
Abstract
The all-iron redox flow battery is an attractive solution for large-scale energy storage because of the low cost and eco-friendliness of iron-based materials. A major challenge to realizing a continuously operable battery is the parasitic evolution of hydrogen at the iron electrode during battery charging. We found that the adsorption of ascorbic acid added to the electrolyte inhibited hydrogen evolution at pH = 0. Elevation of pH near the surface of the electrode during electrodeposition also raised the coulombic efficiency. Thus, electrolyte flow rates significantly influence the coulombic efficiency. Ascorbic acid also served to regulate the pH near the surface of the negative electrode by buffering action. We found that increasing the operating temperature enhanced the kinetics of iron deposition relative to the kinetics of hydrogen evolution, leading to a net rise of coulombic efficiency. Thus, by operating at 60°C and a pH of 3 with ascorbic acid and ammonium chloride, we achieved a coulombic efficiency of 97.9%. While this value of coulombic efficiency is among the highest values reported for the iron electrode in the context of the all-iron flow battery, further improvement in efficiency is needed for supporting repeated cycling. The results presented here provide insights for further improvements.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Reliable, low-cost, durable and eco-friendly methods of storing electrical energy are essential for utilizing the energy generated by large-scale solar photovoltaic arrays and wind turbines. Among the various electrochemical energy storage technologies, redox flow batteries are particularly promising because of their scalability and potentially low cost.1,2 Redox flow batteries based on metal ions and organic redox molecules using both aqueous and non-aqueous electrolytes are being studied widely for grid-scale energy storage applications.3–8 Of the various kinds of redox flow batteries, the all-vanadium system has been developed to a commercial scale.9,10 However, the widespread deployment of vanadium systems has been hindered by the cost of vanadium-based electrolytes. To this end, iron-based redox flow batteries are promising because iron is inexpensive and abundantly available.
The all-iron redox-flow battery is based on the Fe(III)/Fe(II) redox couple as the positive electrode and the Fe(II)/Fe(0) redox couple as the negative electrode (Eqs. 1 and 2) yielding a cell voltage of 1.21 V.
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0002.gif)
This battery system was introduced by Savinell and Hrushka in 1981.11 During the charging of this battery, iron is electrodeposited from solutions of iron (II) chloride at the negative electrode while iron (II) chloride is converted to iron (III) chloride at the positive electrode. During discharge, elemental iron at the negative electrode is returned to the solution as iron (II) chloride, while iron (III) chloride is reduced to iron (II) chloride at the positive electrode.
The earliest experiments on the all iron flow battery system deployed iron chloride solutions and a porous separator between the two electrodes.11,12 Such an arrangement resulted in unavoidable cross-diffusion of Fe (III) and Fe (II) between the positive to the negative sides of the cell and thus reduced the coulombic efficiencies. Further, parasitic hydrogen evolution occurred during charging of the iron electrode leading to increase of electrolyte pH followed by precipitation of iron hydroxides. Therefore, because of the low coulombic efficiency, the pH of the electrolyte had to be maintained below the value of 2 to avoid the precipitation of the iron hydroxides.
In our previous publication we introduced another cell configuration for the all-iron flow battery using an anion exchange membrane (AEM) as a separator between the positive and negative electrodes.13 We demonstrated that the AEM facilitated the transfer of chloride ions between the positive and negative sides of the cell without mixing of the positive and negative electrolytes. Thus, the self-discharge resulting from the transfer of iron (III) ions across the membrane could be avoided. We also demonstrated some approaches to mitigate hydrogen evolution at the iron electrode by the use of complexing agents and additives. Complexation of ferrous and ferric ions with different ligands aimed at enhancing the solubility of iron species at elevated pH has been studied by Savinell et al.14 However, the use of organic ligands shifted the reversible potential for the electrodeposition of iron to negative values, decreasing the coulombic efficiency.12 Further, these researchers demonstrated in experiments using a rotating rod electrode that a combination of pH greater than 2 and a supporting electrolyte of a high chloride concentration could increase the coulombic efficiency of electrodeposition to 97%, significantly reducing the rate of the hydrogen evolution reaction(HER).12 As HER could not be completely prevented, an in-tank electrolyte rebalancing reactor was also introduced to recombine the evolved hydrogen with the excess of accumulated iron(III).7 To address the issue of HER during iron deposition, Yan et al. have been researching an all-soluble, all-iron battery that did not involve the iron deposition reaction. Thus, an all-iron flow battery continues to be a topic of research with the main outstanding issues being the suppression of hydrogen evolution and self-discharge.
The implications of parasitic hydrogen evolution
The hydrogen evolution reaction is thermodynamically favored over the electrodeposition of iron as indicated by the electrode potentials (Eqs. 2 and 3).
![Equation ([3])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0003.gif)
Thus, hydrogen will be evolved not only during charging but also during open-circuit stand leading to the corrosion of iron from a charged negative electrode.15 Most importantly, hydrogen evolution affects the repeated cycling of the redox-flow cell by rapidly changing the composition of the electrolyte in the system (Figure 1).7 As protons are converted to hydrogen the pH of the solution increases. The result is an increase in hydroxide ion concentration. The imbalance of hydroxide ions between the positive and negative side results in diffusion of hydroxide ions through the anion-exchange membrane from the positive to the negative side of the cell as shown in step 1C in Figure 1. Thus, as hydrogen evolution occurs the pH of the negative and positive side will continue to increase (Figure 1 step 2A), finally resulting in the precipitation of iron hydroxides (Figure 1 step 3A). Further, a charging process with the low faradaic efficiencies at the negative electrode leads to a continuous buildup of iron (III) ions in the positive electrolyte leading to a capacity imbalance between the positive and negative sides of the cell (Figure 1 steps 2B and 3B).
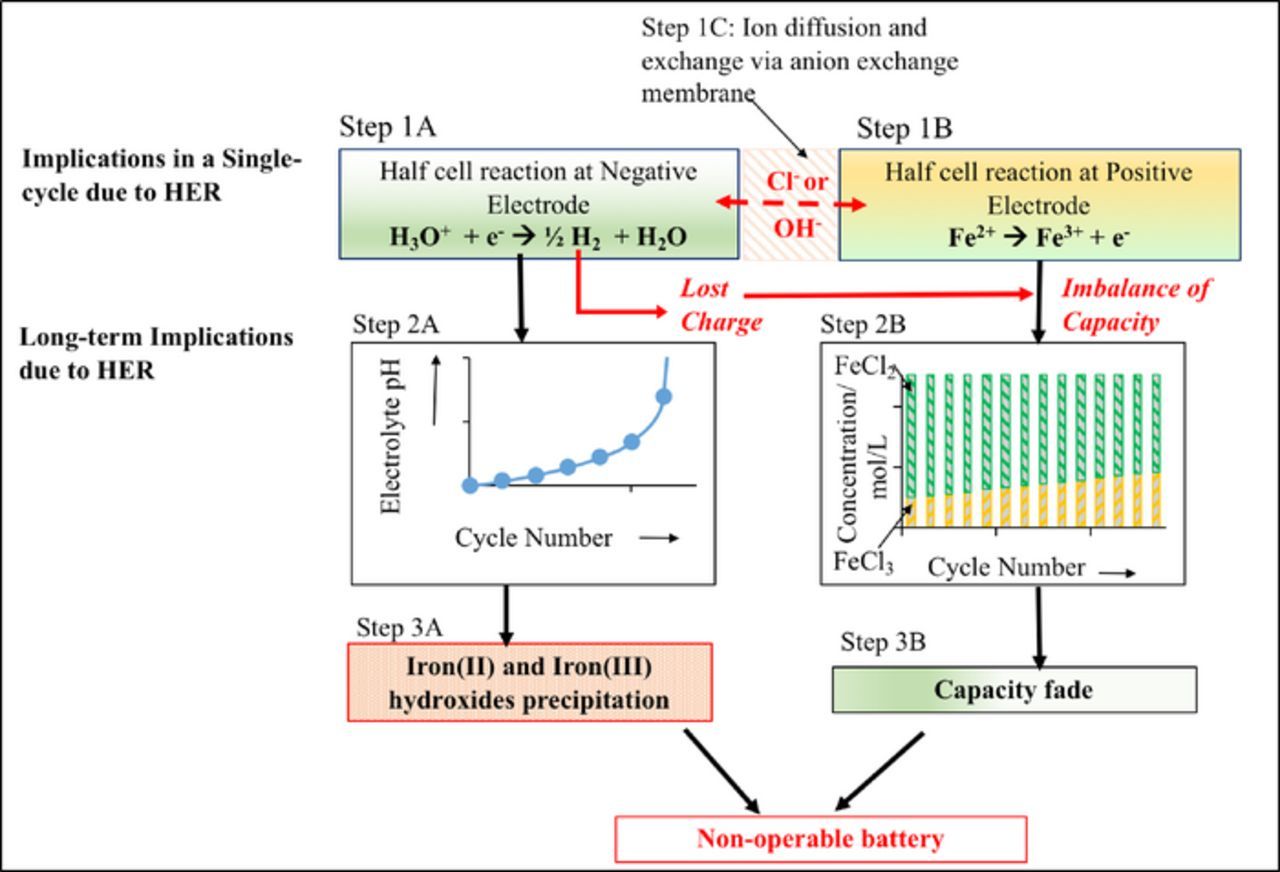
Figure 1. Effects of the hydrogen evolution during the operation of All-iron redox-flow battery.
In Table I we project the impact of hydrogen evolution on the rechargeability of the all-iron battery for various assumed values of coulombic efficiencies. For this calculation, we start with a 2 M solution of iron (II) chloride at its natural pH and simulate the effect of charge and discharge at various values of coulombic efficiency. Then we apply the criteria of capacity imbalance of more than 20% or pH > 3 as the point at which the battery becomes inoperable as a rechargeable system; at pH values greater than 3, precipitation of the hydroxides will start to occur. These simulations were conducted using the relationships in Eqs. 4 and 5.
![Equation ([4])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0004.gif)
![Equation ([5])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0005.gif)
Where α = Coulombic efficiency, F = Faraday constant and Ct = Total capacity
Table I. Predicted Performance of the All-Iron Flow Battery with Coulombic efficiency Losses due to HER.
| Number of Achievable | ||
|---|---|---|
| Charge/Discharge Cycles | ||
| Assumed Coulombic | Criteria 1: Capacity | Criteria 2: pH |
| efficiency % | imbalance | increased to 3 |
| 95.00 | 5 | 2 |
| 96.00 | 6 | 3 |
| 97.00 | 8 | 4 |
| 98.00 | 13 | 6 |
| 99.00 | 25 | 12 |
| 99.99 | 2500 | 1238 |
Total theoretical capacity for 2 M FeCl2 in positive electrolyte: 53.6 Ah L−1; total volume in both chambers = 2 L; starting pH of the electrolyte = 1; pH at which precipitation is projected to occur = 3
From these simulations we find that even when the coulombic efficiency values are as high as 95%, just in a few cycles, the cell operation goes out of capacity balance or leads to precipitation of the hydroxides. Thus, for the continuous operation of this battery for thousands of cycles without onerous capacity re-balancing schemes and re-adjustment of pH, we must suppress hydrogen evolution completely at the negative electrode.
The rate of hydrogen evolution is governed by pH and electrolyte composition (in the bulk and near the surface of the electrode), adsorption of substances, temperature, electrode potential and the specific mechanism of hydrogen evolution operative at the iron electrode.16 Investigation of the kinetics and mechanism of hydrogen evolution from practical solutions used in the all-iron flow battery is challenging because the effect of the foregoing factors is difficult to separate.15,17 In our previous studies, we had demonstrated the benefit of raising the bulk pH and using ascorbic acid as a complexing additive to prevent precipitation of iron hydroxides.13 In the present study, we focus on new findings relating to: (i) the specific effect of ascorbic acid on the overpotential for hydrogen evolution, (ii) the effect of pH near the surface of the electrode on the coulombic efficiency, (iii) the role of ascorbic acid in buffering the pH near the surface of the electrode, (iv) the effect of flow rate on the coulombic efficiency, and (v) the relative effect of temperature on the kinetics of the iron deposition reaction and the hydrogen evolution reaction.
Experimental Methods
Coulombic efficiency of iron deposition and dissolution
To study the coulombic efficiency of iron deposition and dissolution under various operating conditions of pH, current density, temperature and electrolyte composition, we used a three-electrode half-cell (Figure 2). The working electrode (area = 25 cm2) was made of impervious graphite (Graphitestore, catalog number GT001672) to mimic an electrode substrate with high hydrogen overpotential that will be used in an all-iron flow battery. We used a mild steel mesh as the counter electrode and a silver/silver chloride reference electrode. The use of a mild-steel counter electrode ensured that the composition of the solution and pH stays relatively constant because mild steel either underwent dissolution to iron (II) under anodic conditions, or the electrodeposition of iron occurred on mild steel during cathodic conditions. With a platinum counter electrode we generate Fe(III) during anodic conditions and evolve hydrogen during cathodic conditions changing the pH. Thus, a platinum counter electrode in this context results in the change of bath composition. The electrolyte solution consisted of iron (II) chloride (3.25 M) with ammonium chloride (2 M) and ascorbic acid (0.3 M). Ammonium hydroxide was used to adjust the pH of the electrolyte. The coulombic efficiency of electro-deposition of iron on a graphite-working electrode was determined over the range of 4–100 mA/ cm2. Iron was electrodeposited by passing a fixed amount of charge of 60 Coulombs at various current densities of interest, and this was followed by anodic dissolution at 20 mA/ cm2. The coulombic efficiency was calculated, from the ratio of the charge delivered during the anodic stripping step, Qstripping, to the charge uptake during electrodeposition of iron, Qdeposition, as per Eq. 6.
![Equation ([6])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0006.gif)
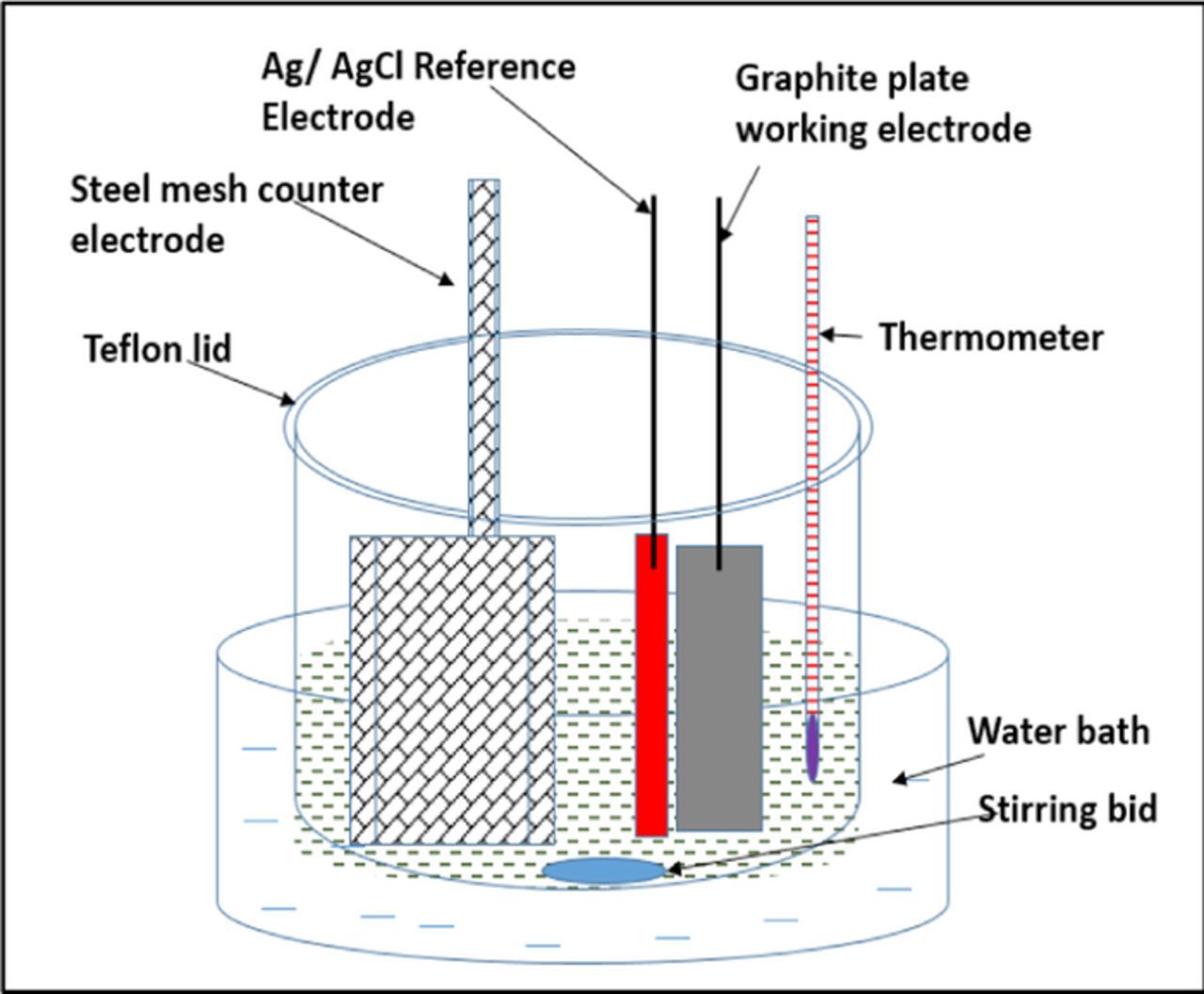
Figure 2. Experimental set-up.
The effect of flow rate (or stirring rate) of the electrolyte on coulombic efficiency was studied for an electrolyte solution that did not contain ascorbic acid. The half-cell experiments were performed using a potentiostat (VMC-4, Princeton Applied Research). During preparation and testing, the electrolyte was de-aerated using argon gas to prevent the air oxidation of Fe (II). The electrolyte was stirred using a magnetic stirrer bar during electrodeposition of iron, to avoid the limitations from the mass transport of iron (II).
Investigations of the hydrogen evolution reaction
To investigate the rate of hydrogen evolution in different electrolytes we conducted polarization studies on an electrodeposited iron surface that mimicked the negative electrode in an all-iron flow battery. To study the effect of cadmium on the surface of the iron electrode, we used a cadmium electrode prepared by electrodeposition using an electrolyte containing cadmium (II) chloride. Additives of interest were dissolved in the electrolyte.
Potentiodynamic polarization studies at a very slow scan rates (5 mV/s) were carried out in a three-electrode cell configuration. The working electrode for this experiment was the relevant metal-covered graphite electrode as described in the foregoing. To study an iron-covered electrode, iron was electrodeposited with 120 Coulombs on a graphite sheet at 60°C using the same electrolyte described in the section Coulombic Efficiency of Iron Deposition and Dissolution at pH value of 3.0. Similarly, for studying a cadmium surface, we electrodeposited cadmium on a graphite sheet at a current density of 40 mA/ cm2 by passing 120 Coulombs from a 0.1 M solution of cadmium chloride in 1 N hydrochloric acid. These electrodes had a smooth surface after electrodeposition. The electrodes were rinsed thoroughly with de-ionized water prior to the hydrogen evolution studies. For the hydrogen evolution studies, we used a platinum wire counter electrode and a silver/silver chloride reference electrode. In these studies, the electrolyte solution was free of the iron or cadmium ions to avoid electrodeposition of the metal; the solution consisted of just ammonium chloride (2.0 M) and ascorbic acid (0.3 M) at pH = 3. We used a pure iron rod (Alfa Aesar, catalog number 40500) as a working electrode to study the effect of pH on HER in the presence of ascorbic acid (0.3 M) in ammonium chloride (2 M) electrolyte.
Correction of electrode potentials for ohmic drop
All the electrode potentials reported here have been corrected for the potential drop across the ohmic resistance between the working electrode and the reference electrode. The ohmic resistance value used for the correction was determined from the real component of the electrochemical impedance at 0.1 MHz, obtained from an impedance spectroscopy experiment.
Impedance spectroscopy at different temperatures
Electrochemical impedance under potentiostatic conditions was measured over the frequency range of 100 kHz to 5 Hz for a pure iron rod working electrode and −0.85 V vs Ag/AgCl at different temperatures in the range of 25°−60°C using the electrolyte described in the section Coulombic Efficiency of Iron Deposition and Dissolution at pH = 3.
Near-surface pH and bulk pH measurement
The pH close to the cathode surface and the pH of the bulk electrolyte during charging, discharging and potentiodynamic polarization studies were measured using two pH-sensing glass electrodes (Thermo Scientific Orion-ROSS Ultra pH/ATC Triode). We have referred to the pH close to the electrode surface as "near-surface pH" in the rest of the manuscript.
Results and Discussion
The effect of electrolyte pH
The coulombic efficiency during electrodeposition of iron increased with increasing pH at all values of current density (Figure 3a). Based on the increase of coulombic efficiency from 85% to 93.6% at 20 mA/ cm2 when the electrolyte pH is raised from 0 to 3.15. We calculated a decrease of 55% in the rate of hydrogen evolution corresponding to this increase of coulombic efficiency. These results are consistent with previous reports of high coulombic efficiency at pH values greater than 2.11,12 Upon changing the pH value from zero to 3.15 the equilibrium potential for the hydrogen evolution reaction (as given by the Nernst Equation) shifts from 0.0 V to −0.185 V vs. NHE. Consequently, at any operating electrode potential for iron deposition, the overpotential applied to the hydrogen evolution reaction would also be reduced by 0.185 V when the pH at the electrode/solution interface is 3.15 compared to a pH value of 0. Based on the electrode potential values observed during iron deposition (Figure 3b), we have calculated the overpotential applied to the hydrogen evolution reaction at various values of pH (Table II).
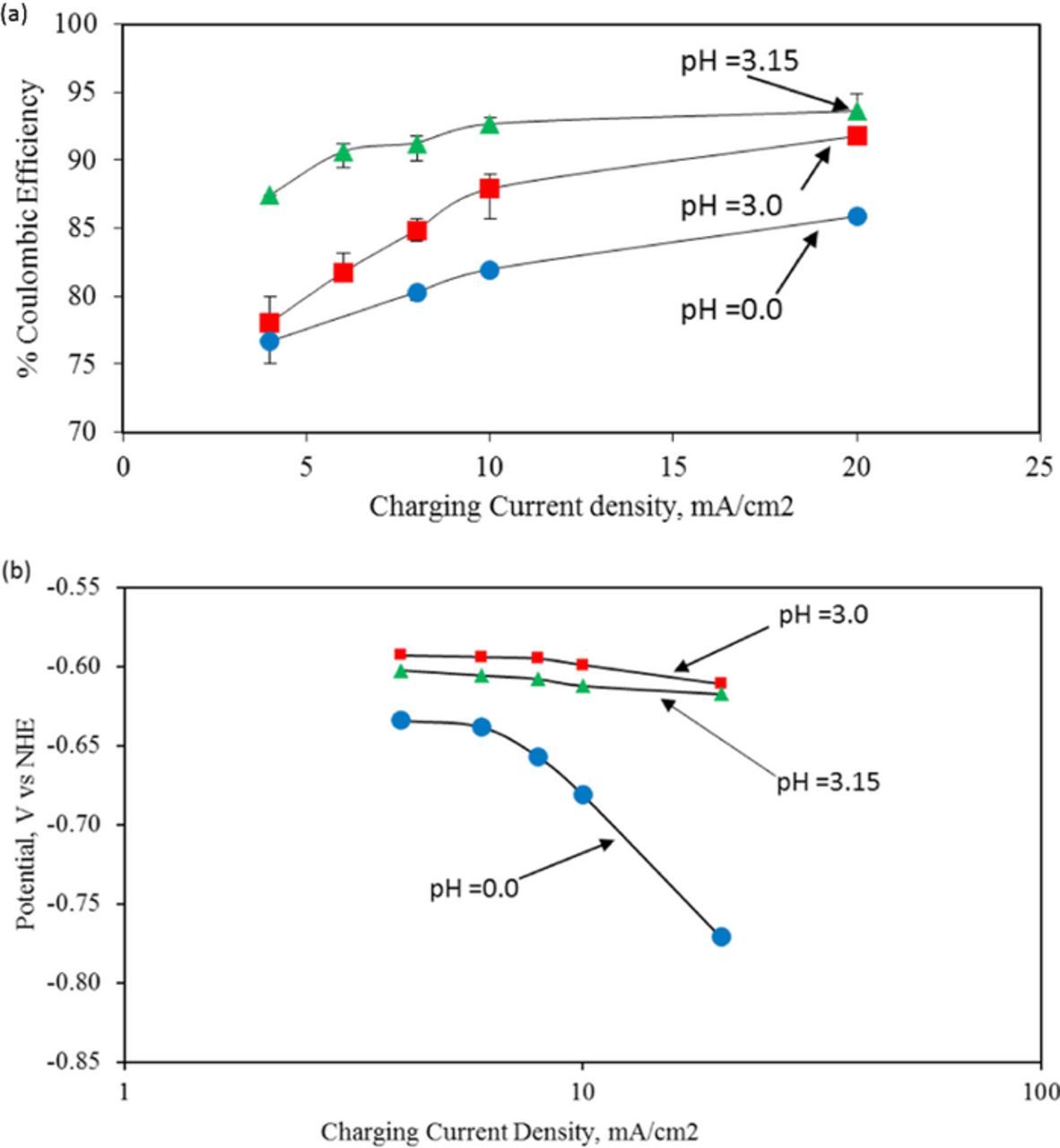
Figure 3. (a) Effect of electrolyte pH on the coulombic efficiency of iron electrodeposition and electrodissolution. (b) Effect of electrolyte pH on electrode potential of iron electrode during electrodeposition; bath conditions (iron (II) chloride (3.0 M), ammonium chloride (2.0 M), ascorbic acid (0.3 M) at temperature = 25°C).
Table II. Electrode potential and HER overpotential at 20 mA/ cm2 at different values of pH.
| Electrode | Equilibrium potential | Calculated | |
|---|---|---|---|
| pH of the | potential, | for hydrogen evolution, | overpotential for |
| electrolyte | V vs NHE | V vs NHE | hydrogen evolution, V |
| 0.00 | −0.771 | 0.000 | −0.771 |
| 3.00 | −0.611 | −0.177 | −0.434 |
| 3.15 | −0.618 | −0.186 | −0.432 |
From the polarization data presented in Table II we can conclude that the overpotential applied to the hydrogen evolution decreased by 0.337 V by changing the pH from 0 to 3. Further, based on the value of Tafel slope for hydrogen evolution on an iron surface of 0.180 V/decade (Figure 4b) we could expect this decrease of overpotential of 0.337 V to result in a reduction in the value of hydrogen current by a factor of 10(0.337/Tafel slope), which is approximately a seventy-five fold reduction in HER current. It is reasonable to assume that any change in roughness of the surface between pH = 0 and pH = 3 affects the hydrogen evolution and iron deposition reaction rates in the same proportion. However, we only obtain a factor of 2 reduction in current and an increase of coulombic efficiency of just 5.9% from pH = 0 to pH = 3 at 20 mA/ cm2. While this may seem surprising, this apparent inconsistency could be attributed to the effect of adsorption of the electrolyte additives, particularly ascorbic acid on the kinetics of hydrogen evolution at the iron electrode. We could expect the effect of adsorption of the electrolyte additives to overshadow the benefit from the shift of equilibrium potentials. We discuss this effect in detail in the following.
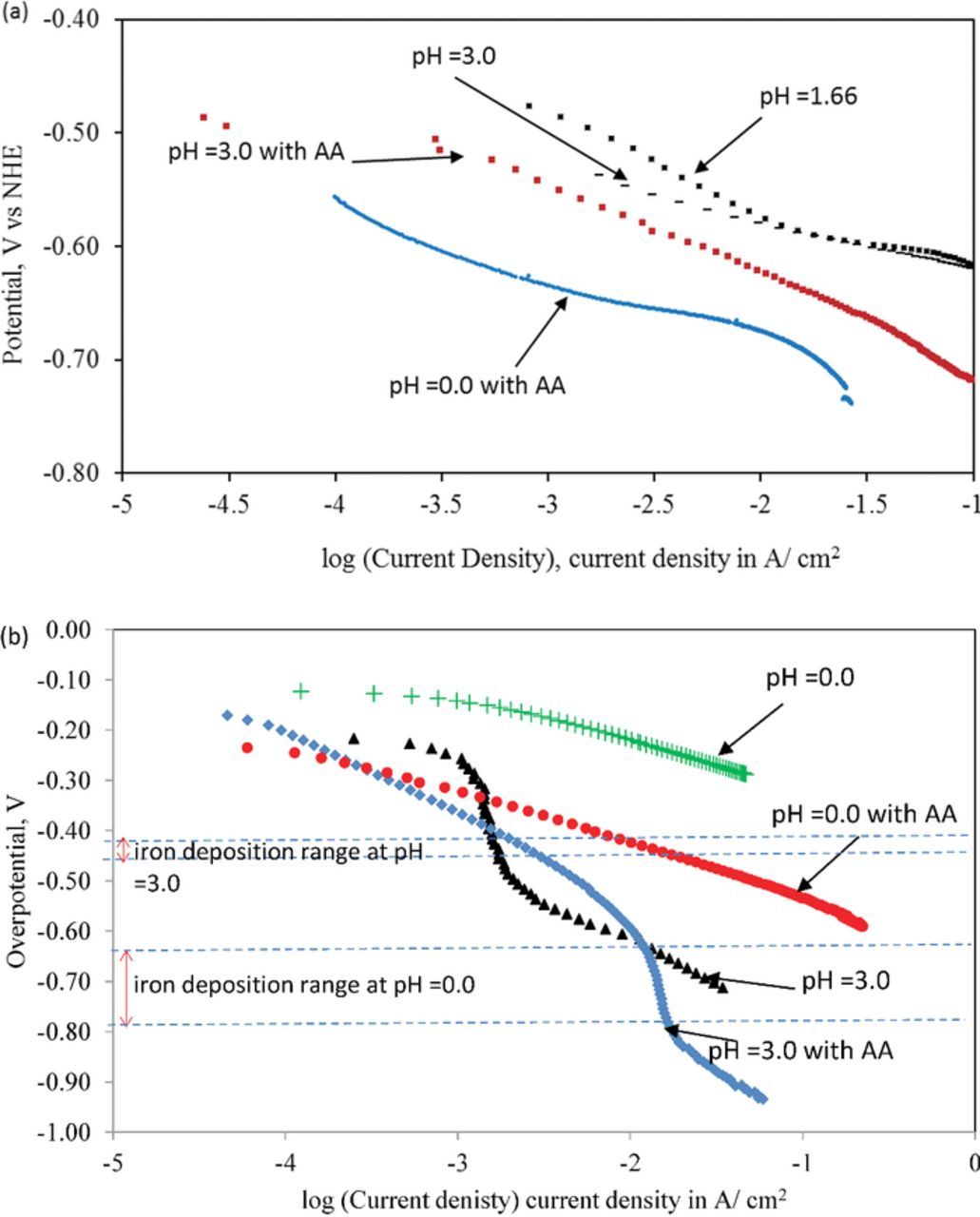
Figure 4. (a) Potentiodynamic polarization studies of iron deposition from a solution of iron(II) chloride(3.0 M), ammonium chloride (2.0 M), T = 25°C in the presence and absence of ascorbic acid (0.3 M) at various values of pH as shown (AA = Ascorbic acid). (b) Overpotential for HER on iron at pH 0 and 3 in the presence or absence of ascorbic acid (0.3 M) and ammonium chloride (2.0 M).
Effect of ascorbic acid on the kinetics of hydrogen evolution
Ascorbic acid has been used as an electrolyte additive in iron plating because ascorbic acid is a strong two-electron reducing agent. Thus, the presence of ascorbic acid served to minimize the air oxidation of iron (II) to iron (III). The formation of iron (III) in the negative electrolyte must be avoided as it will reduce the coulombic efficiency by electrochemical conversion to iron (II) and thus competing with the electrodeposition of iron.18
Spinelli et al. studied ascorbic acid as a mixed type corrosion inhibitor on mild steel in solutions of pH in the range of 2 to 4.19 Ascorbic acid at pH 3 exists as a mixture of the protonated and de-protonated forms and the relative composition of these forms will vary with pH.18,19
Both ascorbic acid and its oxidized form, dehydroascorbic acid, have been shown to adsorb on the iron surface facilitated by the presence of polar groups and pi- electrons.19 The formation of adsorbed hydrogen, a required intermediate step for hydrogen evolution, will now have to compete with the adsorption of ascorbic acid. Consequently, we can expect the kinetics of hydrogen evolution to be hindered in the presence of ascorbic acid. The degree of inhibition will depend on the relative coverage of the surface by ascorbic acid and hydrogen atoms. When the pH is increased from 0 to 3.15, we could expect a decreased adsorption coverage because ascorbic acid would be ionized into a negatively charged species. Such species will be repelled from the surface of the electrode. Thus, ascorbic acid is not likely to have the same strong inhibitory effect at pH = 3.15 as it would at pH = 0. A decrease of electrode potential during iron deposition at 10 mA/ cm2 with increasing pH from 0 to 3.15 in the presence of ascorbic acid is consistent with the reduced adsorption of ascorbic acid (Figure 3b). Further, when we compare the overpotentials for hydrogen evolution at pH = 0 and pH = 3 in the range of potentials for iron deposition (Figure 4b), we find a significantly higher overpotential for hydrogen evolution at pH = 0 compared to pH = 3.
These observations suggest that in spite of the shift of equilibrium potential for hydrogen evolution when we change the pH from 0 to 3, the benefit from a reduction of the applied overpotential is not realized because of the relatively low baseline resulting from the inhibition of hydrogen evolution by the adsorption of ascorbic acid at pH = 0. Consequently, the net effect observed by changing the pH from 0 to 3 was reduced to just a two-fold reduction in hydrogen evolution current at a cathodic current density of 20 mA/cm2.
Effect of ascorbic acid on the kinetics of electrodeposition of iron
Ascorbic acid forms an intensely green complex with iron (II) that is quite stable even on exposure to oxygen.20 The color of this complex changes from green to violet and then to black upon increase of pH.21 Since the complexation of ascorbic acid with iron is facilitated at pH = 3 by the formation of the monoascorbate anion, the overpotential for metal electrodeposition will be increased. Thus, the increase of pH in the presence of ascorbic acid can be expected to have a significant effect on the kinetics of electrodeposition of iron.
In the absence of ascorbic acid in the electrolyte, iron deposition kinetics is not affected significantly by the change of pH from 0 to 3 (Figure 4a). The polarization curves at various pH values fall right on top each other. This observation is not surprising as the equilibrium potential of the Fe(II)/ Fe(0) couple is not expected to change with pH until precipitation of the hydroxides begins to occur.
When 0.3 M ascorbic acid is added to the iron chloride electrolyte, the electrode potentials during iron deposition shifted to more negative values. At pH = 3, at a current density of 10 mA/ cm2, the shift in electrode potential was 41 mV. However, at pH = 0 upon addition of ascorbic acid, the shift increased further to 95 mV. Thus, it was clear that ascorbic acid impacted the kinetics of the iron deposition reaction to different extents depending on the pH value of the electrolyte. Thus, the coulombic efficiency at pH = 0 is dominated by the effect of adsorption of ascorbic acid on the kinetics of hydrogen evolution and iron deposition. However, at pH = 3 and 3.15, we expect complexation reactions of ascorbate with iron to play a significant role (described here below).
Ascorbic acid can interfere with the electrodeposition process by complexing with iron (II) and through the formation of adsorbed layers. The effect of complexation is two-fold: (i) shifting the equilibrium potential to more negative values and (ii) hindering of the charge transfer process by imposing an additional kinetic barrier of "de-complexation" before ion to atom transformation occurs during electrodeposition. Based on the formation constants of iron (II) ascorbate complexes, we expect the shift of equilibrium potential at pH 3 to be about 35 mV (see supplementary information SI1).18 The observed shift in electrode potential at pH = 3 at 10 mA/cm2 is consistent with this shift in equilibrium potential. At pH = 0, ascorbic acid being unionized (pKa = 4.17), complex formation is not significant and we do not expect a shift in the equilibrium potential for the iron deposition reaction. However, at pH = 0 unionized ascorbic acid, as mentioned earlier, is reported to form adsorbed layers on the iron electrode.19,22 Such adsorption will reduce the exchange current density and lead to polarization losses (Figure 4b). Consequently, we attributed the shift of electrode potential by 95 mV during electrodeposition of iron at pH 0 and 10 mA/cm2 to the hindered kinetics resulting from the adsorption of ascorbic acid on the iron surface.
Effect of current density on coulombic efficiency
We observed that increasing the charging current density also improved the coulombic efficiency (Figure 3a). However, this effect was more significant at pH = 3 than at pH = 0. During the electrodeposition of iron, the concentration of iron(II) in the diffusion layer adjacent to the surface of the electrode is lower than in the bulk simply because of the concentration gradient required for mass transport. This lowered surface concentration of iron (II), resulted in a lower extent of hydrolysis of iron (II) as per Eq. 7. The extent of hydrolysis of iron (II) can be calculated using the activity of each of the species (ax, x = Fe(OH)+, H3O+ and Fe2+) using Eq. 8.
![Equation ([7])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0007.gif)
![Equation ([8])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0008.gif)
Consequently, because of lower extent of hydrolysis, the pH at the surface of the electrode during electrodeposition will be higher than the bulk value. Such an increase in pH will result in the reduction in the hydrogen evolution current at any given overpotential and thus an improved coulombic efficiency. Further, at a moderately higher value of pH, both Fe2+ and Fe(OH)+ species would be present in significant concentrations. Dahms et al. have reported that the local pH increase at the electrode surface can influence the charge transfer process by involving Fe(OH)+ in the rate determining step.23
We have verified experimentally that an increase in near-surface pH occurs with increasing charging current densities (Figure 5). The near-surface pH can rise to as high as 6 at 100 mA/cm2 while the bulk pH is still lower than 4. We can expect this rise in near-surface pH with current density to be affected by the diffusion layer thickness which in turn is dependent on the stirring or agitation rate. We have estimated the change in pH in the diffusion layer at any current density using the Nernst diffusion layer model and the hydrolysis constant (Eq. 9) (see supplementary information SI2 for the derivation).
![Equation ([9])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0009.gif)
where Kh = hydrolysis constant, δ = diffusion layer thickness, D = diffusion coefficient of iron(II), I = current density, α = coulombic efficiency for iron deposition, F = Faraday constant and C*Fe(II) = bulk formal concentration of Fe(II) at a given pH.
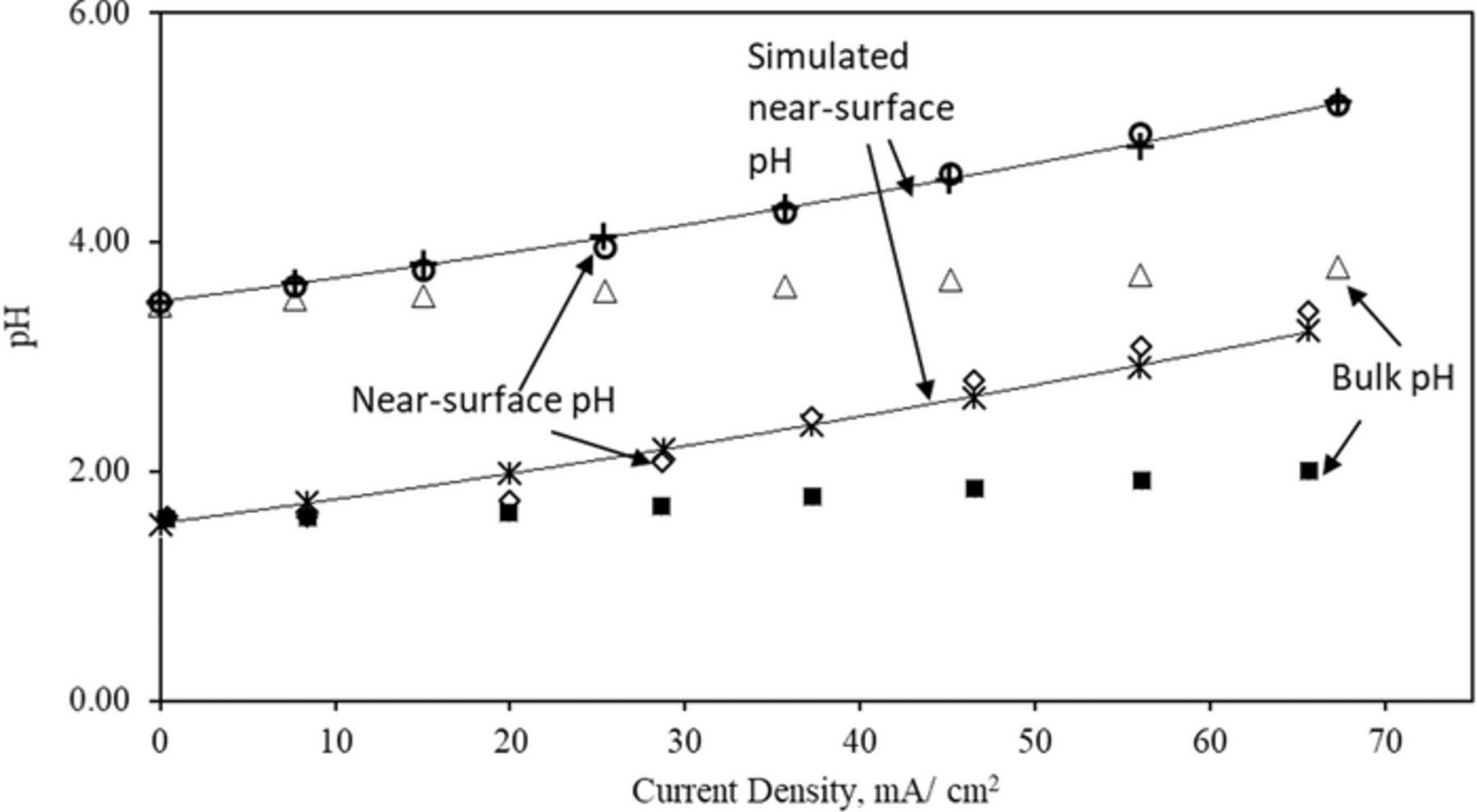
Figure 5. Experimental (◊,●) and simulated (*,+) near-surface pH and experimental bulk pH (■, Δ) deviation during potentiodynamic polarization of metal electrodeposition using an electrolyte composed with iron (II) chloride (3.25 M) and ammonium chloride (2.0 M).
Using Eq. 7 and assuming a diffusion layer thickness of 0.62 mm, we can reproduce the change in near-surface pH with current density (Figure 5). This assumed value of diffusion layer thickness is reasonable to expect under the stirring conditions of the deposition experiment. Since the diffusion layer thickness decreases with increasing flow velocity, we can expect that the extent of stirring of the electrolyte to be an important factor in determining the observed charging efficiencies (see supplementary information SI3 for estimate of diffusion layer thickness as a function of flow velocity). In the experiments where we changed the stirring rate we did observe for charging current densities of 50 and 100 mA/cm2 that the coulombic efficiency of iron deposition decreased with increasing stirring rate (Figure 6). This observation also verified that the coulombic efficiency at a given current density can be a lower value at higher stirring rates due to the decreased diffusion layer thickness. Based on these observations and the simulation we can attribute with confidence the improvements in coulombic efficiency with charging current density to the increase of near-surface pH. Thus, flow velocity or stirring rate is an important factor in determining the coulombic efficiency during battery charging.
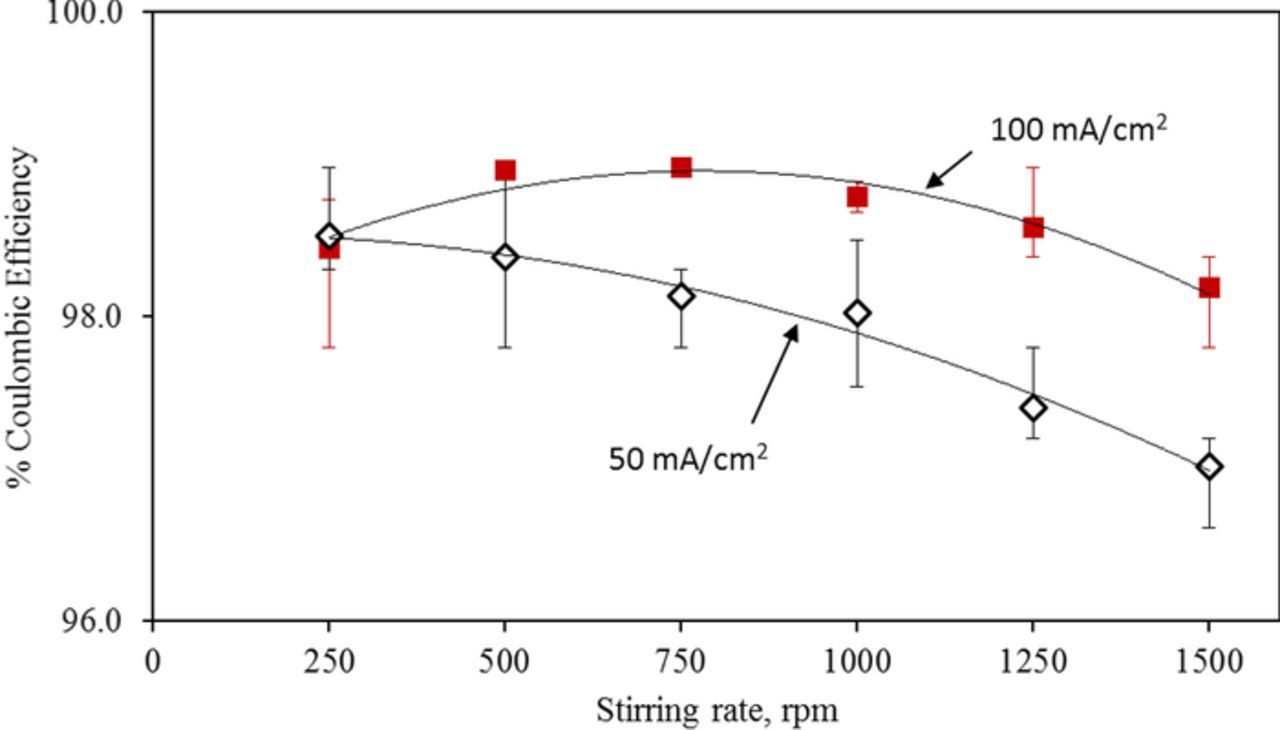
Figure 6. Deviation of coulombic efficiency with stirring rate at constant current densities (◊−50 and ■−100 mA/ cm2) using an electrolyte composed with iron (II) chloride (3.0 M) and ammonium chloride (2.0 M) at pH = 2.00.
While the interaction of charging current density with flow rate and near-surface pH can be exploited to achieve the highest coulombic efficiencies, precipitation of insoluble hydroxides can occur if the optimal current density is exceeded or if the flow rate falls below the set value. The maximum value of pH that we can achieve in the solution of 3.25 M iron chloride without causing formation of the hydroxo complexes of iron is pH = 3.15. Any attempt to raise the pH of the bulk solution where the solution concentration of iron is 3.25 M does not yield a clear solution. For this reason, although bulk values of pH = 3 and 3.15 are not too different, the increase in pH occurring near the electrode surface can be markedly different.
In the following, we describe results with ascorbic acid containing electrolyte that can achieve relatively stable efficiencies over a wide range of current densities.
Effect of electrolyte additives
Increasing the solubility of the electrolyte at higher pH
We have studied the deposition of iron from an electrolyte consisting of a mixture of ammonium chloride and ascorbic acid. Similar compositions were shown in previous studies to increase the stability of iron(II) in air, reduce corrosion of iron, and decrease the rate of hydrogen evolution during the electrodeposition and dissolution of iron.12,24
We have observed that iron (II) chloride solutions exhibit a slightly higher pH in the presence of 2.0 M ammonium chloride (Supporting information SI4). This observation is consistent with the effect of increased ionic strength in reducing the inter-ionic interactions and the tendency to form hydrolyzed products.
Based on the measured pH shift with ammonium chloride we can estimate the change in the activity of the iron (II), using Eq. 10 (readily obtained from the equilibrium constant expressions).
![Equation ([10])](https://content.cld.iop.org/journals/1945-7111/165/9/A1630/revision1/d0010.gif)
We have estimated that the apparent hydrolysis constant in 3.25 M iron(II) chloride is 8.8 times lower with 2.0 M ammonium chloride (Supporting information SI4). The lower apparent hydrolysis constant implies that a higher pH will have to be reached before precipitation occurs in ammonium chloride containing solutions. Thus, the addition of ammonium chloride will extend the pH range of operation of the all-iron flow battery.
Increasing the solubility of iron hydroxide at the electrode surface
We reported in our previous study that ascorbic acid also enhanced the solubility of iron (II) by the formation of metal-ligand complexes allowing the electrolyte pH to be as high as 3.15.13,18 Since the pH near the electrode is even more critical than the bulk pH, we have investigated the effect of ascorbic acid on maintaining the near-surface pH.
In an electrolyte of bulk pH = 0, in the presence of ascorbic acid, the near-surface pH of the iron electrode during electrodeposition rose rapidly with current density (Figure 7a). However, when the bulk pH was 3, the near-surface pH remained close to that of the bulk pH value. This difference is attributed to the buffering action of ascorbic acid. With a pKa of 4.17, ascorbic acid will dissociate in the diffusion layer maintaining a relatively stable pH close to the pKa value.21 Further, the effective pKa of the ascorbic acid in ammonium chloride medium is 2.5 times higher compared to that of the standard value (see supplementary information SI4).18 Thus, the buffering action of ascorbic acid in 2.0 M ammonium chloride shifts to even lower pH values than expected from the standard pKa value of 4.17 (see supplementary information SI5). This buffering action at even a lower pH value is essential for operating at higher current densities as the rapid rise of near-surface pH can cause the precipitation of the iron (II) hydroxo complexes. The near-surface pH in the presence of ascorbic acid at various current densities of operation can be determined by applying the Henderson-Hasselbalch equation. Experimental measurements confirm that in the presence of ascorbic acid, the near-surface pH does not deviate significantly from the bulk value (Figure 7a). As mentioned earlier, in addition to the buffering action of ascorbic acid, the chelation of iron (II) by ascorbate also avoids the precipitation of the insoluble hydroxides (Figure 7b).
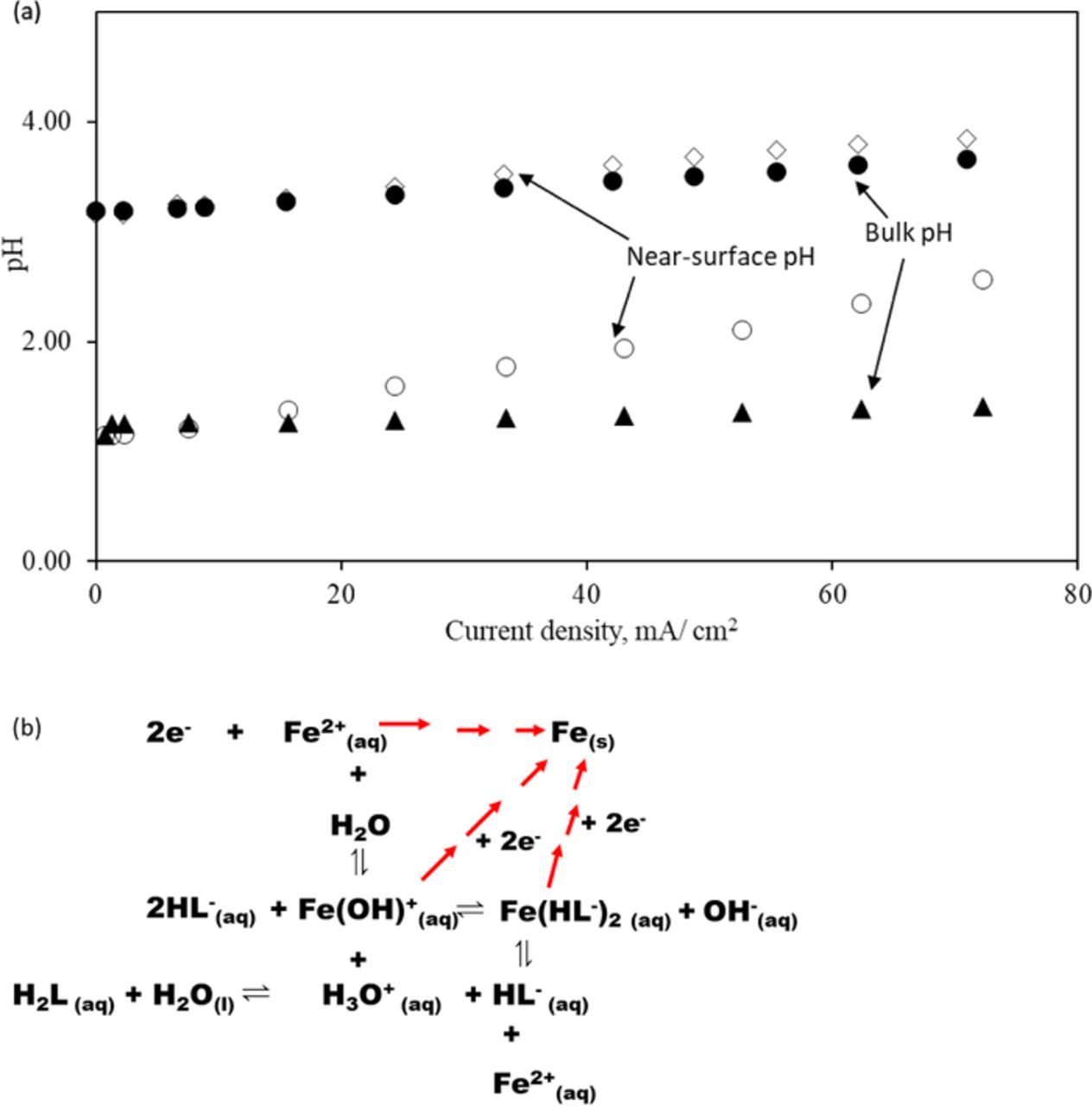
Figure 7. (a) Experimental near-surface pH (◊,○) and bulk pH (●, ▲) deviation during potentiodynamic polarization of metal electrodeposition using an electrolyte composed with iron (II) chloride (3.25 M), ammonium chloride (2.0 M) and ascorbic acid (0.3 M). (b) Representation of the equilibrium reactions near the electrode surface during iron electrodeposition process (Ascorbic acid (H2L) and hydrogen adsorption on iron and HER on iron are not illustrated).
Inhibition of kinetics of HER using a second metal
The composition of the electrode during electrodeposition of iron is expected to affect the rate of hydrogen evolution. In particular, metals such as platinum and nickel facilitate hydrogen evolution because of the enhanced chemisorption of hydrogen resulting from the partially-occupied d-bands of the metal.16 On the contrary, elements with filled d-bands like cadmium, zinc, lead, bismuth and mercury do not support hydrogen evolution readily. Specifically the overpotential for hydrogen evolution at a current density of 10 mA/cm2 in 1 N hydrochloric acid is 1.2 V on cadmium.25 Drazic et al. explained the inhibitory effect of cadmium (II) in the corrosion of iron in sulfuric medium by the increase of the Tafel slope from 130 to 230 mV per decade.26 Thus, if a second metal such as cadmium with high overpotential for hydrogen evolution can be presented to the surface during the electro-deposition of iron, then hydrogen evolution would be suppressed during charging. For such a metal to be always present at the surface, it must be co-deposited with iron and also be immiscible with iron; the immiscibility will ensure that the second metal remains on the surface at all times and does not diffuse into the bulk of the iron electrode. By staying on the surface, the second metal will continue to provide the suppression of hydrogen evolution during the entire course of electrodeposition. Cadmium is one such metal and thus a monolayer of cadmium on the surface of iron can be expected to hinder the kinetics of the hydrogen evolution reaction. Our previous results with small amounts of indium chloride in the bath suggested that the benefit of reduced hydrogen evolution can be realized with the co-deposition of metals with a high ovepotential for hydrogen evolution, indium being one such.13 However, a quantitative understanding of this benefit of co-deposition was not presented. We have therefore explored this effect further with cadmium addition, a metal similar to indium with respect to the high overpotential for hydrogen evolution.
We have observed the doubling of the overpotential for HER at 10 mA/cm2 with cadmium metal (Figure 8a). Further, the Tafel slope for hydrogen evolution on cadmium was higher than that on iron by 73 mV/ decade. These findings implied that cadmium could be exploited to inhibit hydrogen evolution from the negative electrolyte and increase the coulombic efficiency. We expected this effect to be more significant at higher charging current densities. When we electrodeposited iron from solutions containing 20 mM cadmium chloride we found that the coulombic efficiency was consistently higher than for the electrolyte without cadmium when operating higher than 20 mA/cm2 (Figure 8b). The improvement in coulombic efficiency from 90 to 93% resulting from the addition of cadmium, despite seeming small, translates to a 30% reduction in the amount of hydrogen evolved during charging. Thus, even a 3% improvement in coulombic efficiency is significant for increasing the cycle life of the all-iron flow battery. While this comparison proves the viability and basis of this approach of using a second metal to raise coulombic efficiency, the use of cadmium chloride is not desirable for developing an environmentally-friendly battery. A similar strategy relying on bismuth, tin and other non-toxic metals needs to be investigated.27 Inhibition by organic molecules that can adsorb on the iron electrode also needs to be explored.28,29
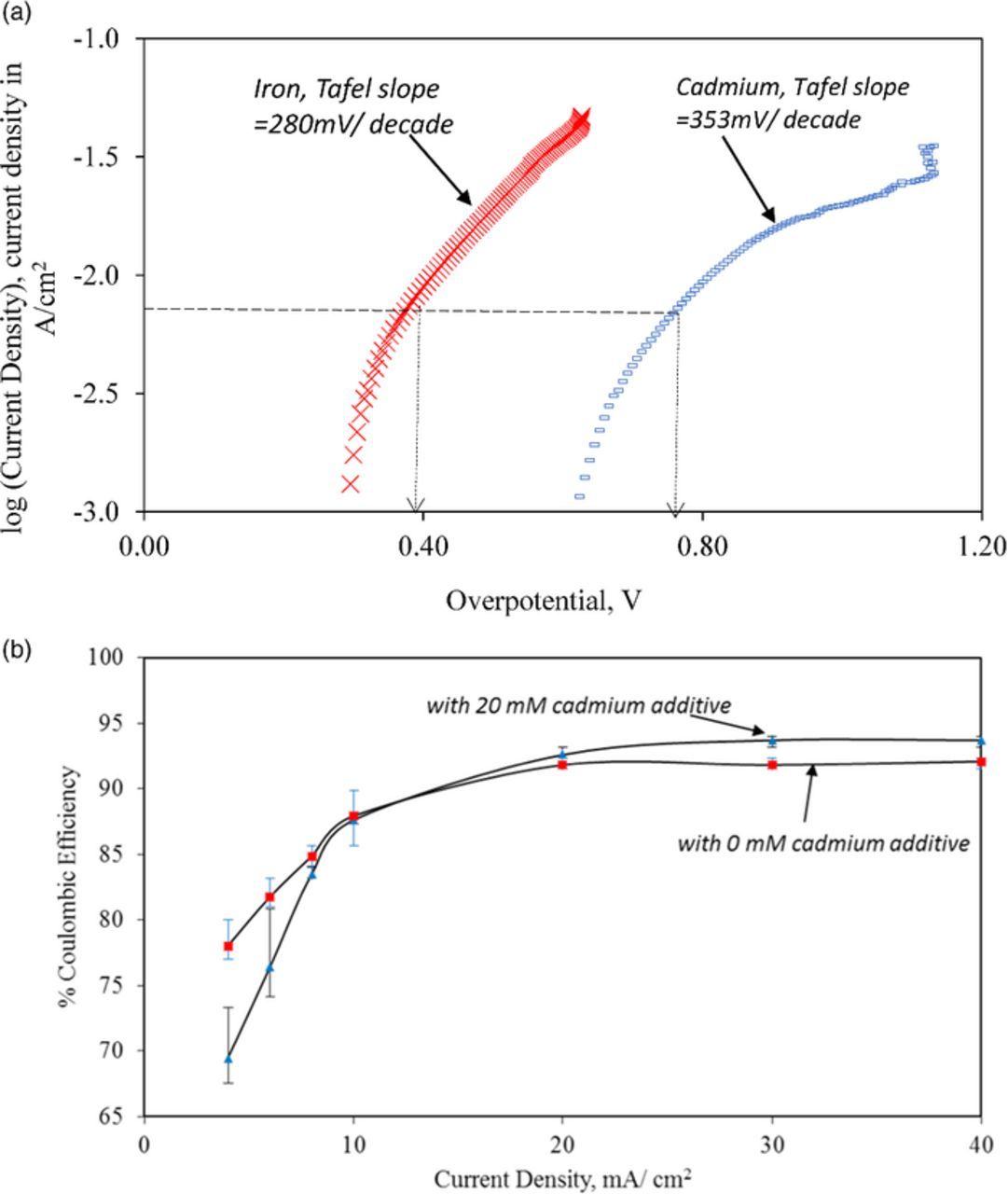
Figure 8. (a) Potentiodynamic studies of HER on iron(x) and cadmium(-). (b) Effect of cadmium chloride additives on the coulombic efficiency of iron electrodeposition and electrodissolution. Bath conditions (iron (II) chloride (3.0 M), ammonium chloride (2.0 M), ascorbic acid (0.3 M) and cadmium chloride (0.0 mM(▲), 20 mM(■) at pH = 3.0 and temperature = 25°C).
Effect of temperature
We found that the coulombic efficiency of iron deposition and dissolution increased by 0.17% per degree Celsius rise of bath temperature in the range of 25–60°C at a charging current of 20 mA/ cm2 (Figure 9). Hrushka et al demonstrated a coulombic efficiency of 90% using ferrous ammonium chloride electrolyte bath operated at 60°C however, their focus was not to study the effect of temperature on the coulombic efficiency.11 We observed a coulombic efficiency of 97.9% at 60°C compared to 91.8% at 25°C. The increase of temperature would increase the solubility of the iron chloride and iron hydroxides, increase the ionic conductivity of the solution, increase the diffusion coefficient and increase the rate constant for charge transfer.30 However, these various effects are not easily separated. The improved kinetics of iron deposition is supported by the decrease of electrochemical impedance with temperature over the entire range of frequencies (Figure 10). We ascribed the real component of the high frequency impedance to the ohmic component arising from the electrolyte and electrode and designated it as the series resistance Rs. Using a Randles' type model, the real component of the impedance at low frequencies (5–10 Hz range) was ascribed to a polarization resistance associated with the charge-transfer process and denoted as Rp (Table III). We find that Rp also decreased with increasing temperature. Absence of no significant difference in the impedance data with and without stirring suggested that the mass transport of the electroactive species is not a limiting process. This finding is consistent with reports in the scientific literature that electrodeposition of iron proceeds via an adsorbed intermediate involved in the rate determining step.15,31–34 Thus, we could attribute the improved kinetics to the desorption and surface diffusion of adsorbed intermediates resulting from an increase of temperature. In addition, we find that upon increasing the temperature from 25 to 60°C the Tafel slope for electrodeposition of iron decreases by about 30 mV/ decade (Supporting information SI6).
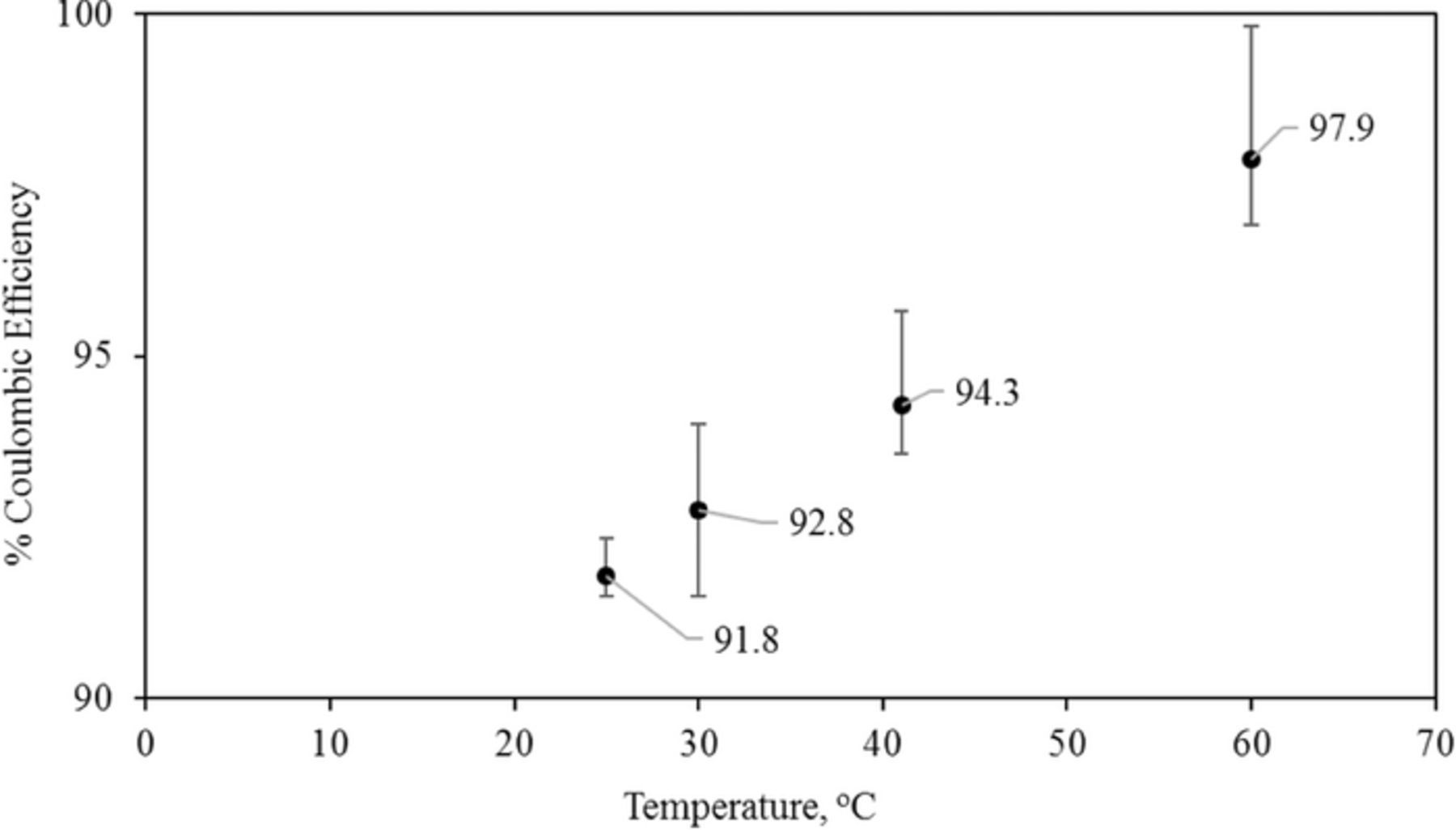
Figure 9. Effect of temperature on coulombic efficiency of iron electrodeposition and electrodissolution. Bath conditions (iron (II) chloride (3.0 M), ammonium chloride (2.0 M) and ascorbic acid (0.3 M) at pH = 3.0). Efficiency values indicated for each point.
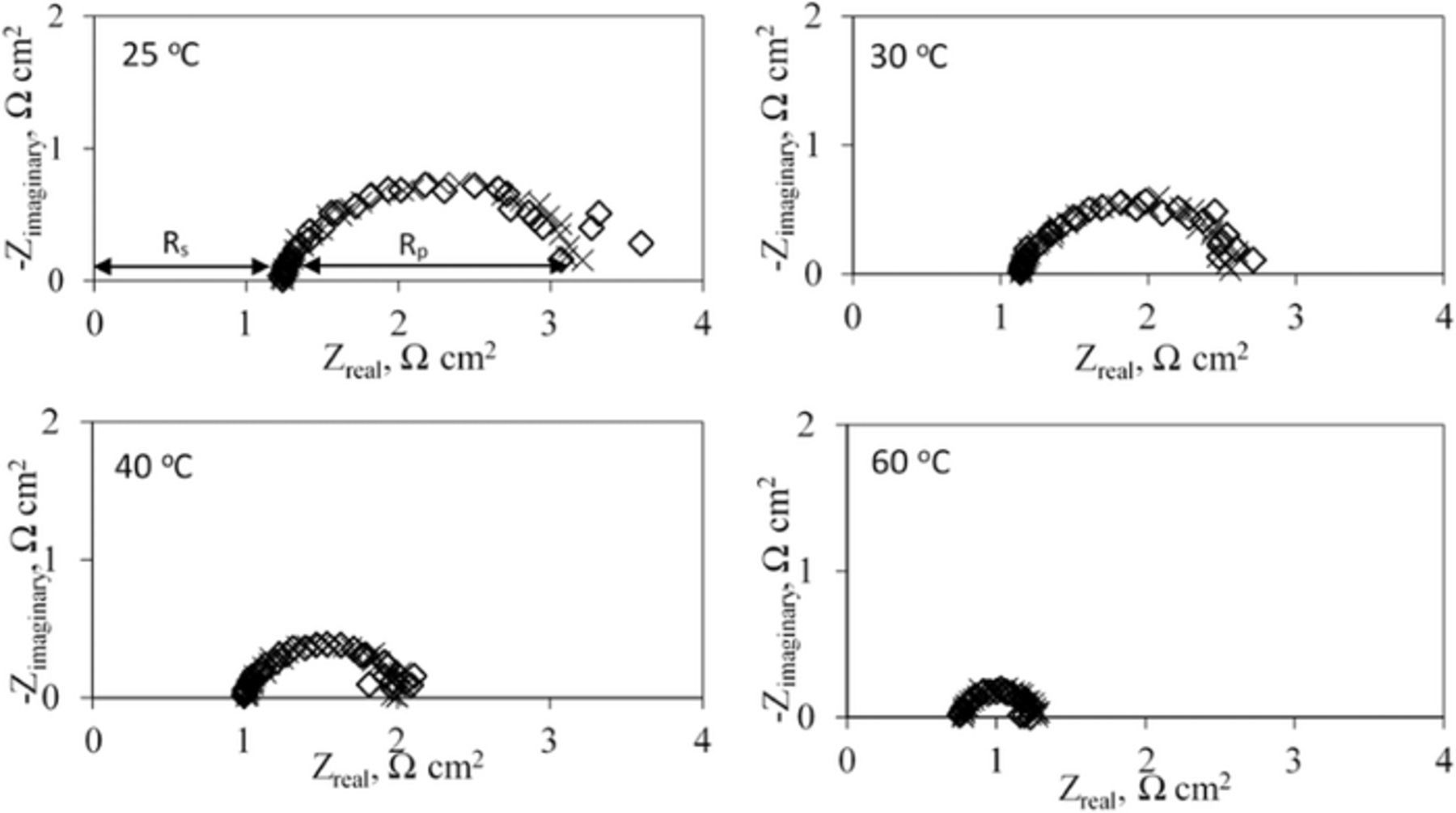
Figure 10. Overlaid Nyquist diagrams at −0.85 V vs Ag/AgCl at 25–60°C at strirring (x) and no stirring (◊) conditions. Bath conditions (iron II) chloride (3.0 M), ammonium chloride (2.0 M) and ascorbic acid (0.3 M) at pH = 3.0). Temperatures indicated on the charts. (Rs -ohmic resistance and Rp -polarization resistance)
Table III. Effect of temperature on electrochemical impedance parameters of iron electrode under potentiostatic conditions at −0.85 V vs Ag/AgCl.
| With no stirring | With stirring | |||
|---|---|---|---|---|
| Temperature,°C | RS, Ω cm2 | Rp, Ω cm2 | RS, Ω cm2 | Rp, Ω cm2 |
| 25 | 1.223 | 2.074 | 1.218 | 1.998 |
| 30 | 1.133 | 1.496 | 1.117 | 1.487 |
| 40 | 1.009 | 1.009 | 0.991 | 1.023 |
| 60 | 0.782 | 0.466 | 0.773 | 0.407 |
Just as the iron deposition process proceeds through adsorbed reaction intermediates, the hydrogen evolution process also involves formation of adsorbed hydrogen.15,31–34 Thus, the deposition of iron and the evolution of hydrogen can be expected to compete for the surface sites. Further, since the electrolyte contains ascorbic acid and ammonium chloride, equilibrium constants of iron hydrolysis and the extent of adsorption of hydrogen and ascorbic acid on the iron electrode surface would also be changed with increasing the temperature. The coulombic efficiency values suggest that the surface energetics of the iron deposition reaction is favored at higher temperatures over that of the hydrogen evolution reaction. We plan to investigate these interesting competitive surface processes in our future studies.
Another benefit of operation at a higher temperature is that the internal stress of the electrodeposit can be relieved. A stress-free deposit will avoid the formation of powdery particles and metal detachment.35 Electroplaters of iron have known these beneficial effects of temperature for some time and the electroplating of iron is always practiced at temperatures greater than 60°C, and even at as high as 80°C.35 Although the overall beneficial effect of temperature on the coulombic efficiency has been measured for the electrolyte containing ascorbic acid and ammonium chloride (Figure 9), additional resolution of the contribution of the other factors affected by increase of temperature requires further investigation.
Conclusions
The all-iron flow battery using iron chloride electrolyte is an attractive solution for large-scale energy storage if the hydrogen evolution that occurs during charging can be suppressed. Our study provides insight into several promising approaches to improving the coulombic efficiency of the iron electrode. We demonstrate a coulombic efficiency of 97.9% using a combination of functional electrolyte additives, pH and elevated temperature. To our knowledge this value of coulombic efficiency is among the highest reported for the "round-trip" coulombic efficiency of charge and discharge of the iron electrode.
The coulombic efficiency during electrodeposition of iron was found to improve with increasing pH at all values of current density. We have found that ascorbic acid has an important role in determining the coulombic efficiency. The adsorption of ascorbic acid on iron inhibits hydrogen evolution over the pH range of 0 to 3. We have found that beyond the pH of the bulk electrolyte, the pH at the surface of the electrode is even more crucial in achieving a high coulombic efficiency; maintaining a high near-surface pH without precipitation is important for increasing the coulombic efficiency. Thus, the coulombic efficiency was also found to be sensitive to the rate of stirring of electrolyte. A key finding is that the near-surface pH can be regulated close to an optimal value of 3 by exploiting the acid-base buffering properties of ascorbic acid. Such an arrangement using ascorbic acid allows the iron electrode to operate at high current densities without the danger of precipitation of the insoluble hydroxides. Thus, ascorbic acid is again shown to be critical in achieving high coulombic efficiency.
We have also demonstrated that a metal such as cadmium that has a high overpotential for hydrogen evolution and that is also immiscible with iron can provide a significant improvement in coulombic efficiency during iron electrodeposition. By the co-deposition of cadmium with iron, we found that the rate of hydrogen evolution can be reduced.
We also conclude that increasing the operating temperature to 60°C has a strong beneficial impact on the coulombic efficiency. We have found that the charge-transfer kinetics of iron deposition was improved relative to that of the hydrogen evolution reaction by increasing the temperature, leading to a higher coulombic efficiency.
With the fundamental insights gained in this study and the improvements in coulombic efficiency demonstrated we believe that the all-iron redox flow battery based on iron chloride will continue to present an attractive pathway for large-scale electrical energy storage and the authors believe that the future will benefit from research on approaches to achieving 100% coulombic efficiency.
Acknowledgments
The authors acknowledge the financial support for this study from the U.S. Army RDECOM CERDEC CP&ID and the Loker Hydrocarbon Research Institute, University of Southern California. We also thank Donald Wiggins and Michael Cowan of the USC Dornsife Machine shop for support with the fabrication of the electrodes.
ORCID
S. R. Narayanan 0000-0002-7259-3728