
Abstract
A combination of classical molecular dynamics (MD) simulation and ab initio fragment molecular orbital (FMO) calculation was applied to a complex formed between the main protease of the new coronavirus and the inhibitor N3 to calculate interactions within the complex while incorporating structural fluctuations mimicking physiological conditions. Namely, a statistical evaluation of interaction energies between N3 and amino acid residues was performed by processing a thousand of structure samples. It was found that relative importance of each residue is altered by the structural fluctuation. The MD-FMO combination should be promising to simulate protein related systems in a more realistic way.
Export citation and abstract BibTeX RIS

Content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
The COVID-19 disease caused by the new severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has still been the worldwide critical issue, as reported by the World Health Organization (WHO). 1) A great amount of works in various fields have been conducted against this difficult situation. In the pharmaceutical industry, several proteins such as the main protease (Mpro) of SARS-CoV-2 have attracted considerable attention because they are potential targets of antiviral drugs, and hence a number of computational studies have been performed to find effective inhibitors against them. For example, Xu et al. 2) showed that nelfinavir may be a promising inhibitor to Mpro, based on a homology modeling and docking simulations of a number of trial ligands. Jin et al. 3) developed a new peptide-like inhibitor named N3 with virtual screening techniques and determined the crystal structure of the Mpro−N3 complex [published as 6LU7 in Protein Data Bank (PDB)]. By this availability of 6LU7 structure, several related studies such as molecular dynamics (MD) simulation were reported. 4–13) Among them, we 13) performed ab initio fragment molecular orbital (FMO) 14–17) based analyses of interactions between the N3 inhibitor and amino acid residues in the pharmacophore, while using pair interaction energy (PIE) 16) or inter-fragment interaction energy (IFIE) 17,18) to identify important residues having site-specific interactions, e.g. hydrogen bond, with N3. We have successfully applied FMO to other SARS-CoV-2 protein systems including the spike protein 19) and RNA polymerase—remdesivir complex, 20) as in the cases of precedent studies for viral proteins. 11,21–24)
Although we have performed even detailed PIE decomposition analyses (PIEDA 25,26)) including an implicit hydration effect through the Poisson–Boltzmann (PB) model 27,28) in Ref. 13, the structural fluctuation effect (which should be essential in physiological situation at room temperature) was not incorporated because we performed only a single structure calculation for the 6LU7 crystal data. 3) Ishikawa et al. 29) pioneered a statistical scheme in which classical MD simulations are first performed to generate structure samples and these series of structures are subjected to FMO calculations so that the structural fluctuation effects on IFIEs can be taken into account. We applied this MD—MO combinative scheme to DNA—uranyl complex 30) and calmodulin—Eu(III) complex 31) as well as the designed peptide—inorganic surface systems. 32–34) Here, we took the same approach for the Mpro—N3 complex, while using a thousand of structure samples, ten times more than those in Refs. 30 and 31, by using a massively parallel computing resource of Fugaku supercomputer. The computational protocol used are summarized as follows.
We started with the crystal structure of the Mpro—N3 complex (PDB ID: 6LU7), 35) from which we extracted a monomer block, and processed it in a standard manner with the MOE software; 36) namely, we added missing hydrogens and optimized them with the AMBER10:EHT 37) force field (FF) (refer also to Ref. 13). This structure is referred to as "static" hereafter. A thousand of "dynamic" structures were prepared by the MD simulation method with the AMBER16 program 38) (with the AMBER10:EHT FF). The MD run durations were 50 ps for the thermal elevation from 0 to 310 K (NVT ensemble), 50 ps for the density relaxation (NPT), 1 ns for the equilibration (NPT), and 100 ns for the production process (310 K, 1 atm, NPT). From trajectories of the production run, a thousand snapshots were sampled with an interval of 100 ps. This long-time and rich sampling should cover larger configurational spaces and thus provide more reliable statistical evaluations of interaction energies, in comparison with our previous studies. 30–34) All the MD simulations were performed on the TSUBAME3.0 computer. Each sample structure was shaped to a droplet form with water molecules within 4 Å of Mpro; this criterion of water layer thickness was determined according to Refs. 39–41. The counter ions (Na+) were retained to electrically neutralize the system.
The electronic energy of two-body FMO expansion 14–17) can be expressed as
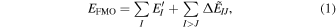
where I and J are fragment indices. The first term of Eq. (1) is the fictitiously isolated monomer energy, and the second term corresponds to the fragment interaction energy termed as PIE 16) or IFIE. 17,18) In PIEDA, 25,26) each interaction energy is further decomposed as
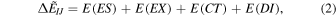
where ES, EX, CT and DI mean "ElectroStatic", "EXchange repulsion", "Charge-Transfer and mix terms", and "DIspersion", respectively. The EX, EX, and CT terms are evaluated at the Hartree–Fock level, 42) whereas the DI term is usually obtained at the second-order Møller–Plesset perturbation (MP2) level including the electron correlation effect. 42–45) We adopted the partial renormalization, 46) to reduce the tendency of overestimation in the MP2 based DI energy. 13) As shown in Fig. 1, the N3 ligand has a covalent bond to Cys145, 3) and N3 was segmented to 5 fragments. 13) The numbers of fragments were 311 for the Mpro—N3 complex and about 1400 for the water layer, respectively. The basis set used was 6-31 G*, 47) a standard choice in recent FMO studies. A series of FMO-MP2/6-31 G* calculations with the PIEDA option were performed with the ABINIT-MP program 17) on the supercomputer Fugaku. Typical job timing for a single structure sample was 0.6 h with 192 nodes (or a half rack) of Fugaku, and the concurrent job processing led to only 6 h for the completion of all the structure samples.

Fig. 1. (Color online) Fragmentation of N3 ligand. The fragmentation point is identified by orange triangle indicating the direction of bond detachment atom (BDA) to bond attachment atom (BAA). 17) There is a covalent bond to Cys145. 3) Refer to related descriptions in Ref. 13 when needed.
Download figure:
Standard image High-resolution imageIn interpretation of IFIE and PIEDA, care is necessary for the fact that the fragmentation of usual FMO is made at the single bond between the carbonyl carbon and the alpha carbon (with side chain) 48) and thus that a misleading assignment shift of interacting fragment can occur when the carbonyl oxygen forms a hydrogen bond. This matter was carefully handled as was done in Ref. 13. We obtained the averages and standard deviations (or 1σ) of IFIE and PIEDA data calculated for the "dynamic" structures.
Below, we present the results and discussion. Figure 2 illustrates a superimposed view of 10 ns interval sampled structures of the N3 ligand and crucial residues. Because of the covalent bond to Cys145 of Mpro, fluctuations of Fragments 3 and 4 are small, whereas those of the terminal Fragments 1 and 5 are larger. Refer also to Fig. S1 (available online at stacks.iop.org/APEX/14/027003/mmedia) in Supporting Information for the root mean square deviation of trajectories.

Fig. 2. (Color online) Superimposed view of MD sampled structures of 10 ns interval for the N3 ligand and crucial residues in pharmacophore. The N3 ligand is depicted in yellow color.
Download figure:
Standard image High-resolution imageThe total IFIE between Mpro and N3 is −199.9 kcal mol−1 for the "static" structure, and the corresponding statistical value of the "dynamic" structures (of a thousand samples) is −145.9 ± 10.8 kcal mol−1, as found in Table S1; see a comparison with another "dynamic" results obtained by the first hundred samples if necessary. As discussed later, the overall decrease of stabilization might be attributed to the increased average distances between the N3 fragments and the interacting residues relative to the "static" or crystal structure. Crystal structures may be generally denser than fluctuated structures in physiological conditions, leading to the closer and stronger interactions with ligands in the pharmacophore. Namely, there are potential overestimations in IFIEs in the present system of Mpro—N3 for the X-ray crystal structure. The IFIE value of each Fragment of N3 is plotted in Fig. 3 (refer to Table S1 again). The stabilization of Fragment 4, which was found to be the most important moiety in Ref. 13, is sizably decreased relative to the "static" value, or the overestimation by the "static" structure is confirmed. As a result, the importance of Fragment 3 becomes comparable to that of Fragment 4 in a statistical view with "dynamic" structures. These two Fragments of N3 are close to a covalent bond to Cys145, and their mobile abilities are rather restricted relative to other Fragments, being consistent with the situation observed in Fig. 2. Interestingly, the relative importance of Fragment 5 is enhanced in the "dynamic" structures. A plausible reason could be considered as follows. Fragment 5 is almost invisible on the electron density map of the crystal (see Fig. S2), so the atomic coordinates of this moiety should be relatively unreliable in the crystal structure. 3) Therefore, the stabilization of interaction energy suggests that Fragment 5 is more precisely positioned by considering "dynamic" structures through MD.
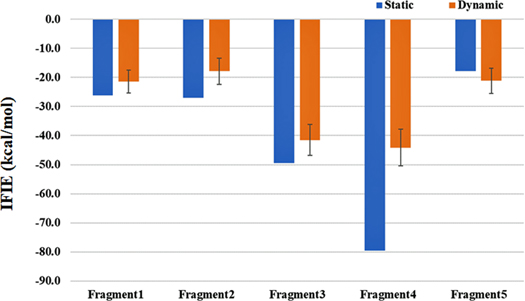
Fig. 3. (Color online) IFIE value of each Fragment of N3 interacting with Mpro. Blue and orange bars correspond to the single value by "static" structure and the averaged value by a thousand of "dynamic" structures, respectively. Vertical line for the latter is standard deviation. Refer to Table S1 for numerical data.
Download figure:
Standard image High-resolution imageNext, we discuss residue specific discussion. Figure 4 shows IFIE plots of leading residues whose stabilization energies are larger than −10 kcal mol−1 (see Table S2 compiling the respective IFIE values and contact distances). As can be seen, His163 is the outstanding residue with the largest stabilization in both "static" and "dynamic" evaluations. Other notable residues in stabilizations are Met165, Leu167, and Gln189. The contribution from these four residues were consistently pointed out in Refs. 3, 6–8, and 13 (ours). A considerable amount of decrease in stabilization (ca. 15 kcal mol−1) is found for His163 (associated with Fragment 4), while those of Met165, Leu167, and Gln189 are lesser. These relations are in accord with the differences in the contact distances listed in Table S2; about 0.6 Å is found for His163. Keller et al. 49) investigated the structural changes of 6LU7 by MD simulation and observed that a loop consisting of Glu166—Gly170 in Mpro moves away from the N3 ligand. The present results are consistent with their observation. More detailed Fragment-wise results of IFIE, are provided in Figures S3–S7 as well as Tables S3–S7; refer to these numerical data when necessary.
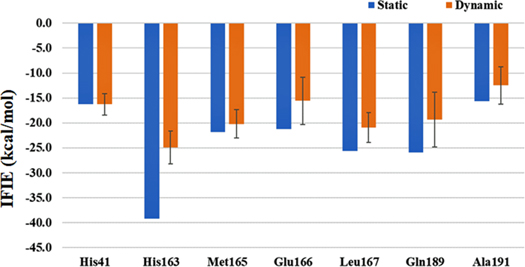
Fig. 4. (Color online) IFIE value of leading residues interacting (larger than −10 kcal mol−1 of total stabilization) with Mpro. Blue and orange bars correspond to the single value by "static" structure and the averaged value by a thousand of "dynamic" structures, respectively. Vertical line for the latter is the standard deviation. Refer to Table S2 for numerical data.
Download figure:
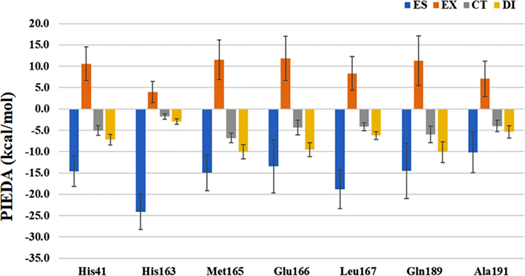
Standard image High-resolution imageFigure 5 plots the averaged PIEDA values (compiled in Table S8) of the leading residues in the interactions between Mpro and N3. The ES term is the main source of stabilization of hydrogen bonds, and its amount of His163 is the largest one. As can be seen in Fig. 2, the N–H part of the ring in His163 is directed toward the carbonyl carbon of Fragment 4, and thus the ES contribution may be enhanced. In comparison with the "static" PIEDA results in Ref. 13, contributions from the CT term are decreased. On the other hand, the DI term plays a supplementary role in stabilization of the "dynamic" situation, especially for Met165 and Gln189. His41 is interacting with Fragment 5 primally by hydrogen bond associated with its N–H part in the ring. This situation is contrasted to another case in which His41 rather has ππ stacking stabilizations with aromatic rings of different (not N3) ligands. 10)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 5. (Color online) Averaged PIEDA value of leading residues interacting (more than −10 kcal mol−1 of total stabilization) with Mpro by a thousand of "dynamic" structures, Blue, orange, gray, and yellow bars correspond to ES, EX, CT, and DI terms, respectively. Vertical line is the standard deviation. Refer to Table S8 for numerical data.
Download figure:
Standard image High-resolution image{kind=link}
As we have seen so far, there are several differences between the "static" and "dynamic" structures in interactions between Mpro and N3. We analyzed the correlation between "dynamic" IFIE values and interacting distances, and the results are given in Fig. S8 and Table S9. As expected, the values of correlation coefficient have sizable values of 0.6–0.7, except for some cases. This fact implies that thermal fluctuations in a realistic condition certainly affect the interactions among the N3 ligand and surrounding residues.
In the present letter, we have reported a combinative approach of classical MD and ab initio FMO for the Mpro—N3 complex, which indicated the importance of statistical evaluation of interactions by employing a thousand of fluctuating structures. The degree of reality to simulate physiological condition was thus enhanced in interaction analyses. Recent publications 6,9–12,49–51) related with SARS-CoV-2 proteins have shed light on the "dynamic" aspects, and our work has been just along this line. The developments of biomaterials 52–58) have attracted attention and interest, and theoretical analyses and simulations could contribute to efficient designs and optimizations (e.g. Refs. 32–34). As demonstrated here, the combination of MD and FMO with an amount of sampled structures has now been made practical by using massive computing resources such as the Fugaku supercomputer. We would hope that this approach will be applied to various systems in the nano-biotechnology field of applied physics. 59)
Acknowledgments
For FMO calculations with ABINIT-MP, computational resources of the supercomputer Fugaku (the evaluation environment in the trial phase) were provided by the RIKEN Center for Computational Science (R-CCS) under a special project against COVID-19. Technical supports on Fugaku were provided by Drs. Kota Sakakura (FOCUS) and Hiromasa Watanabe (HPC Systems Inc.). All MD simulations were performed on TSUBAME3.0 at Tokyo Institute of Technology under a funding by AMED/BINDS (JP20am0101113). The present work was supported also by Rikkyo SFR as well as JSPS Kakenhi (JP17H06353, JP18K03825, and JP19K12010).