
Abstract
The additive effect of a gadolinium ion into a formamidinium lead iodide (FAPbI3) perovskite crystal on electronic structures and molecular dynamics was investigated for improving photovoltaic performance with stability. The electronic structures, band structure, partial density of state, and molecular dynamics were determined by first-principles calculation. The band distribution and charge transfer between the 5d orbital of the gadolinium atom, the 5p orbital of the iodine atom, and the 6p orbital of the lead atom promoted the carrier generation and diffusion related to short-circuit current density. The enthalpy and kinetic energy prompted stabilization of the gadolinium-doped crystal with a slight distortion of coordination structure, as compared with the decomposition of the FAPbI3 crystal. Diffusion coefficients of iodine and lead ions in the FAPbI3 crystal with defect were increased, predicting decomposition. The gadolinium-doped FAPbI3 perovskite crystal has great potential for applications in photovoltaic devices by improving photovoltaic performance.
Export citation and abstract BibTeX RIS
1. Introduction
Fabrication and characterization of lanthanide-doped methyl ammonium lead iodide CH3NH3PbI3 (MAPbI3) perovskite solar cells have been performed to improve photovoltaic performance with long-term stabilities of conversion efficiency based on the quality of perovskite film. 1–3) Crystal engineering 4) and material design have been performed by the composition control of chemical elements such as alkali metal ions 5) and organic cations 6–9) as the A-site, transition metals, 10–13) lanthanides 14–16) as the B-site, and halogen ions 17) as the X-site in the ABX3 type of perovskite crystal. The addition of doping material into the perovskite layer optimized the electronic structure of band dispersion with effective mass related to the carrier mobility, morphology, and crystal orientation, improving short-circuit current density and open circuit voltage and therefore the photovoltaic performance as a result. 18,19) The morphological modification and interface passivation are supported to expand the distance and life of the carrier diffusion in the perovskite layer with the monodispersed crystal grain and suppression of defect and pinhole, improving the photovoltaic performance. 20–22)
The lanthanide-doped perovskite crystal has the advantage for application in commercial products of photovoltaic devices by overcoming various challenges such as photovoltaic performance, wavelength conversion, 23,24) ultra-visible-near-infrared absorption and emission, 25) electron transition between energy levels of f-orbital in lanthanide ion, 26) and morphological modification based on interface passivation with reducing defect and pinhole in perovskite film. 27) For example, the partial substitutions of europium, 28,29) cerium, 30) and neodymium compound 31) promoted carrier diffusion on the uniform internal structure with a reformation of perovskite crystal, yielding long-term stability by a redox reaction. The photovoltaic performance depended on the band structure associated with delocalized 5d and 4f orbitals of the lanthanide ion, and the electron correlation of the lanthanide ion and ligands in the coordination structure. The band dispersion with a charge transfer between the 5d and 4f orbitals of the lanthanide ion and the 5p orbital of iodine ion near the valence and conduction band states cause a decrease of effective mass related to an increase of the carrier mobility, improving the short-circuit current density and conversion efficiency. Car-Parromello molecular dynamics (CPMD) simulations have been used for investing crystal stabilization, the kinetics of crystal formation, 32,33) and interface passivation near grain boundaries, and charge recombination dynamics in perovskite crystal. 34,35)
The purpose of this research is to investigate the additive effect of lanthanides such as europium (Eu) and gadolinium (Gd) introduced into the formamidinium lead iodide (CH(NH2)2PbI3: FAPbI3) perovskite crystal for improving carrier diffusion related to mobility and short-circuit current density as well as photovoltaic performance and stability. The electronic structures and molecular dynamics of lanthanide-doped perovskite crystals were calculated by first-principles calculation. The correlation of band dispersion, the partial density of state (PDOS), the total DOS (TDOS), the electron density distribution, enthalpy, kinetic energy, and structure distortion were considered for clarifying the photovoltaic mechanism and the crystal stability. The diffusion coefficients of lead, iodine, and hydrogen ions at formamidinium (FA) in the FAPbI3 crystal were estimated for determining the crystal stability while suppressing the decomposition by desorption of ions from the crystal. The theoretical analysis predicated the photovoltaic characteristics and stability.
2. Calculation
The Gd2+-doped FAPbI3 perovskite crystals were formed in a cubic crystal phase using experimental lattice constants (FAPbI3: a = 6.3621 Å)
36–38) measured by X-ray diffraction data. The FAPbI3 perovskite crystals (cubic  ) were constructed with supercells (2 × 2 × 2). The mole ratio of the Gd2+ ions to Pb2+ cation ion was adjusted to 1:8, which was equivalent to a replacement of 12.5%. The ab initio quantum calculations were performed using the Vanderbilt ultrasoft pseudopotentials, scalar relativistic generalized gradient approximations, and density functional theory (DFT) without consideration of the spin–orbital coupling effect (Quantum Espresso, v.5.2.1, Quantum Espresso Foundation, UK), and pbe and pz pseudopotentials. Plane-wave basis set cutoffs for the wave functions and charge densities were set at 30 and 320 Rydberg (Ry). The perovskite crystals were composed of the experimental lattice constants. A uniform k-point grid (8 × 8 × 8) in the Brillouin zone was used to calculate the electronic structure and PDOS.
) were constructed with supercells (2 × 2 × 2). The mole ratio of the Gd2+ ions to Pb2+ cation ion was adjusted to 1:8, which was equivalent to a replacement of 12.5%. The ab initio quantum calculations were performed using the Vanderbilt ultrasoft pseudopotentials, scalar relativistic generalized gradient approximations, and density functional theory (DFT) without consideration of the spin–orbital coupling effect (Quantum Espresso, v.5.2.1, Quantum Espresso Foundation, UK), and pbe and pz pseudopotentials. Plane-wave basis set cutoffs for the wave functions and charge densities were set at 30 and 320 Rydberg (Ry). The perovskite crystals were composed of the experimental lattice constants. A uniform k-point grid (8 × 8 × 8) in the Brillouin zone was used to calculate the electronic structure and PDOS.
With the Gd2+-doped perovskite crystal as supercells (2 × 2 × 2), parts of the Pb2+ atom at the B-sites were substituted with Gd2+ ion for one-atom substitution at the center of the cubic structure. For the DFT calculation, a total charge in the Gd2+-doped perovskite crystal was set to 0.0 as a neutral state. The electron density distribution was optimized by a self-consistent field calculation. The atomic populations in the perovskite crystal were calculated by Löwdin population analysis. The number of charges in gadolinium ion was estimated by subtracting the total electron density from the number of electrons. The charge balance of gadolinium, lead, iodine ions, and FA cations were optimized to be at a neutral state in the perovskite crystal. The band structures and effective mass ratios of electrons and hole to free electrons (me*/me and mh*/me) and band gaps (Eg) were analyzed for the Brillouin zone of the perovskite crystal along the direction of the wave vector. The path for the Gd2+-doped FAPbI3 perovskite crystals was set as follows, Γ (0, 0, 0)  X (0, ½, 0)
X (0, ½, 0)  M (1/2, 1/2, 0)
M (1/2, 1/2, 0)  Γ
Γ  R (1/2, 1/2, 1/2)
R (1/2, 1/2, 1/2)  X, M
X, M  R. The Pb2+ cation was set at the position of Γ (0, 0, 0). The energy levels were standardized with the Fermi energy at zero. The PDOS and TDOS were calculated to make the energy level clear for each orbital near the valence (VB) and conduction (CB) band states.
R. The Pb2+ cation was set at the position of Γ (0, 0, 0). The energy levels were standardized with the Fermi energy at zero. The PDOS and TDOS were calculated to make the energy level clear for each orbital near the valence (VB) and conduction (CB) band states.
Car–Parrinello molecular dynamics (CPMD) simulations have been carried out with Quantum Espresso using the PBE exchange-correlation functional with pbe-mt.fhi.upf and pz-sp-high.upf pseudopotentials. Plane-wave basis set cutoffs for the smooth part of the wave functions and the augmented densities were 30 and 120 Ry, respectively. The CPMD simulations were performed using an integration time step of 150 for a total simulation time of 0.018 psec. Thermodynamic behavior based on enthalpy, kinetic energy, and molecular dynamics of Gd2+, Ce4+, Sm2+, and Eu2+-doped FAPbI3 perovskite crystals and VPb, VI, and VFA-FAPbI3 crystals with defect at each position (VPb, VI and VFA defects at the positions of Pb2+ and I− ions, and FA+ cations) were calculated by CPMD. Diffusion coefficient (D) of Pb2+, I− and H+ ions in the FAPbI3 crystal with defect at the positions of Pb2+ and I− ions, and FA+ cations were calculated by Born–Oppenheimer molecular dynamics (BOMD) simulations under a temperature control of around 300 K.
3. Results and discussion
The electron density distribution at the profile plane, cross-section, band structure, and PDOS of the Gd2+-doped FAPbI3 perovskite crystal are shown in Fig. 1. The electron density distributions of the d, 4f orbital of Gd2+ ion and the 5p orbital of iodine ion were delocalized, determining the charge transfer and the carrier generation in the perovskite crystal. The atomic population of 5d and 4f orbitals in gadolinium ion were calculated as follows, 5d (spin up: 5d = 0.73, dz2 = 0.22, dxz = 0.09, dyz = 0.09, dx2–y2 = 0.23, dxy = 0.10, spin down: 5d = 0.63, dz2 = 0.19, dxz = 0.08, dyz = 0.08, dx2–y2 = 0.20, dxy = 0.08), and 4f = 7.10. The number of charges in the gadolinium ion was estimated to be about 1.42 while maintaining the neutral state in the crystal. Recently, the chemical states in the perovskite crystals were investigated by near-edge X-ray absorption of fine structure, high-resolution X-ray photoelectron spectroscopy and Auger electron spectroscopy measurements, electron, and charge density, and ab initio MD simulations. 39–42)
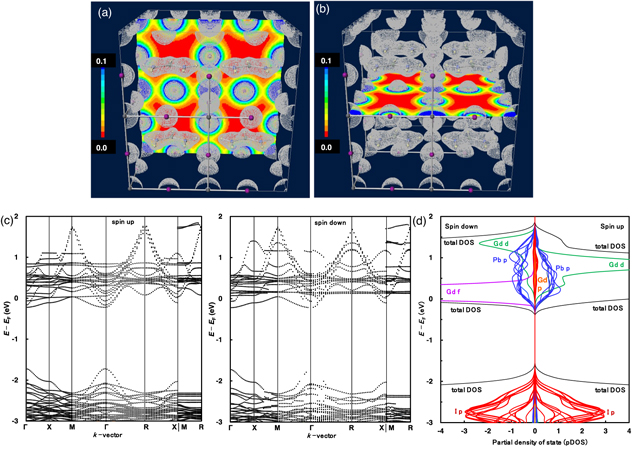
Fig. 1. (Color online) (a) Profile plane and (b) cross-section images of electron density distribution, (c) band structures and (d) PDOS of the Gd2+-doped FAPbI3 perovskite crystal.
Download figure:
Standard image High-resolution imageThe band structure had the narrow band distribution of 5p orbital of iodine ion near VB state, and 6p orbital of the lead ion, degenerated 4f orbital, and split 5d orbitals of Gd2+ ion near the CB state as shown in Fig. 1(c). The band gap of Eg was decreased to 1.52 eV, as compared to that of 1.62 eV in the Eu2+-doped FAPbI3 crystal.
28,29) The narrow band distribution near VB and CB states had an effective mass ratio (me*/me = 0.02 and mh*/me = 0.02) along the direction of the wave vector of Γ  R. These behaviors indicate the semi-conductive characteristics as similar behavior in the Eu2+-doped FAPbI3 crystal with effective mass ratio (me
*/me = 0.03 and mh
*/me = 0.01). These results expect an increase in the carrier mobility related to short-circuit current density as the photovoltaic performance. The total energies of the Gd2+-doped FAPbI3 crystal were decreased to −4662 eV cell−1, as compared with those of Eu2+-doped FAPbI3 crystal and FAPbI3 crystals to −4582 eV cell−1 and −3745 eV cell−1.
28,29) These results expect that the Gd2+-doped FAPbI3 crystal has great potential to apply the photovoltaic devices with the improved performance and the stability.
R. These behaviors indicate the semi-conductive characteristics as similar behavior in the Eu2+-doped FAPbI3 crystal with effective mass ratio (me
*/me = 0.03 and mh
*/me = 0.01). These results expect an increase in the carrier mobility related to short-circuit current density as the photovoltaic performance. The total energies of the Gd2+-doped FAPbI3 crystal were decreased to −4662 eV cell−1, as compared with those of Eu2+-doped FAPbI3 crystal and FAPbI3 crystals to −4582 eV cell−1 and −3745 eV cell−1.
28,29) These results expect that the Gd2+-doped FAPbI3 crystal has great potential to apply the photovoltaic devices with the improved performance and the stability.
Enthalpy and kinetic energy of ions and cells in (a) Gd2+, Ce4+, Sm2+-doped FAPbI3, VPb-, VI- and VFA-FAPbI3 perovskite crystal (VPb, VI and VFA: defect at Pb and I ions, and FA cation position) were calculated as shown in Figs. 2(a)–2(b). As shown in Fig. 2(a), the addition of lanthanide ions and defects in the perovskite crystal increased the enthalpy, suggesting instability as compared with that of the FAPbI3 crystal. The kinetic energies of ions and cells were slightly increased in the Gd2+- and Sm2+-doped perovskite crystal at the final stage. The molecular dynamics of the FAPbI3 perovskite crystal were performed. As shown in Fig. 2(c), the separation between nitrogen and hydrogen atoms in FA was caused at a time of 0.0169 ps. In contrast, the molecular dynamics of the Gd2+-doped perovskite crystal were also performed. A slight distortion of the coordination structure with the Gd-I band was caused at a time of 0.0169 ps. Addition of Gd2+ ions into the crystal supported stabilization of the perovskite crystal while suppressing decomposition based on the desorption of ions from the crystal, as shown in Fig. 2(c). The enthalpy and kinetic energy of the Eu2+-doped FAPbI3 perovskite crystal and the FAPbI3 perovskite crystal were calculated as shown in Fig. 2(b). The CPMD molecular dynamics was performed by using pz pseudopotential. The enthalpies for both crystals were gradually decreased as structure relaxation in the same way. The kinetic energy of ions and cells in the Eu2+-doped FAPbI3 perovskite crystal was slightly increased at the final stage. The kinetic energy was similar to molecular dynamic behavior, as compared with those of the Gd3+-FAPbI3 perovskite crystal. The molecular dynamic behavior exhibited the slight distortion of the chemical band between the Eu2+ ion and iodine ion as ligands in the perovskite crystal, as shown in Fig. 2(c).
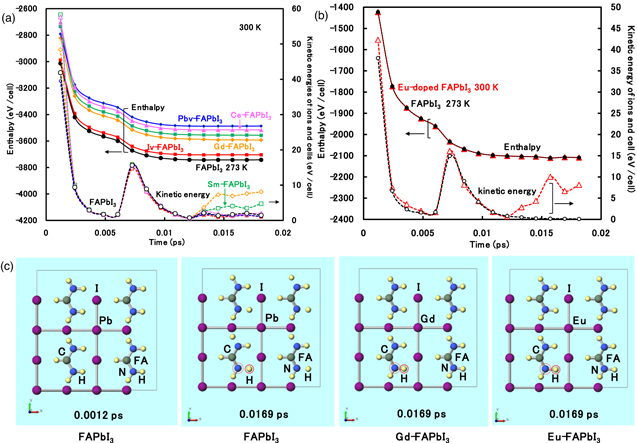
Fig. 2. (Color online) Enthalpy and kinetic energy of Gd2+, Ce4+, Sm2+, and Eu2+-doped FAPbI3 perovskite crystal and VPb, VI and VFA-FAPbI3 crystals with defect at each position (VPb, VI and VFA: defect at the position of Pb2+ and I− ions, and FA+ cation), calculated by CPMD simulations using (a) pbe-mt.fhi.upf and (b) pz-sp-high.upf pseudopotentials, and (c) Molecular dynamics at each moment.
Download figure:
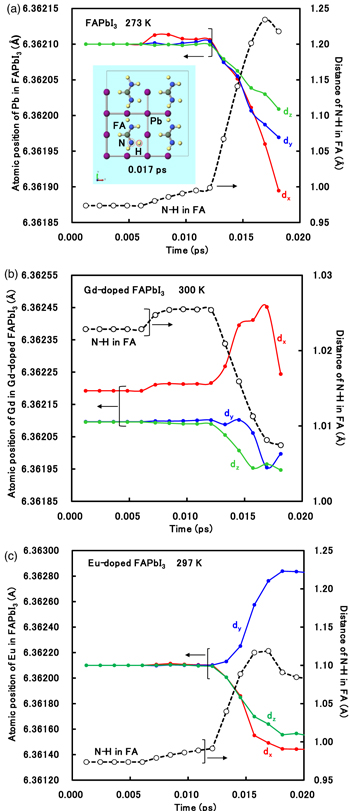
Standard image High-resolution imageThe molecular dynamics of the Gd2+- and Eu2+-doped perovskite crystals as compared to those of the FAPbI3 crystal are shown in Figs. 3(a)–3(c). As shown in Fig. 3(a), the atomic position of the Pb2+ ion in the FAPbI3 perovskite crystal was drastically decreased, shrinking the crystal field in the perovskite crystal. The distance of N-H in FA was increased, as the separation between N-H bands in FA at the final stage. These behaviors indicate the instability and decomposition with the desorption of hydrogen atoms. In the case of Gd2+-doped perovskite crystal, the addition of Gd2+ ion into the perovskite crystal promoted a gradual increase of the atomic position of the Gd2+ ion at x-direction, and a slight decrease of the position at y and z-position, as shown in Fig. 3(b). These behaviors indicate slight distortion of coordination structure based on band interaction between Gd2+ ion and iodine ion as ligands. The distance between N and H atoms in FA was decreased, yielding stability of FA. In the case of Eu2+-doped perovskite crystal, the position of the Eu2+ ion was increased in the y-direction, and decreased in the x and z directions. These behaviors indicate distortion in coordination structure, expecting 5d orbital splitting of Eu2+ ion in the band structure. The distance between N and H atoms in FA was influenced at the final stage in the range of 0.012–0.020 ps. The slight behavior of molecular dynamics of Eu2+ and FA ions was obtained. These results indicate that the perovskite crystal remains a stable structure with suppression of desorption of hydrogen atoms from FA in the perovskite crystal.
{kind=link}
{kind=link}
Fig. 3. (Color online) Molecular dynamics of (a) the FAPbI3 crystal, (b) Gd2+-doped FAPbI3 crystal, and (c) Eu2+-doped FAPbI3 crystal, calculated by CPMD.
Download figure:
Standard image High-resolution image{kind=link}
The D of ions in the FAPbI3 crystal was calculated as listed in Table I. In the case of FAPbI3 crystal at a temperature of 309 K, D of the I−, Pb2+ ions, and H+ ions in FA were calculated to be 8.64 × 10−6, 8.60 × 10−7 and 4.02 × 10−4 (cm2 s−1), respectively. These results indicate that desorption of hydrogen ions and I− ions caused decomposition. In the case with defect near atomic position at Pb2+ and I− ions, D of I− and Pb2+ ions in VPb-FAPbI3 and VI-FAPbI3 crystal was increased, as compared with those values of the FAPbI3 crystals. These behaviors indicate the promotion of diffusion for I− and Pb2+ ions through defects. The diffusion coefficient for H+ ion in FA was slightly decreased by trap near the defect. In the presence of a defect, the decomposition was caused by desorption and diffusion of hydrogen and iodine ions in the perovskite crystal. The addition of Gd2+ and Eu2+ ions into the crystal caused a slight distortion in the crystal field, stabilizing the crystal while suppressing of the decomposition with desorption and diffusion of the ions in the crystal. The stability of the performance would be explained by the thermodynamic characteristics based on molecular dynamics. The addition of lanthanide ions into the perovskite crystal supported the stability of the photovoltaic performance. The calculation prediction expects that the Gd2+-doped perovskite crystals have a high potential to apply for the commercial products of photovoltaic devices with photovoltaic performance and stability.
Table I. Diffusion coefficient of I−, Pb2+, and H+ ions in the FAPbI3 crystal with defect at the position of Pb2+ and I− ions, and FA+ cation, calculated by BOMD simulations under temperature control around 300 K.
| D (cm2 s−1) | ||||
|---|---|---|---|---|
| Perovskites | I− | Pb2+ | H+ (FA) | Temperature (K) |
| FAPbI3 | 8.64 × 10−6 | 8.60 × 10−7 | 4.02 × 10−4 | 309 |
| VPb-FAPbI3 | 9.19 × 10−6 | 3.20 × 10−6 | 3.13 × 10−4 | 307 |
| VI-FAPbI3 | 1.10 × 10−5 | 3.75 × 10−6 | 1.97 × 10−4 | 324 |
| VFA-FAPbI3 | 2.41 × 10−6 | 7.90 × 10−7 | 1.04 × 10−4 | 282 |
4. Conclusions
The additive effect of Gd2+ ion into the perovskite crystal on electronic structure and thermodynamic behavior based on molecular dynamics were investigated. The electronic structures such as the band distribution, PDOS, electron density distribution, enthalpy, kinetic energy, and molecular dynamics were expected by first-principles calculation. Incorporation of Gd2+ ions into the perovskite crystal caused the delocalization of 5d, 4 f-orbital of Gd2+ ion, 6p orbital of Pb2+ ion, and 5p orbital of I− ion as ligand, promoting the charge transfer between 5d orbital of Gd2+ ion and 6p orbital of Pb2+ ion with narrow band dispersion related to carrier mobility, expecting an increase of short-circuit current density as the photovoltaic performance. The enthalpy and kinetic energy based on the molecular dynamics indicate stabilization of the Gd2+-doped perovskite crystal with slight desorption of coordination structure, as compared with the decomposition of the MAPbI3 perovskite crystal. Diffusion coefficients of iodine and lead ions in the crystal with defect were increased, causing the decomposition. The calculation prediction expects that the Gd2+-doped perovskite crystals have great potential for application in the industrial products of photovoltaic devices with photovoltaic performance related to short-circuit current density and stability.
Acknowledgments
This work was supported by JSPS KAKENHI Grant No. JP 21K05261.