CN-p-H2 computed by Kalugina & Lique (2015) and obtained from the Leiden Atomic and Molecular Database (Schöler et al. 2005) and CN− in collision with p-H2 (Kłos & Lique 2011). The rate coefficients for CN-p-H2 have been summed over the hyperfine levels to allow a clear comparison with those of CN−-p-H2.
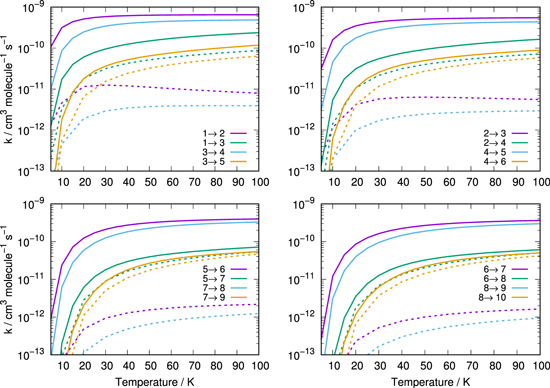
Figure 13. Comparison of rotationally inelastic rate coefficients for H2 scattering from CN− (Kłos & Lique 2011, solid lines) and CN (Kalugina & Lique 2015, dotted lines).
Download figure:
Standard image High-resolution imageWe see clearly in the four panels of Figure 13 that the inelastic rate constants for the excitation of either the CN molecule or its anion are dramatically different as expected from Figure 5. Those involving the ionic species, in fact, are larger by nearly two orders of magnitude with respect to those involving the neutral species for Δj = 1 transitions. It therefore becomes apparent that the relative probabilities for undergoing collisional excitations of rotational states by either He or H2 are invariably much smaller for neutral CN than those involving the anionic species.
5. Einstein Coefficients for Spontaneous Emission
One of the issues in modeling the evolution of rotational state populations in the CSE environment is that of understanding the role of the spontaneous decay path from the levels populated by collisions. This requires assessing the magnitude of the corresponding decay rates by photon emission either spontaneously or by their interaction with the surrounding radiative field.
The transition rate from an excited state k can be written as the sum of stimulated and spontaneous emission rates as
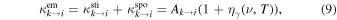
where Ak→i is the Einstein coefficient for spontaneous emission and  is the Bose–Einstein photon occupation number.
is the Bose–Einstein photon occupation number.
The Einstein coefficient for dipole transitions is given as
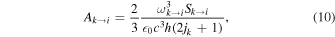
where ωi→k ≈ 2B0(ji + 1) is the transition's angular frequency, and  is the line strength. For pure rotational transitions, Equation (10) simplifies to
is the line strength. For pure rotational transitions, Equation (10) simplifies to
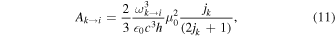
where μ0 is the internuclear electric dipole moment of the molecule.
The LTE assumption holds whenever the population of excited levels is given by Boltzmann's law. This happens when the rate of spontaneous emission is significantly smaller than the rate of collisional de-excitation and therefore can be neglected. This means that the density of gas should be significantly larger than some critical value of that density so that the LTE assumption can be kept. In order to verify the relative importance of the spontaneous emission process viz á vis the collisional state-changing rates, we have calculated Ak→i using Equation (11) for the present molecular anion and for its neutral counterpart. The results are reported by Table 2.
Table 2.
Computed Einstein Spontaneous Emission Coefficients  for CN− and CN
for CN− and CN
| Transition | CN− | CN |
|---|---|---|
|
||
| 1→ 0 | 2.76 × 10−6 | 1.21 × 10−5 |
| 2→ 1 | 2.65 × 10−5 | 1.16 × 10−4 |
| 3→ 2 | 9.57 × 10−5 | 4.19 × 10−4 |
| 4→ 3 | 2.35 × 10−4 | 1.03 × 10−3 |
| 5→ 4 | 4.70 × 10−4 | 2.05 × 10−5 |
| 6→ 5 | 8.24 × 10−4 | 3.60 × 10−3 |
| 7→ 6 | 1.32 × 10−3 | 5.79 × 10−3 |
| 8→ 7 | 1.99 × 10−3 | 8.71 × 10−3 |
Note. All quantities in units of s−1. See the main text for further details. CN− μ0 = 0.709 D, CN μ0 = 1.45 D.
Download table as: ASCIITypeset image
Since the values of the particle densities vary widely in different ISM environments, several models of the conditions in the molecular clouds indicate changes from the situation of the diffuse clouds (around 102 cm−3) to dense clouds (around 103–106 cm−3) as discussed in Snow & McCall (2006) and Agúndez & Cernicharo (2006) and in the references reported there. From our computed collisional rates of, say, Figure 9, we can observe that the above values of expected particle densities can provide a broad range of collisional de-excitation efficiency as given in units of s−1, the same used for the data in Table 2. Hence, in diffuse clouds we obtain collision rotational state de-excitation quantities of the order of about 1.2 × 10−7 while in the dense clouds we have rate coefficients ranging from 1.2 × 10−6 to 1.2 × 10−3. Thus, in the darker environments we see that the collisional de-excitation processes are faster than the spontaneous radiative emissions reported by our calculations in Table 2, while in the more diffuse environments the spontaneous decay channels appear to be faster than the collisional ones. Thus, the validity of the LTE assumption is seen to vary markedly depending on the relative sizes of the local particle densities in the environments of interest and on the rotational level initial state selected for the decay. The above analysis is equivalent to the analysis of critical density values, as recently shown in Lara-Moreno et al. (2019). The definition of a critical density is given as:
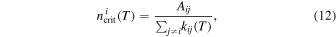
where the critical density for any ith rotational level is obtained by giving equal weight to the effects of the collision-induced and the spontaneous emission processes. We have used the calculated results for the collision rate coefficients for either He or H2 as partners of the present molecular anion, as discussed in detail in the previous sections. We have then employed the radiative coefficients reported in the above Table 2 to obtain the critical densities presented in the two panels of Figure 14. The panel on the left reports the critical densities for the He partner while the panel on the right shows the same critical densities but for the case of the H2 partner for the present molecular anion.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 14. Comparison of computed critical densities for H2 (right panel) and He (left panel) scattering using the Einstein coefficients and the collision rate coefficients discussed in the main text.
Download figure:
Standard image High-resolution image{kind=link}
From the variety of environments discussed earlier, we see that the critical densities associated with all the rotational levels we are considering in both panels are higher than the medium-to-low density environments. This means that we should consider those populated states of the anion not to be under the LTE conditions since the critical density values are all large enough to allow the molecules to radiate before they can collisionally de-excite. Under such conditions, therefore, to know accurately the collision-driven rates calculated in our work would be important since the LTE conditions cannot be reliably employed. It is also interesting to note that the critical densities are larger for the case of the He partner than those for the molecular hydrogen. This is easily understood from the definition in Equation (12) since the collision rate coefficients for H2 are larger than those for He. By the same token, we see that our values in Figure 14 are larger than those discussed earlier for other, but much larger, molecular anions (e.g., see Lara-Moreno et al. 2019 ), thus indicating the importance of the relative magnitudes of the terms contributing to the ratios in Equation (12).
6. Conclusions
The first realistic calculations of the quantum dynamics of CN− interacting with He from accurate ab initio methods has been presented. The CN−–He PES was compared to previously computed surfaces for those of the corresponding neutral system CN–He and for CN−–H2 and CN-H2. We have provided a detailed analysis of the relative efficiencies of collision-driven excitation and de-excitation processes involving the CN− molecular anion interacting with either He atoms or with the H2 molecule, both present in the ISM environment where CN− has been abundantly observed. The relative size and temperature dependence of the inelastic rates for both excitation and de-excitation of CN− rotational states are reported among states considered to be significantly populated in ISM environments, while we have also compared the collisional rates with radiative de-excitation channels to provide additional indicators on their importance in different environments.
From the behavior of the collisions involving H2 as a partner we clearly found that their state-changing efficiency would be higher than that with He atoms. The scaling approximations often used to estimate the behavior of one partner when the rates with the other were also compared and found to be essentially unreliable, a result also confirmed earlier in the literature for the CO neutral case (Walker et al. 2014) and CN (Kalugina & Lique 2015).
The present calculations and comparisons of several state-changing inelastic rates thus provide a broad variety of specific, detailed indicators on the collisional behavior of the CN− molecule with the two most common partners in the ISM environments and also with the expected behavior of its neutral molecule CN, making such rates available for larger kinetic studies of anions in the CSE environments.
We are very grateful to Professor F. Lique for having generously helped us by providing potential energy surfaces for several of the systems we compare our data with in the present study: CN− with H2, CN with He, and CN with H2. We further acknowledge the financial support of the Austrian FWF agency through research grant No. P29558-N36. One of us (L.G-S) further thanks MINECO (Spain) for grants CTQ2015-65033-P and PGC2018-09644-B-100.