Abstract
The electroreduction of peroxodisulfate has been studied on various electrode materials for decades. However, the reduction of peroxodisulfate is not fully understood to date. Many research groups have searched for an electrode material that exhibits a diffusion-controlled response in the electroreduction of peroxodisulfate. However, the voltammetric responses obtained on most electrode materials have shown a kinetically-controlled reaction. Reaction mechanisms of peroxodisulfate reduction have also been proposed by a few groups. This review paper focuses on the electrochemical responses for the reduction of peroxodisulfate obtained on platinum, gold, platinized platinum, and bismuth-adsorbed platinum electrodes. This paper will not review the polarograms obtained on mercury drop electrodes for the reduction of peroxodisulfate anions because we believe that the results do not contribute to the understanding of the electrochemical reactions. We propose a simplified version of the reaction mechanisms. This paper also reviews the kinetic models proposed by several groups which could assist the readers to apprehend the kinetic behaviors of peroxodisulfate reduction. This paper discusses several issues related to peroxodisulfate from the natural decomposition of peroxodisulfate to the reaction mechanisms of peroxodisulfate reduction.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Peroxodisulfate (S2O82−), also known as peroxydisulfate or persulfate, is a very strong oxidizing agent.1–5 Peroxodisulfate is available in the form of three inorganic salts: ammonium peroxodisulfate, potassium peroxodisulfate, and sodium peroxodisulfate.6,7 Peroxodisulfate salts are non-hygroscopic, highly soluble in water, and have a relatively long shelf-life.7
More than 65% of peroxodisulfate produced is being used as a polymerization initiator.6,8–11 It has also been utilized in hair bleaches and lighteners,12–22 wastewater and water treatment, soil and groundwater remediation,7,23 and etching of copper on circuit boards.6,24–28 Peroxodisulfate has also been reported to be employed in the preparation of soluble starch, medical drugs, cellophane, rubber, soaps, detergents, adhesive papers, dyes for textiles, and as a reducer or retarder in photography.13,28–30 Peroxodisulfate is cheaper than other oxidants but it is more expensive than hydrogen peroxide for large-scale operations.7 However, hydrogen peroxide may not be as effective as persulfate or as suitable for use in some processes. For instance, persulfate is preferred in the remediation of groundwater as it is relatively stable in solution, but can be heat activated to produce sulfate radicals; these radicals are very reactive with a variety of contaminants.31
Wacławek et al.7 claimed that peroxodisulfate anions are stable in purified water for several months. The decomposition reactions could be very slow even though the standard free energy change is large;1 this is reflected in the long life of persulfate anions in aqueous solutions.32 House1 states that the half-life of S2O82− in acidic aqueous solutions at room temperature is about 80 days for 0.05 M H+ based on the first-order rate constant.32 Bartlett and Cotman also claimed that the decomposition of peroxodisulfate is first order.33 Even though persulfate anions are stable in purified water for a long time, it is recommended that peroxodisulfate solutions are prepared fresh on each day prior to conducting experiments. Nonetheless, peroxodisulfate anions can decompose at 25°C even in the absence of a catalyst.1,32
The decomposition reaction can be instigated by dust, impurities, and contaminants as well as light1,34 since persulfate anions have been reported to be photosensitive.35 The reaction is initiated by the homolysis of the peroxide bond as follows:1,36
![Equation ([R1])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0001.gif)
where hv represents energy imparted by photons. This means that every piece of apparatus and glassware has to be thoroughly cleaned before conducting experiments, especially when surfactants such as Decon 90 are involved in the cleaning procedure. One of the possible ways to remove surfactants is to use highly-pressurized water when washing the glassware.
Persulfate anions can directly oxidize contaminants. This process, however, tends to be slow.32 Thus, sulfate radicals produced from the decomposition of persulfate are of interest for decontamination purposes as they are very reactive with various contaminants.31 The decomposition rate of persulfate anions can be accelerated by using several activation methods: hydrogen peroxide activation, heat activation, alkaline activation, low-valent metal activation, and chelated-metal activation.32 However, Robinson et al.37 suggest that activation via hydrogen peroxide might actually proceed as a consequence the heat released from the decomposition of hydrogen peroxide. Thus, hydrogen peroxide activation can be excluded from the list due to the ambiguity of the reaction. Nonetheless, the decomposition of peroxodisulfate anions, Reaction R1, can be accelerated by heat.36 Thus, Reaction R1 should be replaced by:31
![Equation ([R2])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0002.gif)
where Δ is heat. The decomposition of persulfate anions can also be accelerated by hydrolysis or radiolysis of water as described by the following reactions:1,32,34,38
![Equation ([R3])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0003.gif)
in dilute acid or
![Equation ([R4])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0004.gif)
in concentrated acid or
![Equation ([R5])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0005.gif)
in neutral or basic solutions.
Persulfate anions can also undergo decomposition catalyzed by hydrogen cations in acidic solutions as follows:39,40
![Equation ([R6])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0006.gif)
Other than these reactions, Siegrist and colleagues32 explained the activation of persulfate in an alkaline environment (> pH 11); yet, the precise mechanism is still unknown. In addition, the decomposition reaction can be catalysed by a redox process with an electron donor including low-valent metals, Mn+, such as Fe2+ or Ag+;41 such catalysed reactions can be expressed as:31
![Equation ([R7])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0007.gif)
Every decomposition reaction of peroxodisulfate may play a major role in distorting electrochemical results because the products of these reactions could interfere with the electroreduction of persulfate.
Bartlett and Cotman33 suggest that the sulfate radicals produced from the decomposition of persulfate do not seem to react with persulfate anions. They claim that autocatalysis is not observed in thermal decomposition. However, the increase in the decomposition rate of peroxodisulfate ions in the presence of reducing agents could be due to other radicals produced from the reaction between the persulfate anions and the reducing agents; the radicals react with the peroxodisulfate anions and this increases the rate of persulfate dissociation.1 For example, the reaction between sulfate radicals from the dissociation of peroxodisulfate, Reactions R1, R2, and R7, and hydroxide ions in alkaline solutions can produce hydroxide radicals as follows:32
![Equation ([R8])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0008.gif)
Therefore, the produced hydroxide radicals could increase the rate of peroxodisulfate decomposition. Although the rate of natural decomposition might be slow, it could still affect the electroreduction of persulfate anions which will be discussed in Electrochemical behavior of peroxodisulfate and Diffusion coefficient of peroxodisulfate sections. Nonetheless, this suggests that additional care must be taken when designing experimental work.
This paper reviews on the electrochemical behavior of peroxodisulfate studied on various materials that have been reported in the literature throughout the years. To the best of our knowledge, no group has yet reviewed the electrochemistry of peroxodisulfate aqueous solutions, despite a long history. Several review papers have been written on the applications of peroxodisulfate in the environmental field,42,43 however, the electrochemical behavior of peroxodisulfate still remains a mystery waiting to be solved. We believe that this review article will be beneficial in illuminating the pathway to a better understanding of the reduction of peroxodisulfate at electrode/electrolyte interfaces for a range of readers. This article provides guidance and insights on performing electroreduction of peroxodisulfate. The objective of this review is to summarize the results obtained from these studies mainly for platinum (Pt), gold (Au), nanostructured Pt, and bismuth-adsorbed Pt electrodes as well as to propose a simplified version of reaction mechanisms for peroxodisulfate reduction. The next section briefly describes the preparation of persulfate. Following this, the electroreduction of persulfate on several types of electrodes is discussed in Electrochemical behavior of peroxodisulfate section, starting with Pt electrodes in Platinum electrodes section, Au electrodes in Gold electrodes section, platinized Pt electrodes in Platinized Pt electrodes section, bismuth-adsorbed Pt electrodes in section Bismuth-adsorbed Pt electrodes and finally, discussion of other electrode materials can be found in Other electrode materials section. In The role of metal oxides section we briefly describe how the presence and formation of metal oxides can affect the rate of reduction. The measurement of peroxodisulfate diffusion coefficients, with particular attention on experimental design is described in Diffusion coefficient of peroxodisulfate section. We then describe the mechanisms of the electroreduction of persulfate and present a simplified picture of the reaction mechanisms in Mechanism of peroxodisulfate reduction section. Kinetic behavior of peroxodisulfate at metal electrode/electrolyte interface section explains thoroughly the work that has been done in determining the kinetic behavior of peroxodisulfate at the metal-electrode/electrolyte interface. Detection of peroxodisulfate anions section discusses the methods that have been used to detect persulfate anions in solution. Finally in Future goals section, we present an outlook for the future of this important area of research.
Preparation of Peroxodisulfate
Peroxodisulfate salts can be produced by means of chemical or electrochemical reactions. Ammonium peroxodisulfate salts are manufactured by applying very positive overpotentials, between 5 and 7 V, or high current density, between 0.5 and 1 A cm─2, in solutions containing ammonium sulfate and sulfuric acid.6 The concentration of sulfate anions in the solution has to be > 2 M.44 As for the electrodes, lead (Pb) or graphite is employed for cathodes and Pt was conventionally employed for anodes.6 The concentration of peroxodisulfate produced in the solution after electrolysis is between 1 and 1.5 M.6 Two major drawbacks of utilizing Pt anodes for this purpose are: the production inefficiency of peroxodisulfate because of oxygen gas evolution and the inability to maintain a chemically pure solution due to the accumulation of corrosion products leads to the ultimate failure of this approach;44 it is possible that these issues were not considered during the early period of mass production. According to Pourbaix,45 Pt metals dissolve as Pt2+ cations in strong acidic aqueous solutions during anodic polarization. Since a high concentration of sulfate anions is required and very positive overpotential is applied to produce peroxodisulfate salts, Pt corrosion will occur.
To address these setbacks, boron-doped diamond (BDD) anodes have been proposed for the synthesis of persulfate salts as they have a very wide potential window, ∼ 3 V in 1 M sulfuric acid.44 Khamis and co-workers44 reported that the overpotential needed to generate peroxodisulfate anions using BDD electrodes in sulfuric acid is > 2.1 V vs. Ag/AgCl. Since oxygen evolution for BDD electrodes only occurs beyond 3 V in 1 M sulfuric acid, their efficiency to produce persulfate anions should be high. Unfortunately, BDD electrodes do not always have a wide solvent window. The potential window or solvent window of stability is the working potential range of an electrode before the electrolysis of water takes place.46 Macpherson46 reported that the potential window of BDD electrodes depends on the amount of non-diamond carbon (NDC) and the pH of electrolytes; this is because the electrolysis of water requires the presence of catalytic sites on the electrode surface as it is an inner sphere redox process.46 NDC on BDD electrodes provides the binding sites needed for the reaction to proceed; BDD electrodes with a large amount of NDC exhibit a higher electrocatalytic activity toward water electrolysis than those with smaller amounts of NDC.46 Therefore, the potential window of BDD electrodes with high amounts of NDC is shorter than those with a lower quantity of NDC. Hence, the production of peroxodisulfate using BDD electrodes with high boron concentration could be inefficient due to oxygen gas evolution, as they tend to contain high amount of NDC. Macpherson46 suggests the use of BDD electrodes with low boron concentration for any processes. Nevertheless, the incorporation of NDC into BDD electrodes during film growth is unavoidable.46 There are reports that suggest several methods to reduce the amount of sp2 carbon on BDD electrodes.46 The quantity of NDC on BDD electrodes could be decreased by acid washing followed by anodic polarization or re-hydrogenation of the surface through continuous potential cycling in acid solutions.47,48 Another approach to reduce the amount of NDC is to use a slow cool-down procedure in the presence of atomic hydrogen after depositing BDD films.49 However, it is now possible to prepare BDD electrodes with high concentration of boron, containing insignificant amounts of NDC, due to the improvements in film deposition techniques, over the years;50 these electrodes are likely to exhibit large potential windows. BDD electrodes with large potential windows should be utilized to ensure high efficiency in producing peroxodisulfate anions or salts.
Potassium peroxodisulfate and sodium peroxodisulfate salts can also be produced via electrochemical reactions using potassium sulfate and sodium sulfate respectively,6 instead of ammonium sulfate. Peroxodisulfate salts can also be made by a metathesis reaction using ammonium peroxodisulfate as follows:6
![Equation ([R9])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0009.gif)
![Equation ([R10])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0010.gif)
The three types of persulfate salts are commercially available from most chemical suppliers. Peroxodisulfate anions have been produced using BDD electrodes under an acidic environment to treat water and wastewater.51 Some researchers are interested in investigating the efficiency of BDD electrodes in generating persulfate anions.44 However, some important issues need to be addressed in advance, which will be explained in Gold electrodes and Diffusion coefficient of peroxodisulfate sections. The next section discusses the electrochemical behavior of peroxodisulfate probed using various electrode materials starting with Pt electrodes.
Electrochemical Behavior of Peroxodisulfate
Platinum electrodes
The electroreduction of peroxodisulfate gained popularity after T. A. Kryukova reported polarograms with an abnormal shape in 1949.52 The reduction of peroxydisulfate is relatively complicated and remains poorly understood. Many groups have studied the electroreduction of persulfate using rotating and stationary disk electrodes as well as several spectroelectrochemical techniques.
The investigation of peroxodisulfate reduction on Pt electrodes goes as far back as 1954 with the work of Nikolaeva and Grossmann;52–54 however, the experimental results were explained in terms of the "psi-prime" effect.52 Müller and colleagues55 were the first to conclude that the reduction of peroxodisulfate on Pt electrodes occurs via a slow dissociative chemisorption pathway relating to the splitting of the O─O bond; this phenomenon leads to the formation of adsorbed species or atoms, termed as adspecies or adatoms respectively hereafter, on the electrode surface.52 The reaction seems to be an inner sphere redox process that needs an adequate amount of adsorption sites. The proposed reactions are as follows:55
![Equation ([R11])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0011.gif)
![Equation ([R12])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0012.gif)
To date, this suggestion is well accepted by the electrochemistry community. Müller et al.55 also reported that the surface coverage of persulfate adspecies on Pt electrodes is 12% for 1 mM of peroxodisulfate.
The electroreduction of peroxodisulfate proceeds within the potential ranges where hydrogen adsorption usually occurs on Pt electrode surface.52 Cyclic voltammograms recorded using Pt electrodes demonstrate only one reduction peak assigned to the electroreduction of peroxodisulfate. Interestingly, cyclic voltammograms obtained using bare Pt electrodes possess similar characteristics regardless of the crystal structures of electrodes, pH of solutions, potential sweep rate, rotation speed, and concentrations of peroxodisulfate and supporting electrolytes: 1) there is no plateau region observed on the cyclic voltammograms despite the fact that rotating disk electrodes were employed, 2) the hysteresis between the forward and reverse scans is relatively large, 3) the currents obtained are decaying as the potential is swept to more negative values, and 4) the experimental currents are smaller than the theoretical currents; these electrochemical behaviors most probably indicate that the reactions are kinetically- or mixed-controlled. Other than that, the currents for positive-going/backward sweep are often larger than those for negative-going/forward sweep. Kokkinidis and colleagues56 suggest that the larger currents for the backward scans could be due to the surface activation after hydrogenation of adsorbed peroxodisulfate and its intermediates, Reaction R11, explained by the following reaction:
![Equation ([R13])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0013.gif)
However, this explanation is arguable as the currents for positive-going scans recorded using Au electrodes are also larger than those for negative-going scans as shown by Samec and co-workers5,57 in Figure 1, refer to the original papers. Unlike Pt electrodes, Au electrodes do not demonstrate hydrogen adsorption and desorption processes.58 Since the reaction is pH dependent, one possible explanation is that SO4─ adspecies react with hydrogen ions, H+, in the solution rather than hydrogen adspecies on the electrode surface as proposed by Reaction R13. This means that Reaction R13 should be replaced with SO4─(ads) + H+ → HSO4─. According to Reaction R13, SO4─ adspecies is released from the electrode surface after reacting with hydrogen ions making the electrode surface devoid of the adspecies; this event leads to the increase in currents obtained for the positive-going sweep. Since the reduction of peroxydisulfate on Pt electrodes occurs at the hydrogen adsorption region, Reaction R13 could still be taken into account. The protonation of sulfate has to be considered, as pH plays an important role in the reduction of peroxodisulfate.44 Yet, Zhutaeva and Tarasevich59 suggest that HSO4─ anions could strongly adsorb on Pt electrode surface; this behavior could also complicate the reduction of persulfate. Nevertheless, the HSO4─ anions might have desorbed afterwards since the potential is swept to very negative values. Nonetheless, the electrode surface is activated when cycling the potential to more negative values. The surface activation leads to higher currents for positive-going scans.
Climent and co-workers53 suggest that the current decay seen on cyclic voltammograms recorded using bare Pt rotating disk electrodes could be the result of the reaction between SO4─ and hydrogen adspecies on the electrode surface, Reaction R13. Botukhova and Petrii52 also speculate that the surface coverage of adsorbed hydrogen may have an effect on the reduction of peroxodisulfate. This means that the state of the electrode surface could be the reason for the current decay seen on the cyclic voltammograms acquired using Pt electrodes when sweeping the potential to more negative values as shown in the literature;52,53,56 both hydrogen and peroxodisulfate ions are competing for the adsorption sites on the electrode surface. However, current decay can also be seen for the cyclic voltammograms acquired using Au rotating disk electrodes as shown by Samec et al.5 As mentioned earlier, Au electrodes do not exhibit hydrogen adsorption and desorption processes.58 This means that the current decay might not be caused by the competition for adsorption sites between hydrogen and persulfate ions. Interestingly, Kokkinidis and colleagues56 reported that the applied overpotential decreases and the current density increases for the electroreduction of peroxodisulfate after holding the potential in the hydrogen adsorption region for a period of time; this demonstrates the electrocatalytic response of hydrogen adsorbed Pt rotating disk electrodes toward the reduction of persulfate. In addition, the authors showed that a plateau region was seen on the linear sweep voltammograms recorded for the reduction of peroxodisulfate after holding the potential at 0.05 V vs. standard hydrogen electrode (SHE) for 6 minutes in perchloric acid, HClO4 solution, Figure 2 in the original paper. Kokkinidis et al.56 claim that the limiting currents acquired using the hydrogen underpotential deposited Pt electrodes are diffusion-controlled, indicated by the sigmoidal linear sweep voltammograms and the linearity of the Levich plot, a plot of current density vs. square root of rotation speed. These electrochemical responses are obtained presumably because the adsorption sites are blocked by adsorbed hydrogen which forces peroxodisulfate anions to be reduced mainly via the direct electron transfer pathway; this implies that the current decay might be due to the complications coming from the adsorption of persulfate and its intermediates, Reaction R11, on the electrode surface. Once the peroxodisulfate and its intermediates are adsorbed on the electrode surface, the removal of these adspecies could be extremely difficult. Climent and co-workers53 reported that the onset of persulfate reduction shifts toward more positive potentials as the rotation speed is increased i.e. less overpotential is needed to electrochemically reduce persulfate anions at higher rotation rate. This behavior could be due to higher efficiency in removing the peroxodisulfate and its intermediate adspecies, Reaction R11, from the electrode surface at higher rate of mass transport.53 This suggests that the reduction of peroxodisulfate is fairly complex when using electrodes with low rate of mass transport due to the adsorption of persulfate and sulfate anions, Reaction R11, on the electrode surface.
Climent et al.53 showed that the crystal structure of the electrode does affect the reduction of peroxodisulfate but only in terms of the reduction potential. Their results showed that the peaks corresponding to the reduction of persulfate shifted to more negative potentials in the following order: Pt (111) (ca. 0.5 V vs. reversible hydrogen electrode, RHE) > Pt (100) (ca. 0.42 V vs. RHE) > Pt (110) (ca. 0.18 V vs. RHE); this implies that less overpotential is needed for Pt (111) to drive the reduction of peroxodisulfate. They also found that the reduction of persulfate is completely inhibited in the hydrogen adsorption region as the hydrogen adsorption and desorption peaks remain unchanged, Figure 1 in the original text; this is acceptable except for Pt (111) since the current decay starts at the double layer region.
To conclude this section, the electroreduction of persulfate is fairly complicated on Pt electrodes. The complications are thought to arise from the adsorption of persulfate anions on the electrode surface. It has been proposed that the reduction of persulfate proceeds via a dissociative chemisorption pathway. Other electrode materials could be considered to probe the electrochemical behavior of persulfate reduction. The next section reviews the electrochemical response obtained using Au electrodes.
Gold electrodes
The electroreduction of persulfate has been investigated on Au electrodes specifically because the interfacial structure of this electrode is well understood.5 Most of the studies on peroxodisulfate were done on Au rotating disk electrodes.3,5,60–62
The electrochemical response of persulfate reduction on Au electrodes is quite different than that on Pt electrodes. Samec et al.5 showed that the reduction of persulfate on Au electrodes occurs at two different regions, Figure 1 in the original paper; peaks attributed to the reduction of peroxodisulfate were seen at positive potentials, 0.4 V vs. saturated calomel electrode (SCE), and at more negative potentials, 0 V vs. SCE, in 5 mM HClO4. They suggest that the reduction peak for peroxodisulfate at 0.4 V vs. SCE could be related to the dissociative chemisorption of persulfate anions, Reactions R11 and R12, since the peak is affected by the scan rate but it stays unchanged when applying different rotation rates. Meanwhile, the reduction peak at 0 V vs. SCE was attributed to the direct electron transfer to peroxodisulfate anions as the peak is influenced by the rotation speed but it is unaffected by the scan rate.5 However, like Pt electrodes, the four characteristics of cyclic voltammograms for the reduction of persulfate mentioned in Platinum electrodes section can also be seen on cyclic voltammograms recorded using Au electrodes; this suggests that the reduction of persulfate on Au electrodes is either kinetically or mixed-controlled.
For the peak at 0 V vs. SCE related to the direct electron transfer to persulfate anions, Samec and colleagues5 ascribed the peak to the chemical equation proposed by Frumkin and Florianovich as follows:
![Equation ([R14])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0014.gif)
![Equation ([R15])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0015.gif)
The change in the reduction pathway from dissociative chemisorption on positively charged electrode surfaces to direct electron transfer on negatively charged electrode surfaces is associated with the electrostatic repulsion of divalent persulfate anions from the surface.5
Samec and co-workers5 reported that the influence of pH on the position or the magnitude of both persulfate reduction peaks is negligibly small. This is contrary to the findings that have been reported by Khamis and colleagues.44 However, Samec et al.5 reported that the Au oxide reduction peak may interfere with the persulfate reduction peak at more positive potentials, 0.4 V vs. SCE, when performing the experiments in electrolytes with neutral pH. This is certainly not an issue for acidic solutions such as perchloric or sulfuric acid as the oxide reduction process on Au electrodes under acidic environment occurs at more positive potentials than the reduction of persulfate.5
Samec et al.60 highlighted the shape of oxide formation region on cyclic voltammograms acquired using Au (111) electrodes in the presence and in the absence of peroxodisulfate. They showed that the oxide formation peaks on the cyclic voltammograms obtained in the presence of peroxodisulfate anions are similar to those recorded in the presence of sulfate anions even though sulfate ions are initially absent. This proves that an unknown amount of persulfate anions undergo natural decomposition to form sulfate anions (as discussed in Introduction section) before they get to be reduced electrochemically; assuming that the cyclic voltammograms shown are from the first cycle. Otherwise, sulfate anions could be coming from the electroreduction of persulfate anions, Reactions R11, R12, R14, and R15.
Samec and co-workers60 also showed the effect of employing single crystal Au electrodes exposing a (111) crystal plane. They demonstrate that the reduction peak current at 0.4 V vs. SCE associated with fast dissociative chemisorption was larger than that obtained with polycrystalline Au electrodes. However, the persulfate reduction peak at 0 V vs. SCE related to the direct electron transfer remains the same. This suggests that the reduction of peroxodisulfate is a surface sensitive reaction when it involves the adsorption of peroxodisulfate and its intermediates, Reaction R11, on the electrode surface. Samec et al.60 suggest that the reduction rate of adsorbed persulfate anions is higher on Au (111) compared to polycrystalline Au, due to a better matching of the peroxodisulfate structure with the trigonal structure of Au (111). The distance between oxygen atoms in various tetrahedral anions such as ClO4─ and SO42─ anions: 0.243 nm,63 and in SO4 groups in peroxodisulfate, 0.244 nm64 is quite similar to that between the centers of Au atoms, 0.288 nm.60 In other words, there are two causes for the enhancement of the current. Either it arises from an increased amount of peroxodisulfate anions adsorbed on the electrode surface and/or increased rate of electron transfer to the persulfate adspecies due to better matching of the persulfate anions with the Au (111) structure.60 Samec et al.57 went on to confirm that the improvement in the current is due to the increased rate of electron transfer to persulfate adspecies.
To summarize this section, the reduction of persulfate on Au electrodes is also complicated similarly to Pt electrodes. Two reduction peaks can be seen on the cyclic voltammograms recorded with Au electrodes in the presence of persulfate. The first persulfate reduction peak is located at more positive potentials, 0.4 V vs. SCE in HClO4 solution, and this peak is associated to the dissociative chemisorption pathway. The second peroxodisulfate reduction peak is situated at more negative potentials, 0 V vs. SCE in HClO4 solution, which is linked to the direct electron transfer to persulfate anions. The voltammograms recorded with Au electrodes share similar characteristics with those obtained using Pt electrodes. It is essential to search for an electrode material that can exhibit a diffusion-controlled reaction for the reduction of persulfate. The reduction of persulfate on platinized Pt electrodes is reviewed in the next section.
Platinized Pt electrodes
Most of the studies on the reduction of persulfate have been performed using platinized Pt rotating disk electrodes; these electrodes are quite different from mesoporous or nanostructured Pt electrodes. Nanostructured Pt thin films are electrodeposited in the presence of a template such as lyotropic liquid crystalline phases of surfactants. Lyotropic liquid crystalline phases contain structures that have long range periodicities and repeat distances ranging from 2 to 15 nm.65 In water, surfactants normally clump together into micelles or lyotropic liquid crystalline phases depending on the concentrations.66 Surfactant aggregates can be utilized as templates to prepare inorganic porous nanostructures.66 The electrodeposition of Pt confined to this aqueous environment results in the formation of Pt films with ordered, well-defined, long-ranged porous nanostructures and high specific surface areas.65 These films are recognized as mesoporous or nanostructured Pt films. Mesoporous materials are of high interest as catalysts because of their specific high surface areas and larger pore sizes than the conventional zeolites.65 Zeolites are categorized as microporous solids67 that contain pores with diameters of not more than 2 nm.68 To the best of our knowledge, the electroreduction of persulfate has never been studied on nanostructured Pt electrodes.
For platinized Pt electrodes, a Pt thin film is electrodeposited from chloroplatinic acid solutions in the absence of any templates.52,69 Yet, both mesoporous and platinized Pt electrodes provide high surface areas and large amount of adsorption sites. These features could result in higher electrocatalytic behavior since the reduction of peroxodisulfate on Pt electrodes proceeds with dissociative chemisorption of persulfate anions that requires adsorption sites.
Botukhova and Petrii52 showed that the cathodic currents for the reduction of persulfate recorded with platinized Pt rotating disk electrodes are higher than those obtained with bare Pt electrodes; this could be due to the higher surface area and large amount of adsorption sites. Since the reduction of peroxodisulfate is a surface process or inner sphere redox process, it demands a large number of catalytic sites to facilitate the interaction between the analytes and the electrode surface for an efficient electron transfer.46 The number of binding sites for platinized Pt electrodes is higher than that for bare Pt electrodes due to larger surface area; this feature aids the electron transfer between metal surface and peroxodisulfate. Thus, higher amounts of adsorption sites enable more peroxodisulfate molecules to interact with the metal surface; this leads to higher currents. Unfortunately, there is still no plateau region on the cyclic voltammograms recorded in the presence of persulfate and the hysteresis between the forward and backward scans is quite large as shown by Botukhova and Petrii,52 refer to Figure 6 in the original paper. Unfortunately, they did not compare the currents acquired using the platinized Pt electrodes with the theoretical currents. Similar electrochemical responses are shown by Nikiforova and Petrii70 in Figure 2 in their published work. Furthermore, the peaks attributed to the surface processes for Pt electrodes such as hydrogen adsorption and desorption as well as oxide formation and removal are enhanced due to higher amount of adsorption sites.52 Slow scan rates and high concentrations of persulfate have to be employed in order to suppress the increased background currents as a result of a larger surface area and greater amount of adsorption sites.52
These observations show that the reduction of peroxodisulfate is fairly complicated even on electrodes containing a large number of adsorption sites. However, this could be due to high concentrations of persulfate, 50 mM Na2S2O8, used in order to suppress the other surface processes such as hydrogen adsorption/desorption and oxide formation/stripping. The large number of adsorption sites cannot compensate the high concentration of persulfate anions in the solution. Platinized Pt electrodes are not suitable to be employed as persulfate sensors because they can only detect high concentrations of peroxodisulfate.
The large number of adsorption sites and high surfaces area might not be beneficial for the reduction of peroxodisulfate as discussed in this section. High concentration of persulfate is needed in order to suppress the other surface processes. The next section discusses the results obtained using bismuth-adsorbed Pt electrodes.
Bismuth-adsorbed Pt electrodes
The underpotential deposition of sub-monolayer amounts of foreign metal adspecies on the electrode surface through the immersion of Pt electrodes in a solution containing metal cations has attracted attention due to the interesting electrocatalytic properties.53,71 In 1985, Shibata and Motoo72 probed the likelihood of retaining the deposited metal adspecies, lead and bismuth, even after rinsing and transferring the electrodes to a solution in the absence of the metal cations. They claim that the loss of metal adspecies is less than 3% during the transfer which is virtually negligible. This means that metal adspecies can be deposited on electrode surfaces even without applying an overpotential, and remain adsorbed even after rinsing the electrode and transferring it into a solution in the absence of the metal cations of interest.53 Several metal adspecies such as lead,56,70 cadmium (Cd),70 thallium,56 and bismuth53,56 have been used to study the effects of adsorbed species on the reduction of persulfate. All of the groups have employed rotating disk electrodes for their work. However, two methods have been employed by researchers to decorate the electrode surface with metal adspecies: 1) the immersion of Pt electrodes in a solution containing metal cations of interest before being rinsed and transferred to a test solution in the absence of the metal cations and 2) the presence of metal cations in the test solutions.72 The latter method is not recommended as natural decomposition by low-valent metal could affect the reduction of persulfate, Reaction R7.
Kokkinidis et al.56 have reported that bismuth-adsorbed Pt electrodes are the most active electrocatalyst for the reduction of peroxodisulfate amongst thallium- and lead-adsorbed Pt electrodes in acidic solutions. The authors suggest that this is because bismuth adspecies tend to desorb from the electrode surface at more positive potentials than the other metal adspecies. Thus, the bismuth adspecies is still present when the potential is cycled to where the reduction of peroxodisulfate occurs.
A sub-monolayer amount of bismuth can be deposited on Pt electrodes simply by immersing the electrodes in a solution containing bismuth cations for a period of time (15 s – 3 min).53,73,74 It has been suggested that the electrons at the electrode surface are transferred into bismuth adsorbed atoms when they approach each other so as to reduce the electrostatic interaction; this phenomena changes the charge state of the bismuth adatoms.75,76
Clavilier et al.73 reported that hydrogen adsorption peaks on the cyclic voltammograms recorded using bismuth-adsorbed Pt electrodes in H2SO4 solutions are suppressed, Figure 1 in the original text. This implies that bismuth adspecies is hindering the adsorption of hydrogen at the electrode surface;73 this is probably because most of the binding sites have been occupied by bismuth adatoms. Thus, the surface coverage of bismuth adspecies on the electrode surface can be calculated from the hydrogen adsorption or desorption peaks using this equation:74
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0016.gif)
where θbi is the fractional coverage of bismuth adatoms, QHb is the charge involved in the hydrogen adsorption or desorption before modifying the electrode surface with bismuth adatoms and after the modification, QHa. The surface coverage of bismuth adspecies can be altered by changing the concentration of bismuth solutions, contact time between electrode surface and bismuth solution, and roughness factor of the electrode surface.
Climent and colleagues53 claimed that bismuth adatoms stay on the electrode surface even when sweeping the potential to more positive values, given that the cyclic voltammograms are relatively stable after continuous potential cycling. However, Salvatore and co-workers74 reported that the fractional coverage of bismuth adspecies on the electrode surface declines from ca. 0.7 to ca. 0.35 after 30 potential cycles from −0.25 to 0.9 V vs. Ag/AgCl (saturated KCl) in H2SO4 solutions; this response is attributed to the gradual bismuth desorption from the electrode surface upon constant potential cycling. It has been reported that bismuth adspecies can desorb from the electrode surface when cycling the potential to more positive values.56,73 Clavilier and co-workers73 suggest that metal adsorbates could completely desorb from the electrode surface in the order of tens of seconds in a slow kinetic process when scanning the potential beyond 0.95 V vs. RHE in H2SO4 solutions.
One thing to note is that bismuth adspecies can still be oxidized via the following reaction:53
![Equation ([R16])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0017.gif)
A bismuth oxidation peak is seen at 0.62 V vs. RHE shown in Figure 1 in the original text.73 This could change the local pH at the proximity of the electrode surface if sufficient overpotential is applied to oxidize bismuth adatoms, which could affect the electrochemical behavior of peroxodisulfate. The change in local pH could also promote the decomposition of persulfate, Reactions R3, R4, and R6.
The electrochemical response of persulfate reduction is significantly improved on bismuth-adsorbed Pt rotating disk electrodes. A plateau region is seen on the linear sweep voltammograms recorded with bismuth-adsorbed Pt electrodes in HClO4 solutions as shown by Kokkinidis and co-workers,56 see Figure 5 in the original paper. Bismuth-adsorbed Pt rotating disk electrodes are thought to be electrocatalytic toward the reduction of persulfate in an acidic environment because the experimental limiting currents are close to the theoretical limiting currents and less driving force is needed for the reaction to proceed.53,56 This could be because bismuth adatoms are inhibiting the persulfate anions from adsorbing on the electrode surface which results in a faster electron transfer to peroxydisulfate anions.56 Kokkinidis et al.56 claimed to have achieved diffusion-controlled limiting currents, since their currents are proportional to the concentration of peroxodisulfate and to the square root of scan rates. These observations indicate that the direct electron transfer pathway should prevail since the adsorption of persulfate is blocked by bismuth adatoms.53 Nonetheless, Kokkinidis and co-workers56 argued that the reaction is actually under mixed-control considering that the hysteresis between the forward and backward scans is quite large.56 Other than that, the potentials at half the current of the linear sweep voltammograms obtained using bismuth-, lead- and thallium-adsorbed Pt rotating disk electrodes in the presence of peroxodisulfate under acidic condition seem to shift to more negative potentials respectively, according to the potentials where the metal adatoms desorb from the electrode surface. It is known that bismuth adspecies desorb from the electrode surface at more positive potentials than thallium and lead adspecies;77,78 thus, the potential at half the current for the reduction of persulfate on bismuth-adsorbed Pt electrodes is more positive than those on lead- and thallium-adsorbed Pt electrodes. However, the effect of metal adatom desorption on the reduction of peroxodisulfate has not been explained.
Climent and co-workers53 claimed that the reduced form of bismuth (elemental bismuth) is more electrocatalytic toward the reduction of peroxodisulfate than the oxidized form of bismuth, Reaction R16, under an acidic environment; this suggestion was made considering that the peak for the reduction of peroxodisulfate coincides with the reduction peak of bismuth adatoms. However, the reduction peak for bismuth adatoms is not seen on the linear sweep voltammograms shown by Kokkinidis et al.56 Moreover, it is practically impossible to confirm the electrochemical response of peroxodisulfate reduction on an oxidized form of bismuth adatoms, since the reduction of persulfate coincides with the reduction of bismuth adspecies. Bismuth adspecies will always be in reduced form when probing the electroreduction of persulfate.
The conclusion of this section is that bismuth-adsorbed Pt electrodes exhibit sigmoid shaped voltammograms which are expected for diffusion-controlled redox reactions. However, the cathodic reaction may still be mixed-controlled as the hysteresis between the forward and backward scans was quite large. Nonetheless, bismuth-adsorbed Pt electrodes could be the best persulfate sensors to date. It would be interesting to study the electroreduction of persulfate using platinized Pt electrodes decorated with bismuth adatoms. Salvatore and colleagues74 showed encouraging results of formic acid oxidation obtained using platinized Pt microdisk electrodes modified with bismuth adspecies. Other electrode materials that have been used to study the electroreduction of persulfate anions are discussed in the next section.
Other electrode materials
The electroreduction of peroxodisulfate has also been investigated using other electrode materials: lead- and thallium-adsorbed silver (Ag),79 Cd-adsorbed platinized Pt,70 riboflavin/graphene quantum dots-modified glassy carbon,80 ruthenium oxide/thionine- and ruthenium oxide/celestine blue-modified glassy carbon,81 mercury,82–84 Cd,85–87 bismuth,86 Prussian blue,88 and BDD.89 However, none of these materials exhibit diffusion-controlled currents. El-halim and colleagues79 claimed that the electroreduction of persulfate on thallium- and lead-adsorbed Ag single crystal is diffusion-controlled and an outer sphere redox process, but their data were obtained in concentrated solutions.53
Provent and colleagues89 attempted to measure the amount of peroxodisulfate in solutions using BDD electrodes with different levels of boron doping. Two types of BDD electrodes were employed: rotating disk and microdisk array electrodes.89 In Figure 4 in their published article, they showed sigmoid shaped linear sweep voltammograms obtained using BDD rotating disk electrodes in different concentrations of peroxodisulfate anions. Unfortunately, the scan rate employed was 100 mV s─1 which is deemed to be too fast to observe limiting currents on rotating disk electrodes. Typically, limiting currents are achieved using rotating disk and microdisk electrodes at low scan rates (1–10 mV s─1).90 Thus, the limiting currents shown by them could constitute other unknown processes. Nonetheless, the plateau region was not as wide as that seen on linear sweep voltammograms recorded using Pt electrodes modified with metal cations as discussed in Bismuth-adsorbed Pt electrodes section. Provent and colleagues89 claim to have observed a linear relationship between the current density and concentration of S2O82─ even in concentrated solutions.
The electrochemical responses for the reduction of persulfate on these materials are not as good as those obtained using bismuth-adsorbed Pt electrodes in acidic environments. Most of the voltammetric responses recorded using these electrode materials display the characteristics of voltammograms recorded using pristine Pt electrodes as stated in Platinum electrodes section. Thus, their electrochemical results will not be discussed in this paper. The next section discusses the role of metal oxides on the electroreduction of peroxodisulfate.
The role of metal oxides
The concentration of oxides on the electrode surface could determine the reduction rate of persulfate. Samec and colleagues5 claimed that Au oxides do not affect the reduction of peroxodisulfate tremendously, since the reduction rates in neutral and strong acid solutions are almost equal. However, the formation of metal oxides is not affected by the pH of solutions. More metal oxides are formed as the potential is swept to more positive values. Thus, in order to study the effect of metal oxides on the reduction of persulfate, different upper potential limits can be set. Yet, Samec et al.57,60 showed that the formation and stripping of Au oxides are neither facilitating nor inhibiting the reduction of peroxodisulfate, Figure 4 in the original text shows that the shape of the peaks for the reduction of persulfate remains the same regardless of the direction of potential sweep.
The role of metal oxides on the reduction of peroxodisulfate in KOH solution has been studied using surface-enhanced Raman spectroscopy by Desilvestro and Weaver.3 The spectra showed that Au oxides are retained on the electrode surface at 0.3 to 0.4 V more negative than in the absence of persulfate anions. It is believed that Au oxides accelerate the dissociative chemisorption of persulfate anions,55,91 act as reaction intermediates,3 or inhibit the electroreduction of peroxodisulfate.2,61 It is important to remember that the reduction of persulfate anions is sensitive to electrode surface and to materials. Different electrode materials and crystal structures might produce different forms of oxides. Thus, the formation of oxide layers on various materials may affect the electrochemical behavior of persulfate reduction differently. As a conclusion, it is beneficial to study the effect of oxide formation on the electroreduction of peroxodisulfate before conducting further investigation. The next section reviews the diffusion coefficient of peroxodisulfate.
Diffusion coefficient of peroxodisulfate
Many groups have reported values of the diffusion coefficient for persulfate reduction. However, the values of the diffusion coefficient vary widely in the literature. Values of the diffusion coefficient were typically determined from Levich plots. Most of the published works employ a similar value of kinematic viscosity, 0.01 cm2 s─1, for the calculation. Table I shows the diffusion coefficient of peroxodisulfate electroreduction that have been reported in the literature. The values shown for Au electrodes, regardless of the crystal structures, were taken from the slope of Levich plots for the peak at more negative potentials which is attributed to the direct electron transfer to persulfate anions. For Au electrodes, Samec et al.5 reported that only the persulfate reduction peak at more negative potentials is affected by the rotation speed. Thus, it would make sense to only consider the values of diffusion coefficient determined from the peroxydisulfate reduction peak at more negative potentials considering that Levich plots were used. As mentioned in Platinum electrodes section, Levich plots are graphs of current vs. square root of rotation speed.
Table I. Diffusion coefficient values calculated from Levich plots for the reduction of peroxodisulfate obtained on different electrode materials.
| Electrode | Solution | Diffusion coefficient/cm2 s─1 | Ref. |
|---|---|---|---|
| Bismuth-adsorbed Pt | Na2S2O8 + HClO4 | 1 ± 0.05 × 10─5 | 52 |
| K2S2O8 + HClO4 | 0.83 × 10─5 | 53 | |
| Bismuth single crystal | Na2S2O8 + NaF | 0.62 × 10─5 | 105 |
| Bare smooth Au | Na2S2O8 + HClO4 | 1.2 ± 0.2 × 10─5 | 5 |
| 0.4 mM Na2S2O8 + 1 M HClO4 | 1.4 × 10─5 | ||
| 1 mM Na2S2O8 + 0.1 M HClO4 | 1.3 × 10─5 | ||
| 1 mM Na2S2O8 + 1 M HClO4 | |||
| 1 mM Na2S2O8 + 5 mM H2SO4 | |||
| 1 mM Na2S2O8 + 5 mM NaF | |||
| 2 mM Na2S2O8 + 5 mM H2SO4 | 0.97 × 10─5 | ||
| Bare roughened Au | K2S2O8 + KOH | 0.8 × 10─5 | 3 |
| Cd (0001) and Bi (hkl) | Na2S2O8 + Na2SO4 | 1.15 × 10─5 | 86 |
| Cd (0001) | Na2S2O8 + NaF | 1.2 × 10─5 | 87 |
| Hg/Cu | (NH4)2S2O8 + H2SO4 | 0.34 × 10─5 | 84 |
| Au (111) | Na2S2O8 + HClO4 | 0.9 × 10─5 | 60 |
| Au (110) and Au (111) | Na2S2O8 + HClO4 | 0.8 – 1.1 × 10─5 | 57 |
| Lead-adsorbed Pt | K2S2O8 + HClO4 | 0.8 × 10─5 | 70 |
| Hydrogen-adsorbed Pt | K2S2O8 + HClO4 | 0.92 × 10─5 | 56 |
The wide range of diffusion coefficient values could arise from various factors. The reduction of persulfate is a surface sensitive reaction,53 thus, it could be affected by the state of electrode surface, electrode materials, roughness factor, surface coverage of modifying species, crystal orientation, thickness of electrodeposited thin film electrodes, electrode surface pre-treatment (chemical etching, electrochemical polishing, mechanical polishing, flame-annealing, etc.), and functional groups on the electrode surface. The electrochemical response of persulfate reduction could also be influenced by the concentration, pH and type of test solutions (aqueous/organic media, cations/anions, etc.), scan rate, temperature, and the presence of contaminants.
These external factors can change the electron transfer kinetics of the persulfate reduction. To resolve this problem, one should find an electrode material that can exhibit a diffusion-controlled response for the reduction of persulfate, preferably with a pristine electrode surface; this can eliminate a number of external factors from affecting the rate of persulfate reduction. Other than that, the temperature of solutions should be regulated at room temperature using a thermostatic water bath to avoid heat activation of persulfate. The experiments should be performed inside Faraday cages to prevent persulfate activation from direct exposure to light and electromagnetic radiation interference. To avoid the activation of persulfate, the glassware and apparatus used for the experiments have to be thoroughly cleaned of any contaminants and dust. Peroxydisulfate has to be free from impurities to prevent persulfate activation. The activation of peroxodisulfate was explained in Introduction section. Mechanical polishing should be performed using diamond pastes instead of alumina slurries as alumina could result in the formation of oxides on the electrode surface during polishing. Low concentrations of peroxodisulfate and supporting electrolytes should be employed as suggested by Botukhova and Petrii.52 They claimed that the persulfate reduction currents obtained using Pt electrodes are relatively small in concentrated supporting electrolytes. In addition, high concentration of persulfate anions would require more adsorption sites for an efficient electron transfer. Botukhova and Petrii52 suggest that the type of anions could affect the hysteresis between the forward and reverse scans for the electroreduction of persulfate anions using Pt electrodes. They explained that it could be due to the specific adsorption of anions such as perchlorate and sulfate on the electrode surface. They showed that the cathodic currents for the reduction of peroxodisulfate recorded using Pt electrodes in perchloric acid are larger than those obtained in sulfuric acid; this could be because sulfate anions have a higher specific adsorption on Pt electrodes than perchlorate anions since specifically adsorbed anions can hamper the peroxydisulfate reduction process.52 Samec et al.5 added that adsorbates other than persulfate anions such as perchlorate adspecies could hamper the reduction of persulfate. Thus, the type of supporting electrolyte should be chosen wisely. As a conclusion, experimental design should take into account all aspects to ensure that the results are robust. The next section reviews the mechanism of persulfate reduction.
Mechanism of peroxodisulfate reduction
The electroreduction of persulfate anions occurs in two parallel pathways.5 The first pathway could proceed with the adsorption of peroxodisulfate anions on the electrode surface.5 The fast dissociative chemisorption with O─O bond breaking leads to the formation of adsorbed SO4─ anions on the electrode surface, Reaction R11.53 This is followed by a slow one-electron reduction, Reaction R12.55 The reduction peak associated with the fast dissociative chemisorption pathway is visible at more positive potentials, 0.4 V vs. SCE, on Au rotating disk electrodes in HClO4 solutions.5,60 This phenomenon can be described as an inner sphere redox mechanism as it proceeds with the adsorption of persulfate anions on the electrode surface.52 The persulfate reduction peak at more positive potentials is claimed to be affected by scan rate.5,60
The second pathway could comprise of a two-step direct electron transfer mechanism with a slow uptake of the first electron explained by Reactions R14 and R15.5 The peroxodisulfate cathodic peak assigned to the direct electron transfer pathway is located at more negative potentials, 0 V vs. SCE, when recorded using Au rotating disk electrodes in HClO4 solution;5 this cathodic peak is influenced by rotation speed. The direct electron transfer mechanism could arise from the electrostatic repulsion of peroxodisulfate anions from negatively charged electrode surface.5 The proposed overall reaction for the electroreduction of persulfate is as follows:79,84
![Equation ([R17])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0018.gif)
This overall reaction was probed by Frumkin et al.92 on various electrode materials under the influence of a negative outer Hemholtz potential.79
Because pH affects the kinetics of the reaction, the protonation of sulfate anions has been suggested by Stark:44
![Equation ([R18])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0019.gif)
and/or:
![Equation ([R19])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0020.gif)
However, it is unknown whether the protonation of persulfate only involves the free moving anions or the chemisorbed species as well, analogous to SO4─(ads) as expressed in Reaction R19. It is possible for both chemisorbed and free moving persulfate and its intermediate to react with protons. Nonetheless, Reactions R18 and R19 should also be taken into account based on the electrochemical results obtained using Au rotating disk electrodes.5,60 Thus, we propose a simplified version of the reaction pathways through the following illustration for easier understanding as summarized in Figure 1:
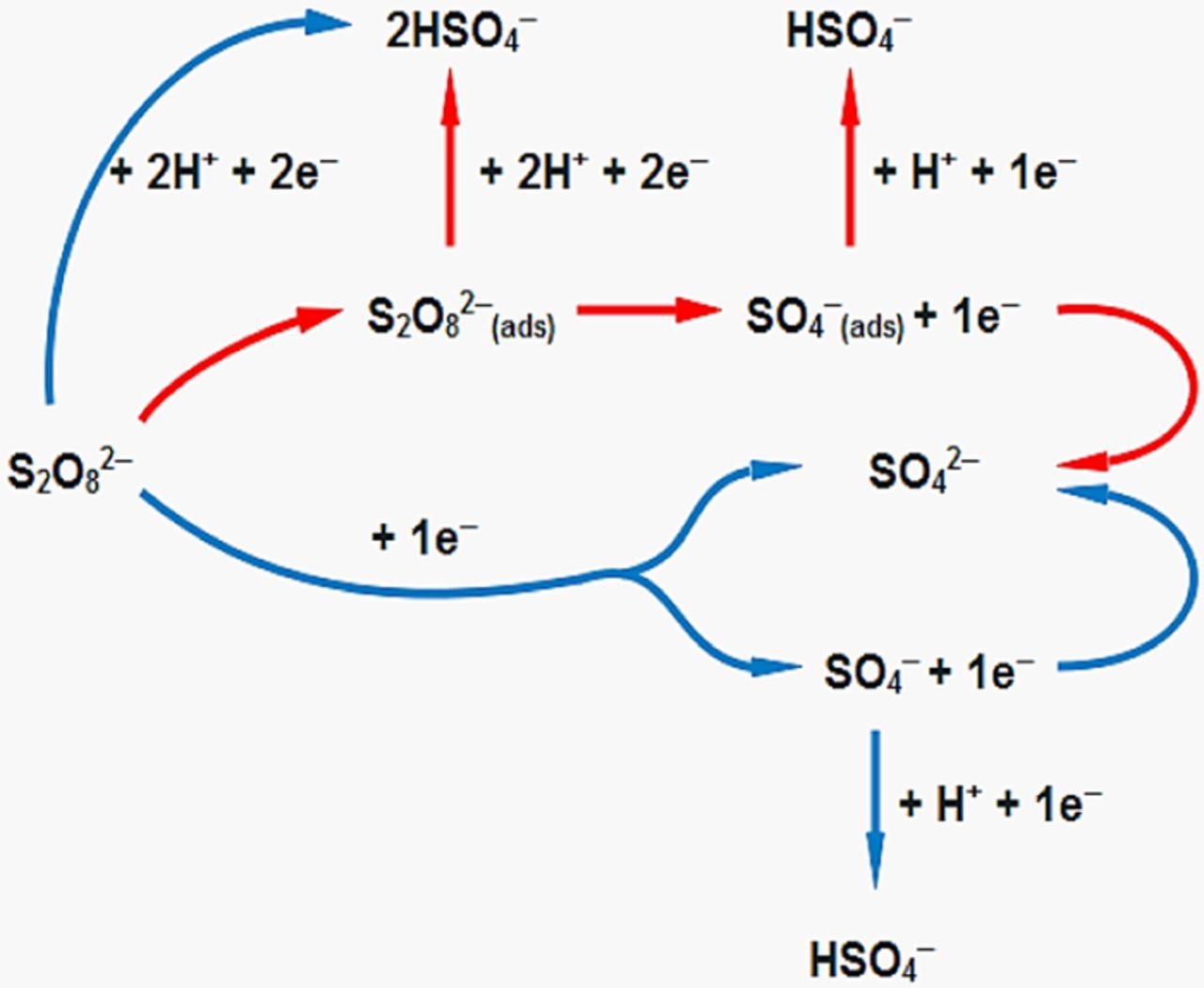
Figure 1. Reaction mechanisms for the electroreduction of peroxodisulfate. Red arrows ( ) indicate the reaction mechanism related to the dissociative chemisorption pathway and blue arrows (
) indicate the reaction mechanism related to the dissociative chemisorption pathway and blue arrows ( ) are linked to the direct electron transfer mechanism.
) are linked to the direct electron transfer mechanism.
It can be noted that SO42─ is more favorable; it is a more stable form of sulfate anion. There are several possible pathways for HSO4─ anions. HSO4─ ions could undergo decomposition as follows:93
![Equation ([R20])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0021.gif)
or
![Equation ([R21])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0022.gif)
This reaction will increase the concentration of SO42─ and H+ or H3O+ ions in the solution. On Pt electrodes, HSO4─ anions could bind strongly on the electrode surface.59 This phenomenon could prevent the adsorption of peroxodisulfate which should result in higher reduction currents; this response is akin to having bismuth adatoms on the electrode surface as discussed in Bismuth-adsorbed Pt electrodes section. However, this explanation is unacceptable as the persulfate cathodic currents obtained using bare Pt electrodes decay as the potential is swept to more negative values as shown in Figure 1 in the paper by Kokkinidis and colleagues;56 this means that the adsorption of HSO4─ anions only adds complications to the reduction of persulfate. Yet, the adsorption of HSO4─ might not be the only reason behind the current decay as this electrochemical response can also be seen on Au electrodes as shown by Samec and colleagues.5 Kokkinidis and colleagues56 showed that hydrogen underpotential deposition on Pt rotating disk electrodes can improve the electrochemical behavior of peroxodisulfate reduction. If the underpotential deposition of hydrogen on Pt electrodes is performed in the presence of sulfate anions, it is possible that HSO4─ adatoms could assist hydrogen adspecies in blocking the adsorption of persulfate; this event leads to higher currents. However, more experiments need to be conducted to confirm this hypothesis.
Kinetic Behavior of Peroxodisulfate at Metal Electrode/Electrolyte Interface
As already discussed in detail in Diffusion coefficient of peroxodisulfate and Mechanism of peroxodisulfate reduction sections, it can be concluded that the kinetic activities of the peroxodisulfate are greatly influenced by several factors, as the reduction of S2O82─ anions is a surface sensitive reaction. This electrochemical feature complicates the discussion of S2O82─ kinetic behavior as the S2O82─ ions tend to adsorb at the metal surface. To date, there are undeniably extensive articles reporting the kinetics of peroxodisulfate reduction at metal/electrolyte interfaces. Yet, there are only a few articles in the literature that we feel could be of interest to readers in order to easily understand the kinetic behavior of peroxodisulfate electroreduction at metals/electrolyte interfaces.
Samec and Doblhofer5 reported that the rate constant of peroxodisulfate at a polycrystalline gold electrode is significantly contributed to different reaction pathways: surface catalytic (Reactions R11 and R12) and direct electron transfer mechanisms (Reactions R14 and R15); these mechanisms lead to the formation of chemically adsorbed and free-moving intermediates respectively, as already summarized in Figure 1. In addition, the desorption of adsorbed S2O82─ anions from the surface due to the negatively charged electrode surface alters the electrical double layer; this effect significantly changes the magnitude of the kinetics. From the investigation of peroxodisulfate reduction in various electrolytes (NaF, HClO4, and H2SO4) using hydrodynamic voltammetry, they found that the charge transfer coefficient, α, is insignificantly affected by the various cations (H+ and Na+) or anions (F─, ClO4─, and SO42─) where the reactions occurred via a two-step electron transfer (Reactions R14 and R15), where the first electron exchange is a slow uptake electron transfer. Essentially, the two-step electron transfer is characterized by the first-order rate constant, k, as expressed by:
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0023.gif)
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0024.gif)
where kt is the rate constant after correcting for the double layer effect through Frumkin theory, z is the ionic charge, ψ1 is the electrical potential at an optimum point for ion discharge, ko is the potential independent rate constant, and E is the applied electrode potential. R, T, and F are the gas constant, temperature, and Faraday constant respectively.
On condition that electrons are transferred to peroxodisulfate anions located in the outer Helmholtz plane for Reaction R14 which is the rate-determining step, the electrical potential can be assessed using the Gouy-Chapman theory as follows:
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0025.gif)
where σ is the charge of metal, σi is the charge produced by the adsorbed film, c is the concentration of electrolyte, and ɛ is the dielectric constant of solvent. However, Eq. 4 is only valid for 1:1 electrolytes such as sodium chloride, NaCl, where the ions are singly-charged; this means that the ionic strength of electrolyte is equal to the concentration of the electrolyte.94 Moreover, based on Eq. 3, when the applied potentials are more positive than the potential of zero charge (pzc), E > Epzc, peroxodisulfate or sulfate anions that are produced from the reduction process are specifically adsorbed on gold electrode surfaces. As a result, σi compensates for σ. Nonetheless, the kt from Eq. 2 can be eliminated when three criteria are met: 1) the σi is more negative than the σ, 2) the ψ1 is close to 0, and 3) the concentration of electrolyte affects the ψi. In the case of Samec and Doblhofer,5 the kinetic data presented in Figures 3 and 4 in the original paper represent the true rate constant as the aforementioned criteria were met; thus, it is not necessary to correct the kinetic data. They reported that the value of α was found to be 0.3 obtained from Tafel slope, log k vs. E, at approximately 0.2 V vs. SCE. By contrast, the adsorbed anion at the electrode surface will desorb from the surface when E < Epzc; this leads σi to approaching 0. Thus, the values of the ψi can be determined using Eq. 4 where σ can be obtained by integrating the capacitance curves. However, overestimation of the effect of the electrical double layer is likely when performing this procedure as the desorption of S2O82─ ions shifts the Epzc to more negative values (Epzc = ─ 0.18 V). Therefore, Samec and Doblhofer5 plotted a graph of kt as a function of the corrected electrode potential by subtracting ψ1 from E, so instead using, E ─ ψ1. Through this approach, they found that the apparent kinetic data differ from those obtained when E > Epzc by a factor of 3. They also reported that the value of α was found to be ∼ 0. The value of α seems to be strongly dependent on E. The value of α shifted from ∼ 0.3 at E > Epzc to ∼ 0 at E < Epzc. This response indicates that a more complex mechanism of electron transfer may have taken place during the reduction process of peroxodisulfate at E < Epzc; this could involve a cationic bridge or an ion pair discharge which can be supported by data for sp metals.95
Samec and co-workers60 compared the kinetic behavior of peroxodisulfate reduction at Au (111) electrodes in HClO4, investigated using two different techniques: cyclic and rotating disk electrode voltammetry. They also employed polycrystalline Au electrodes as working electrodes for comparison. They discovered that the rate of electroreduction of S2O82─ at Au (111) electrodes is relatively large compared with that at polycrystalline Au electrode by two orders of magnitude. They believe that this is due to a better matching between the structure of S2O82─ and the trigonal symmetry of the Au (111) plane; this leads to higher amounts of S2O82─ being adsorbed at the electrode surface and much more efficient electron transfer to peroxodisulfate anions. In addition, there are two significant findings in this work: 1) the catalytic effect of protons on the kinetic activity of S2O82─ electroreduction and 2) the inhibition of the S2O82─ reduction by ClO4─ adatoms. Interestingly, both effects can be treated by a simple electrocatalytic model as follows:
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0026.gif)
where i is the current, n is the number of electrons transferred, A is the electrode area and co is the concentration of peroxodisulfate at the electrode surface. As discussed in Gold electrodes section, the reduction of S2O82─ anions on Au electrodes exhibit two reduction peaks. Thus, k in Eq. 5 takes into account the total of two parallel charge transfers from the peaks denoted as c1 and c2 shown in Figures 4 and 5 in the original text.60 Since the overall reaction of peroxodisulfate reduction, Reaction R17, occurs at potentials more negative than the standard potential, E°, the backward reaction is not considered. The value of k can be calculated from the measured current at the stationary electrode as expressed by the following equation:
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0027.gif)
where co is the concentration of peroxodisulfate in the bulk of the solution. m is the convolution integral as follows:
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0028.gif)
t is time. Meanwhile, m1 refers to the following mathematical equation:
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0029.gif)
where D is the diffusion coefficient.
For rotating disk electrodes, the k value can be determined using the following mathematical expression:
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0030.gif)
where iL is the limiting current. Note that Eq. 9 can be simplified to yield the Koutecky-Levich equation, Eq. 10:
![Equation ([10])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0031.gif)
where current arising from the kinetics
![Equation ([11])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0032.gif)
From their results, both k values, determined using cyclic and rotating disk electrode voltammetry, were obtained after subtracting the current of peak c1 from the voltammograms.
As mentioned in Gold electrodes section, the electroreduction of peroxodisulfate anions on Au electrodes at more positive potentials proceeds via single dissociative chemisorption mechanism, Reaction R11. However, Samec et al.60 proposed two parallel reactions in the potential range of ∼ 0.4 V vs. RHE as shown in Figure 2.
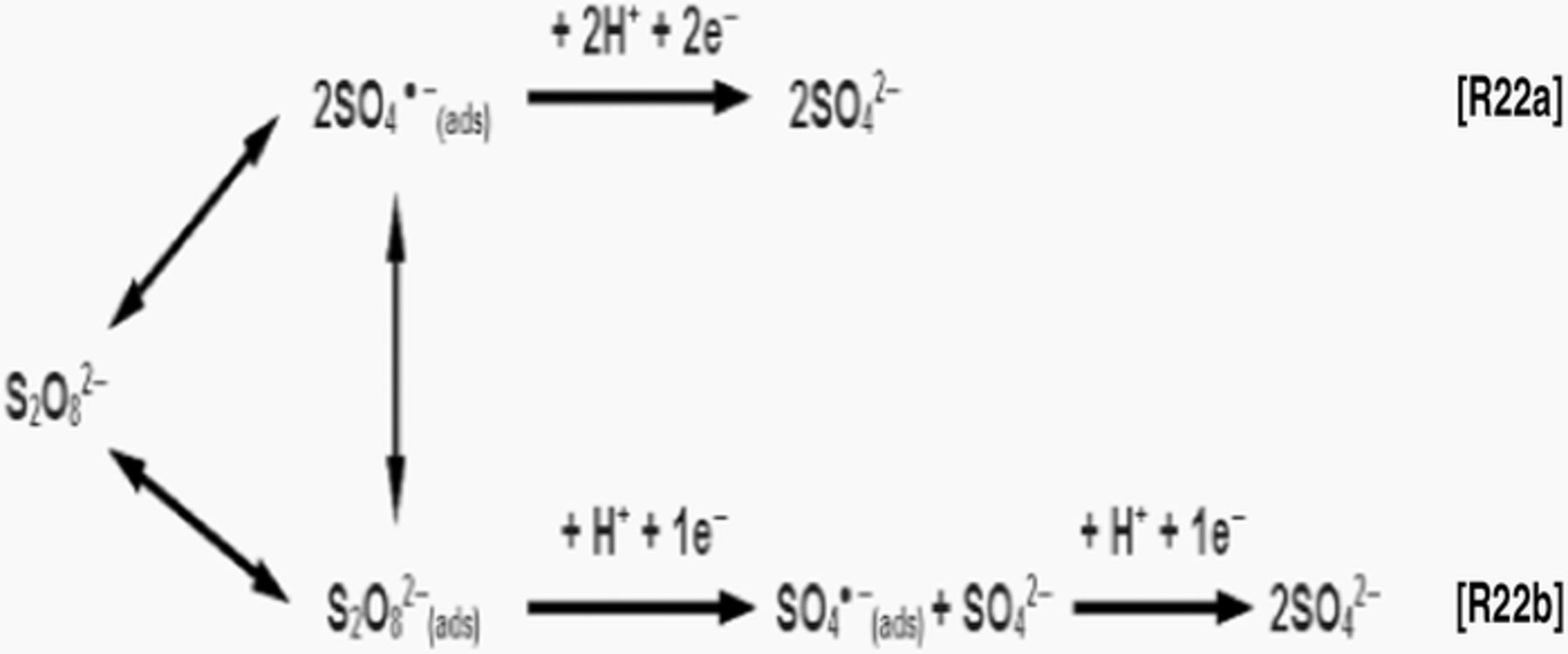
Figure 2. Two parallel reaction mechanisms related to the dissociative chemisorption process for the reduction of peroxodisulfate anions on metal surface redrawn from Samec and colleagues.60
Nevertheless, they reported that experimental data cannot distinguish between Reactions R22a and R22b i.e. it is unknown whether Reactions R22a or R22b prevails for the reduction of peroxodisulfate reduction at more positive potentials. (SO4●─)ads radicals are known to be less stable than (S2O82─)ads anions. Thus, the relative surface coverage, θ, for the (SO4●─)ads should be lower than that for (S2O82─)ads. Yet, the reaction involving (SO4●─)ads radicals should be fast since they are highly reactive. Therefore, Reactions R22a and R22b could proceed at a fairly similar reaction rate. In the case of Reaction R22b, the higher reactivity of (SO4●─)ads would imply that the first electron uptake [(S2O82─)ads + 1e─ + H+ → (SO4●─)ads] should be the rate-determining step.
From the reaction models above, when a steady state is achieved on the time scale of the experiments, the relative surface coverage, θ, of the electrode by persulfate adatoms follows from balancing the rates of the adsorption (va), desorption (vd), and electron transfer (ve) steps as explained by the following equations:
![Equation ([12])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0033.gif)
![Equation ([13])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0034.gif)
![Equation ([14])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0035.gif)
![Equation ([15])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0036.gif)
![Equation ([16])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0037.gif)
where θ can be determined by obtaining the ratio of adsorbate surface excess to the maximum surface excess (θ = Г / Гmax). c0B is the concentration of HClO4. ka and kd are the rate constant for adsorption and desorption steps respectively. koa, kdo, and koe are the rate constants for adsorption, desorption, and electron transfer steps respectively at E = 0. Σθi is the ratio of the relative surface coverage between ClO4− and S2O82− adatoms, which is explained in more detail following Eq. 23. γ and γ' are the coefficient for the dependence of adsorption and desorption rate on E respectively. The third power in Eqs. 13 and 14 is proposed by assuming that one (SO4●─)ads or (S2O82─)ads adatom replaces three molecules of water on the electrode surface. The ikin is then given by:
![Equation ([17])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0038.gif)
The combination of Eqs. 11 and 16 with Eq. 17 enables the determination of the rate constant as expressed in the following equation:
![Equation ([18])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0039.gif)
where θ can also be given by
![Equation ([19])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0040.gif)
with
![Equation ([20])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0041.gif)
![Equation ([21])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0042.gif)
and
![Equation ([22])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0043.gif)
In order to consider the co-adsorption of ClO4─ anions at the Au (111) electrode surface, they further expanded the mathematical equations that allow the calculation of k; this was done by assuming that the ratio of the relative surface coverage between ClO4─ and S2O82─ adatoms, expressed as θ' and θ'' respectively, is proportional to the ratio of their concentrations as described using these equations:
![Equation ([23])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0044.gif)
![Equation ([24])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0045.gif)
Thus, the constant Kaa expresses the ratio of the adsorption rate constants of the two anions on the free surface (Σθi → 0) or the ratio of the corresponding adsorption coefficients in equilibrium. This mathematical consideration is to show that the constant Kaa can vary with time; this means that Kaa can have a higher value in experiments involving rotating disk electrodes where the time scale is fairly long. Therefore, the values of α, β, koe, Kad, Ked, and Kaa must be determined prior to calculating the value of k for Eq. 18; the values of some parameters can be estimated from experiments.
They concluded that their kinetic model is able to predict the various effects observed in the potential range of c1. It is worth mentioning that the assumption about the co-adsorption of ClO4─ anions is rather crude and seems illogical since an unrealistic value of its ratio to sulfate adsorption needed to be introduced (Kaa = 1). Yet, these types of inhibitory effects appear to be common and worth considering.
By contrast, the c2 peak is thought to arise from a direct reduction of S2O82─ anions and is probably controlled by a single one-electron transfer rate determining step as explained by Reactions R14 and R15. Through this pathway, the electron is directly transferred to the peroxodisulfate anions in the solution close to the vicinity of the electrode surface. Essentially, the rate constant can be evaluated using Eqs. 18 and 19. From their findings, the rate constant for the direct electron transfer pathway of peroxodisulfate reduction on Au (111) electrodes is fairly similar to that on polycrystalline Au electrodes. Nevertheless, the rate constant strongly depends on the ion distribution at the solution side of the electrolyte interface; for instance, the excess charge on the metal surface, the specific ion adsorption, or the hysteresis between the forward and backward scans.
Another interesting piece of work on the kinetic behavior of S2O82─ anions at metal electrodes was reported by Thomberg and Lust.87 In their work, they studied the electroreduction of S2O82─ anions using polished Cd (0001) rotating disc electrodes in a weak base solution of sodium fluoride, NaF. In this work they found that kinetics of electroreduction of S2O82─ depends on the E and the concentration of NaF solution. They found that the concentrations of NaF affect the diffusion layer thickness. They reported that the values of α decreased from 0.22 to 0.18 as the concentration of NaF increased. However, when the concentration of NaF is constant, α value is practically independent on the varying concentration of S2O82─ ions. They managed to determine kinetic current densities at constant potential using the Koutecky-Levich method. The kinetic current densities were subsequently utilized to obtain the apparent rate constant of persulfate reduction. Analogous to the work that was reported by Samec and his associates,5,60 Thomberg and Lust87 normalized the current as the current density to obtain the apparent rate constant of the heterogeneous reaction of the peroxodisulfate reduction, khet, as follows:
![Equation ([25])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0046.gif)
where jk is the kinetic current density at constant E that can be obtained from the intercept of Koutecky-Levich plots as explained by:
![Equation ([26])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0047.gif)
Meanwhile, j is the current density and jD is the limiting current density. In addition, jk can also be obtained using the following expression:
![Equation ([27])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0048.gif)
From their work, log khet increased as the concentration of NaF increased but it was completely unaffected by the concentrations of Na2S2O8. The corrected Tafel plots (cTps) were made with respect to jk by using the potential acquired from a GSCG model, ψo, based on impedance measurements. From their linear Tafel plots, the slopes slightly decreased when the concentrations of NaF were varied from 2 × 10─3 to 1 × 10─2 M; this means that α values also decreased. Nevertheless, α is independent to the concentration of S2O82─ anions; this trend was also seen for khet.
Another finding from their work is the non-linearity of cTps as well as the deviation of cTps toward smaller j at E > ─1.1 V vs. SCE. Consequently, these effects contributed to the deviation of their system from the Frumkin model; these effects could arise from the weak specific adsorption of F─ adatoms which inhibited the adsorption of S2O82─ anions at electrochemically polished Cd (0001) electrodes. This is in good agreement with the kinetic model proposed by Samec and co-workers.5,60
Another significant work on the kinetic behavior of S2O82─ reduction using Cd and bismuth, Bi, electrodes was also reported by Thomberg and colleagues.86 In their work, they investigated the kinetic behavior of S2O82─ ions at electrochemically polished Cd (0001), Bi (111), and Bi (011) electrodes using cyclic voltammetry, rotating disk electrode voltammetry, and electrochemical impedance spectroscopy techniques in alkaline electrolytes of NaF and sodium sulfate, Na2SO4. They reported that the khet for the electroreduction of S2O82─ ions was practically dependent on the electrode polarization and the concentration of base electrolyte solutions. In addition, the α value was obtained after taking into account the double layer effect. The α value was found to be ≤ 0.15 and the value was weakly dependent on the electrode potential and the concentration of electrolyte solutions. They obtained the jk values from the Koutecky-Levich plots. They found that the jk values obtained on Bi (111) electrodes were somewhat higher than that on Bi (001) electrodes; the values of jk obtained in Na2SO4 were also higher compared to that in NaF. This trend obeys the Frumkin slow discharge theory and is in a good agreement with the results obtained in the higher concentrations of Na2SO4.
The values of khet for electroreduction of S2O82─ for Cd and Bi electrodes were calculated using Eq. 25. It seemed that the values of jk and log khet decrease with the dilution of the base electrolyte solution. Unfortunately, the decrease of khet is relatively small compared with the magnitude projected by Frumkin slow charge transfer theory, especially for Bi (011) electrodes. Moreover, they found that the log khet did not depend on the concentration of S2O82─ anions when it is less than 5 × 10─5 M for Bi (111), less than 1 × 10─4 M for Bi (011), and less than 2 × 10─4 M for Cd (0001). At higher concentrations of persulfate than mentioned previously for the respective electrodes, they discovered a small increase in the log khet at fixed E particularly at more negatively charged Bi (hkl) electrodes; this indicates that the surface is blocked due to the weak adsorption of (S2O82─)ads or (SO4─)ads anions, Reaction R11, at the bismuth electrode/electrolyte interface. At even higher concentrations of peroxodisulfate, they reported that a small increase in khet was seen; this suggests that the reaction mechanism has changed. For electrochemically polished Cd (0001) and Bi (001) electrodes, khet is weakly dependent on the ionic strength of electrolyte in moderate concentration of electrolyte, between 1 × 10─3 and 1 × 10─2 M. The khet values obtained on Bi (011) and Bi (111) are weakly affected by the concentration of NaF and Na2SO4; this implies the deviation of the experimental system from the Frumkin slow discharge theory. They have also determined the values of α from cTps. They showed that the values of the corrected kinetic current density significantly decrease with the increase in the concentration of base electrolyte at fixed potential (E ─ ψ0; where ψ0 is the classical Gouy-Chapman diffuse layer potential for the base electrolyte). In addition, the cTps slopes for Cd and Bi (hkl) obtained from Na2SO4 electrolytes are lower than those acquired from NaF electrolytes. As a result, lower α values were obtained when conducting experiment in Na2SO4 solutions. The cTps show a non-linear trend near the Epzc and the deviation of the cTps toward the higher values of corrected current density can be formed which is non-conforming to the Gouy-Chapman and Frumkin discharge theories. In their studies, they also investigated the ionic association process of poly-charged complex anions such as S2O42─ and S2O82─ with base electrolyte cations in the solution phase. The association constant, KA, was simulated based on the Fuoss equation:
![Equation ([28])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0049.gif)
![Equation ([29])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0050.gif)
where eo is the elementary charge and NA is the Avogadro number; the value of KA was determined by assuming the distance from center to center of the ions in a pair, a = 0.36; 0.6 and 0.8 nm, are relatively low (KA = 0.118 for 0.36 nm). In these conditions, the kinetic current density can then be expressed as follows:
![Equation ([30])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0051.gif)
where zi is the charge of peroxodisulfate anions (−2), zi + 1 is the charge of associated persulfate anions with either Na+, K+, or NH4+ (base electrolyte cations), ψo is the potential at the outer Helmholtz plane, kA is the relative rate constant for the reduction of the reactants with (zi + 1) = ─1 (NaS2O8─), c1 and c2 are the effective volume concentrations of the S2O82─ and NaS2O8─ ion complex respectively. The difference between the k values calculated for Cd (0001) and Bi (hkl) electrodes using Eq. 30 and those determined from cTps in the case of 0.002 and 0.03 M NaF and Na2SO4 solutions was quite large. Thus, the ionic association evidently does not influence the shape and coincidence of cTps for Cd (0001) and Bi (hkl) electrodes in the Na2S2O8 + NaF or Na2S2O8 + Na2SO4 solutions.
In 2008, Jäger et al.96 developed a new kinetic analysis model to study the effects of nanoscopic roughness of electrode surfaces on the kinetic parameters of S2O82─ reduction at chemically etched as well as electrochemically polished Bi (111) electrodes. They reported that the values of root mean square roughness for chemically etched and electrochemically polished Bi (111) electrodes were 143 nm and 0.45 nm respectively based on in situ scanning tunnelling microscopy measurements. They also presented similar results to the work of Samec et al.5,60 and Thomberg et al.86,87 where khet and jk decreased with the decrease in the concentration of base electrolytes. Unfortunately, the decrease of khet is relatively small compared with that predicted from Frumkin slow charge transfer theory; this response is akin to the findings reported by Thomberg and co-workers.86 In addition, the khet values for electrochemically polished Bi (111) electrodes are larger than those for chemically etched Bi (111) electrodes particularly at E << Epzc; this result could be due to a higher nanoscopic surface roughness of chemically etched Bi (111) electrodes compared to electrochemically polished Bi (111) electrodes. This response could also arise due to higher adsorption energy of peroxodisulfate and its intermediates, Reaction R11, at chemically etched Bi (111) electrodes which have more defects than electrochemically polished Bi (111) electrodes. The fact that base electrolyte concentration can affect the khet can be justified by the differences in the effective Debye screening length as well as surface charge density at σ << 0. They also found that the khet values decreased at higher concentration of S2O82─ anions as a result of the blocking of Bi (hkl) surfaces. However, this data trend is predominant on the chemically etched Bi (111) electrodes which highly favor the adsorption of S2O82─ ions due to the presence of defects on their surface. They utilized the cTps to obtain the value of α by assuming that σ for chemically etched Bi (111) electrodes at minimum potential diffuse layer, Emin, is close to zero. Thus, the corrected kinetic current density via the constructed cTps was calculated using Eq. 31 at ψ1 = ψo condition:
![Equation ([31])](https://content.cld.iop.org/journals/1945-7111/165/13/H785/revision1/d0052.gif)
According to the data obtained, the cTps slopes decreased when increasing the concentration of NaF electrolyte in both cases of chemically etched and electrochemically polished Bi (111) electrodes. Consequently, the α value also decreased; for instance, α = 0.14 for 0.002 M NaF and α = 0.012 for 0.02 M NaF. On the other hand, the α value is independent of the concentrations of S2O82─ ions at constant concentration of NaF. Nevertheless, a significant difference of the cTps slopes was observed in the case of chemically etched Bi (111) electrodes when compared to cTps slopes obtained using electrochemically polished Bi (111) electrodes; this could be due to the energetic inhomogeneity of the chemically etched Bi (111) electrode surface where more defect sites are present.
As discussed herein, we believe that the kinetic activities of peroxodisulfate at the metal electrodes/electrolyte interface clearly cannot be easily understood by a simple kinetic mechanism. To better analyse the kinetics of peroxodisulfate reduction at electrode/electrolyte interface, there are some factors that have to be considered: the adsorption and desorption of S2O82─ ions and its intermediates on the electrode surface, dielectric layers, electrode polarization (surface charge density), nanoscopic roughness of the electrode surface, the concentration and type of electrolyte (acid or base), the type of anions present in electrolyte solutions, the concentration of S2O82─ ions, the ionic association between S2O82─ and S2O4─ with cations in electrolyte and other factors. Due to the nature of peroxodisulfate reduction being a surface sensitive reaction, it is believed that the weak adsorption of S2O82─ anions is the major factor that influences the kinetic activities of peroxodisulfate reduction at metal/electrolyte interfaces. The next section discusses the methods utilized to detect the presence of persulfate anions in solutions.
Detection of Peroxodisulfate Anions
Clark et al.4 reported a variety of chemical substances that can be used to detect persulfate anions in acidic solutions and they are: Zwikker reagent (4 ml. of 10% copper sulfate, 1 ml. of pyridine, and 5 ml. of water), 2,7-diaminofluorene, aniline, and benzidine. The presence of peroxodisulfate in acidic solutions is marked by either the presence of precipitates or coloration of solution after the addition of the specified chemical substance.4 Other techniques are spectrophotometry, titration, and ion chromatography.7,97 However, these methods do not provide information on the exact quantity of peroxodisulfate anions in a solution. In addition, there are several drawbacks for these methods as sample pre-treatments are needed, the procedures are time-consuming, and skilled workers are required, particularly for the ion chromatography technique. The detection of persulfate using electrochemistry can address this problem. Electrochemistry is one of the most sensitive techniques in analytical chemistry since there are a few electrochemical approaches that could detect as low as 10−8 M of analyte concentration; they are differential pulse voltammetry (DPV) and square wave voltammetry (SWV).98,99 Khamis et al.44 have employed SWV to detect peroxodisulfate using bare Au microelectrodes because of its low detection limit. To our knowledge, there is no other group that has utilized SWV and DPV to detect peroxodisulfate anions. Electrochemical sensors are attractive due to their quick detection speed, experimental simplicity, less requirement for chemical and physical sample treatments, and low cost.100 A good electrochemical sensor should have high sensitivity, low detection limit, and good linearity for the calibration curve over a wide range of the concentrations of analyte. Thus, peroxodisulfate electrosensors would also have to exhibit these features as well as be able to give reliable data. Two electrochemical techniques that have been used to detect the presence of persulfate anions are amperometry and voltammetry using various modified electrodes.7 There are several reports that provide the sensitivity and limit of the detection (LOD) of a few developed peroxodisulfate sensors which have been presented in Table 1 in the original papers.80,81,101 Some groups claim that their developed electrosensors for the detection of S2O82− anions offer higher sensitivity as well as lower LOD values with a wider linear range for the calibration curve of the concentrations of S2O82− anions. However, most of them utilized Pb or organic dyes (Prussian blue,88 natural red,102 poly brilliant cresyl blue,101 naphtol green,97 celestine blue81) in which case they are not eco-friendly. When utilizing immobilized organic dyes, the detection of S2O82− anions was achieved in the presence of mediators. They have also modified the electrode surface with carbon nanocomposites such as multi-walled carbon nanotubes, graphene, and carbon paste by direct physical deposition of the composite materials. The unstable deposited nanocomposite films and immobilized organic dye films may desorb from the electrode surface when performing electrochemical experiments. As a result, several electrosensors for the detection of S2O82− ions seem to have several issues especially in terms of stability (the modified electrodes are less robust because the modified surfaces are not stable), reduced linearity (non-linear response at higher concentrations), significant baseline drift, and extremely clean electrode surface is needed. Therefore, it is believed that these issues will decrease the sensitivity and increase the LOD of the electrodes as well as shorten the linear range of calibration curves in detecting the S2O82− anions. By contrast, as mentioned in Platinum electrodes and Gold electrodes sections, the electroreduction of peroxodisulfate on a bare metal surface is a surface reactive reaction, thus the weak adsorption of S2O82─ anions and its intermediates will alter the dielectric layer at the interface during electrochemical experiments. This tends to enhance the capacitive current. Consequently, it will interfere with the faradaic current for the reduction of persulfate. This will also result in low sensitivity, high LOD, and short linear range of calibration curves for peroxodisulfate detection. Moreover, more S2O82− anions tend to adsorb onto the electrode surface at higher concentrations of peroxodisulfate; this event will block the active sites of the electrode. Then, fouling of electrode surface could occur. A prominent consequence of this effect is a non-linear response at high concentrations will be observed, resulting in a reduced sensitivity as well as shortened linear range of calibration curves for the concentrations of analytes, and increased LOD of the developed methods. The detection of peroxodisulfate using voltammetry has been extensively discussed in Electrochemical behavior of peroxodisulfate section.
As mentioned in Preparation of peroxodisulfate section, peroxodisulfate can be electrochemically produced by oxidizing sulfate anions, at > 2 V vs. Ag/AgCl, in > 2 M of sulfate anions, preferably using BDD electrodes.44 Khamis and colleagues44 studied the efficiency of BDD electrodes with low boron concentrations to produce peroxodisulfate using the scanning electrochemical microscopy (SECM) technique. SECM was developed by Bard and co-workers in 1989.103 This technique is one of the most sophisticated methods to study the generation of peroxodisulfate on any materials. For the SECM technique, a microelectrode employed as a SECM tip is brought toward a substrate, in this case BDD electrodes, using a micro-positioner.104 Khamis and co-workers44 used Au microelectrodes as a SECM tip. The substrate is biased at a potential where sulfate anions are oxidized to persulfate, > 2 V vs. Ag/AgCl. Meanwhile, the potential of the SECM tip is held where the reduction of persulfate occurs. However, we have discussed in Gold electrodes section that Au electrodes are not the best electrode to detect persulfate anions. The experimental currents shown are smaller than the theoretical limiting currents.5,44 Moreover, there is no plateau region seen on the cyclic voltammograms and the hysteresis between the forward and backward scans is relatively large. Khamis et al.44 employed 50 mV s─1; this scan rate is deemed to be quite fast for a complicated reaction such as the electroreduction of persulfate. The scan rate should be below 10 mV s─1 to ensure sufficient time for persulfate anions to be reduced. Moreover, it is impossible for a microelectrode to achieve a true steady state when the time scale of the experiment is short as discussed in Other electrode materials section. At the moment, the most likely electrode to be used as a probing tip for SECM experiments so as to detect persulfate would be bismuth-adsorbed Pt microelectrodes. However, more experiments have to be performed considering that bismuth-adsorbed Pt microelectrodes have never been used for the electroreduction of persulfate. Nonetheless, the sensitivity and LOD of bismuth-adsorbed Pt electrodes for peroxodisulfate anions are still unknown. The lowest concentrations of peroxodisulfate employed by Kokkinidis et al.56 and Climent et al.53 were 0.5 and 0.8 mM respectively. They did not attempt to use lower concentrations of peroxodisulfate. It would be beneficial for electrosensors to have high sensitivity and low LOD. The LOD and sensitivity of bare Pt and Au electrodes should not be considered since the currents obtained for the electroreduction of peroxodisulfate is lower than the theoretically predicted currents; this could lead to false conclusions. The next section provides our perspective of what we believe the future goals for the electroreduction of peroxodisulfate should be.
Future Goals
There are many research groups that have investigated the electrochemical behaviors of peroxodisulfate on various electrode materials. Yet, the complicated electrochemical process is still poorly understood. Hence, there is still a need for further research to unravel the mystery.
The primary objective should be to find the best material that can exhibit diffusion-controlled currents for the reduction of peroxodisulfate, preferably without the need to modify the electrode surface. Although bismuth-, lead-, and thallium-adsorbed Pt rotating disk electrodes demonstrate diffusion-controlled limiting currents,56 the surface modification has made the data analysis more complicated; many external factors may affect the reduction of peroxodisulfate such as the surface coverage, roughness factor, desorption of metal cations, and the amount of adsorption sites. Then, the electrodes can be utilized to further understand the reaction mechanisms and electrochemical behavior of persulfate reduction. Once the electrochemical behavior of peroxodisulfate reduction have been thoroughly understood, the electrodes can be used to detect persulfate anions; they could be useful in water and wastewater treatment other than soil and groundwater remediation. By knowing the exact concentration of persulfate anions, it could reduce the operational cost of treatment plants. The electrode material would need to have high sensitivity and a low LOD for persulfate anions. The efficiency of BDD electrodes in generating persulfate anions can also be investigated. Considering that peroxodisulfate salts are employed in various processes, researchers should also find an alternative material that can produce persulfate anions with high efficiency. This is because BDD electrodes can be quite expensive and their efficiency in producing persulfate anions depends on the concentrations of sp2 carbons incorporated into the electrodes. Thus, the efficiency in generating peroxodisulfate anions varies amongst BDD electrodes.
Overall, the electroreduction of persulfate is very interesting. It is undeniable that the reaction is fairly complicated, however, persulfate anions have been used for many processes which made it very valuable.
List of Symbols
| a | Distance from center to center of the ions in a pair |
| A | Electrode area |
| c | Concentration of electrolyte |
| co | Concentration of peroxodisulfate in the bulk of the solution |
| c0B | Concentration of HClO4 electrolyte |
| cO | Concentration of peroxodisulfate at the electrode surface |
| c1 | Effective volume concentrations of the S2O82─ complex |
| c2 | Effective volume concentrations of the NaS2O8─ complex |
| D | Diffusion coefficient |
| eo | Elementary charge |
| E | Applied electrode potential |
| E° | Standard potential |
| Emin | Minimum potential diffuse layer |
| Epzc | Potential of zero charge |
| F | Faraday constant |
| ikin | Current arising from the kinetics |
| i | Current |
| iL | Limiting current |
| j | Current density |
| jk | Kinetic current density at constant E |
| jD | Limiting current density |
| k | First-order rate constant |
| ka | Rate constant for the adsorption step |
| kd | Rate constant for the desorption step |
| ko | Rate constant at potential independent |
| khet | Rate constant of the heterogeneous reaction of the peroxodisulfate reduction |
| kt | Rate constant after correcting for the double layer effect through Frumkin theory |
| koa | Rate constants for the adsorption steps at E = 0 |
| kod | Rate constants for the desorption step at E = 0 |
| koe | Rate constants for the electron transfer steps at E = 0 |
| kA | Relative rate constant for the reduction of the reactants with (zi + 1) = ─1 (NaS2O8─) |
| Kaa | Ratio of the adsorption rate constants of the two anions on the free surface (Σθi → 0) |
| Kad | Ratio of the rate constants of the adsorption and desorption steps |
| Ked | Ratio of the rate constants of the electron transfer and desorption steps |
| KA | Association constant |
| m | Convolution integral |
| n | Number of electrons transferred |
| NA | Avogadro number |
| QHa | Charge involved in the hydrogen adsorption or desorption after modifying the electrode surface with bismuth adatoms |
| QHb | Charge involved in the hydrogen adsorption or desorption before modifying the electrode surface with bismuth adatoms |
| R | Gas constant |
| t | Time |
| T | Temperature |
| va | Rate of the adsorption step |
| vd | Rate of the desorption step |
| ve | Rate of the electron transfer step |
| z | Ionic charge |
| zi | Charge of peroxodisulfate anions (−2) |
| zi + 1 | Charge of associated persulfate anions with either Na+, K+, or NH4+ (base electrolyte cations) |
Greek
| α | Charge transfer coefficient |
| γ | Coefficient for the dependence of adsorption rate on E |
| γ' | Coefficient for the dependence of desorption rate on E |
| ɛ | Dielectric constant of solvent |
| θ | Relative surface coverage |
| θ' | Relative surface coverage ClO4─ adatoms |
| θ'' | Relative surface coverage S2O82─ adatoms |
| θbi | Fractional coverage of bismuth adatoms |
| σ | Charge of metal |
| Σθi | Ratio of the relative surface coverage between ClO4− and S2O82− adatoms |
| σi | Charge produced by the adsorbed film |
| ψ1 | Electrical potential at an optimum point for ion discharge |
| ψo | Potential at the outer Helmholtz plane |
| β | Charge transfer coefficient (1 ─ α) |
Acknowledgment
The authors thank University of Science Malaysia (USM) for the financial support to cover the publication fee. Our highest gratitude goes to Dr. Guy Denuault and Prof. Andrew L. Hector for their insights.
ORCID
Saiful Arifin Shafiee 0000-0003-0907-624X
Jolyon Aarons 0000-0002-1290-5494
Hairul Hisham Hamzah 0000-0002-8296-1360
Errata
Errata was published for this paper on January 23, 2019. See http://jes.ecsdl.org/content/166/2/X2.