Abstract
Enzymatic fuel cells (EFCs) utilize enzymatic catalysts to convert chemical energy to electrical energy, typically by performing a 2e− oxidation of saccharides. In the case of sugars, a single 2e− oxidation does not fully exploit this energy-dense fuel that is capable of producing 24e− from its complete oxidation to CO2. Here, we propose an efficient approach to design a versatile EFC that can produce electrical energy from 12 (oligo)saccharides by combining two enzymes that possess diverse substrate specificities: pyranose dehydrogenase (PDH) and a broad glucose oxidase (bGOx). Additionally, PDH is able to perform single or two sequential oxidations of glucose (at C2 and/or C3) yielding up to 4e−, whereas bGOx only performs a single 2e− oxidation at the anomeric (C1) position. By combining PDH and bGOx, we demonstrate the ability to achieve deep oxidation of glucose and xylose, whereby each is able to undergo sequential oxidations by PDH and bGOx. Additionally, we demonstrate that this deep oxidation can yield improved performances of EFCs. For example, an EFC comprised of a bi-enzymatic PDH/bGOx bioanode using xylose as a fuel yields a maximum current density of 586 ± 3 μAcm−2 whereas mono-enzymatic PDH or bGOx EFC bioanodes result in current densities of 440 ± 4 μAcm−2 and 120 ± 1 μAcm−2, respectively.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: oa@electrochem.org.
Owing to their high selectivity, enzymatic fuel cells (EFCs) are devices that offer multiple advantages over traditional fuel cell systems (i.e. H2/O2) such that they are able to operate under physiological temperature and pH and are able to produce electrical energy from common energy-dense fuels, such as glucose.1–5 In contrast to traditional fuel cells that operate on precious metal catalysts, EFCs employ enzymes as biocatalysts at the anode and cathode. In addition to mild operational requirements, inherent substrate specificity of enzymes can allow for the operation of EFCs in the absence of a membrane separator between the anodic and cathodic compartments.6
Theoretically, the complete oxidation of a single glucose molecule to CO2 yields 24e− that could be harnessed within an EFC.7 While many examples of high power EFCs can be found,8–12 the vast majority of glucose EFCs report only a single 2e− oxidation, thereby operating at <10 % efficiency. In the recent past, alternative enzymes to the commonly used glucose oxidase (GOx) for glucose oxidation have been explored, namely flavin adenine dinucleotide-dependent glucose dehydrogenase (FAD-GDH),8,13–15 nicotinamide adenine dinucleotide-dependent glucose dehydrogenase (NAD-GDH),16–18 pyrroloquinoline quinone-dependent glucose dehydrogenase (PQQ-GDH),19,20 cellobiose dehydrogenase (CDH),21,22 pyranose oxidase (POx)23 and pyranose dehydrogenase (PDH).24–27 FAD-GDH and CDH, which both oxidize their substrates at the anomeric (C1) position, are promising alternatives to GOx, because they do not utilize molecular oxygen as their electron acceptor, thus bioanode efficiency is retained in the presence of O2 (required for the biocathode of most membrane-less glucose/oxygen EFCs).28,29 As an alternative to utilizing an O2-insensitive enzyme (such as FAD-GDH), Mano and coworkers were able to almost completely suppress the ability of GOx to reduce O2 by the replacement of the FAD cofactor with a riboflavin derivative.30 Furthermore, CDH exhibits a larger degree of promiscuity toward substrates that can be oxidized, adding versatility to EFC devices.31 PDH is fast emerging as a favorable anodic enzyme over its oxygen-dependent counterpart (POx) whereby recent studies have delivered large current densities from low concentrations of glucose.24–26 Additionally, PDH can oxidize a range of saccharides wherein multiple oxidative reactions can even occur on each substrate molecule; glucose can effectively be oxidized at the C2 and C3 positions whereas GOx and FAD-GDH exclusively oxidize glucose at the C1 position.32 The combination of two anodic enzymes to improve the coulombic efficiency of EFC was reported between 2010–2013 using PDH and CDH.33–35 Photometric measurements showed that possibility of performing a 4e− oxidation of glucose by PDH bioelectrodes and a 6e− oxidation of glucose at PDH + CDH bioelectrodes. The first constructed hybrid EFC (using a Pt black electrode as the cathode) yielded an open circuit voltage of 0.5 V and maximum power density of 135 μW cm−2 in 100 mM glucose at pH 7.4.36 However, no stability tests were performed to evaluate the real suitability of the approach. In addition, the versatility is limited by CDH that cannot oxidize a large range of substrates. The same bioelectrodes (bi-enzymatic) were able to produce an approximate maximum power density of 30 μW cm−2 in 5 mM glucose when coupled to a Pt-based O2-reducing cathode and operating in a compartmentalized (membrane-separated) configuration. Expansion of this work by Shao et al. resulted in the formation of a bi-enzymatic device that was able to operate as a single-compartment device that utilized an enzymatic O2-reducing biocathode, resulting in an approximate maximum power density of 20 μW cm−2.35
We recently reported the use of an engineered GOx (broader-substrate GOx (bGOx)) that was able to oxidize a range of (oligo)saccharides; however, it was assumed that oxidation remained limited to the anomeric position of each substrate.37 We therefore hypothesize that the combination of bGOx and PDH within the same electrode architecture could produce a bioelectrode that would facilitate multiple oxidations of a range of saccharides. Herein, we demonstrate the ability of a ferrocene redox polymer (FcMe2-C3-LPEI) to facilitate the mediated bioelectrocatalytic oxidation of multiple (oligo)saccharides by PDH (from A. meleagris). The ability of the resulting PDH bioelectrodes to perform multiple oxidative reactions was also demonstrated by product analysis (following bulk bioelectrocatalysis). Furthermore, PDH was combined with the previously-reported bGOx and the resulting bi-enzymatic bioanodes were evaluated for their ability to oxidize multiple (oligo)saccharides. We demonstrate that the simultaneous combination of bGOx and PDH within the same bioelectrode architecture yields a platform that is able to facilitate up to three sequential 2e− oxidations of single glucose and xylose molecules. Additionally, a single EFC configuration (employing bilirubin oxidase at the biocathode, enabling the 4e− reduction of O2 to H2O) prepared using the bi-enzymatic bioanode was able to produce electrical energy from a total of 12 (oligo)saccharides without modifying the configuration of the EFC.
Experimental
Chemicals and materials
D-glucose, D-galactose, D-gluconolactone, D-fructose, D-mannose, D-ribose, tetrabutylammonium bromide (TBAB), ferrocenium hexafluorophosphate (Fc+PF6−), hydrochloric acid (HCl, 37%), Nafion perfluorinated resin, dimethylferrocene (FcMe2), 3-bromopropanoyl chloride, aluminum chloride, sodium bicarbonate, sodium chloride, sodium hydroxide, magnesium sulfate, borane-tertbutylamine, poly(2-ethyloxazoline), and paraffin wax were purchased from Sigma-Aldrich (St. Louis, MO) and used as-received, unless specified otherwise. Sucrose, D-xylose, D-maltose monohydrate, D-cellobiose, β-D-lactose, and 2-deoxy-D-glucose were purchased from Alfa Aesar (U. S. A.) and used as received. Citric acid monohydrate and sodium phosphate dibasic (anhydrous) were obtained from Fisher-Scientific (U. S. A) and used as received. Sodium phosphate monobasic (monohydrate) was purchased from Macron and used as received. Untreated Toray carbon paper was purchased from Fuel Cell Earth (TPG-H-060) and used as received. Ethylene glycol diglycidyle ether (EGDGE) was purchased from Polysciences, Inc. and used as received. Water used was from a Millipore Type 1 (Ultrapure) Milli-Q system (18.2 MΩ cm at 25°C). Anthracene-modified multi-walled carbon nanotubes (Ac-MWCNTs) were prepared as previously reported.38 Glucose oxidase (bGOx, broader substrate glucose oxidase, Lot N0151331R) and bilirubin oxidase (BOx, E.C.: 1.3.3.5, Myrothecium sp., Lot B0L0852601) were obtained from Amano Enzyme Inc. (Japan) and used as received. Pyranose dehydrogenase (PDH, E.C.: 1.1.99.29) from Agaricus meleagris was obtained from Huvepharma (Sofia, Bulgaria), recombinantly expressed in Pichia pastoris and purified as reported elsewhere.39 Dimethylferrocene-functionalized linear poly(ethylenimine) (FcMe2-C3-LPEI) and tetrabutylammonium bromide-modified Nafion (TBAB-Nafion) were synthesized as previously reported.38,40
Enzymatic activity assays
The specific enzymatic activity of PDH (21.5 ± 0.3 U mg−1) was determined at pH 7.5 using a modified procedure41,42 by following spectrophotometrically the D-glucose dependent reduction of the ferricenium ion (Fc+) to ferrocene at 300 nm (e300 = 4.3 mM−1 cm−1) for 3 min in a standard reaction mixture containing 200 μL Fc+PF6− (1 mM), 50 μL glucose (50 mM, pH 7.5), 1 mL potassium phosphate buffer (pH 7.5, 100 mM) and 2 μL of the appropriately diluted PDH sample (Figure S1). The solution of Fc+PF6− was freshly prepared by dissolving the salt (3.3 mg) in 10 mL of 5 mM HCl. Prior to the UV-Vis assay, all the stock solutions are incubated at 30°C and one unit of PDH activity is defined as the amount of enzyme necessary for the reduction of 2 μmol ferrocenium ion per min under the conditions described above. The specific enzymatic activity of bGOx (30.0 ± 2.0 U mg−1) was determined at pH 6.5 as previously reported.37 A Thermo Scientific Evolution 260 Bio UV-visible spectrophotometer was used for enzymatic activity assays.
Bioelectrode preparation and fuel cell tests
Bioelectrode preparation
For all bi-enzymatic bioelectrodes, the optimized conditions for the preparation of the bioanodes containing both enzymes consist on the vortex mixing of bGOx (4 mg mL−1, 4.5 μL) and PDH (20 mg mL−1, 4.5 μL) with FcMe2-C3-LPEI (10 mg mL−1, 21 μL) and EGDGE (10% vol.%, 1.125 μL). For figures using only a single enzyme (such as Figure 1), a doubled quantity of the enzyme was used (i.e., 9 μL of the above enzyme solution). Denatured enzyme controls were performed throughout, whereby denatured enzymes (dPDH or dbGOx) are obtained by heating at 100°C for 20 min; in the case of bi-enzymatic bioelectrodes employing denatured enzymes, the denatured enzyme served as a direct replacement for its active counterpart. Figure S1a (red spectrum) shows that this initiated denatured method is efficient to suppress the biocatalytic activity of the enzyme. Then, a suitable volume (30 μL) of this mixture is coated on an L-shape Toray carbon electrode surface. The modified bioelectrodes were left to dry overnight at room temperature and were rinsed with ultrapure water before use. BOx biocathodes were prepared (as previously reported) by adding Ac-MWCNTs (7.5 mg) to BOx (20 mg mL−1, 75 μL) in citrate/phosphate buffer (0.2 M, pH 6.5). The covalent modification of graphite electrodes and MWCNTs with hydrophobic anthracene was previously demonstrated to improve the efficiency of DET with BOx.43,44 The mixture was exposed to a series of successive vortex mixing/sonication steps, followed by the addition of 25 μL of TBAB-Nafion and an additional vortex mixing/sonication step.8,38 In all experiments, 33 μL of the resulting biocathode mixture was deposited on a Toray carbon paper electrode, which was cut into L-shape and coated with paraffin wax at the connecting ends to have 1 cm2 exposed electrode area.
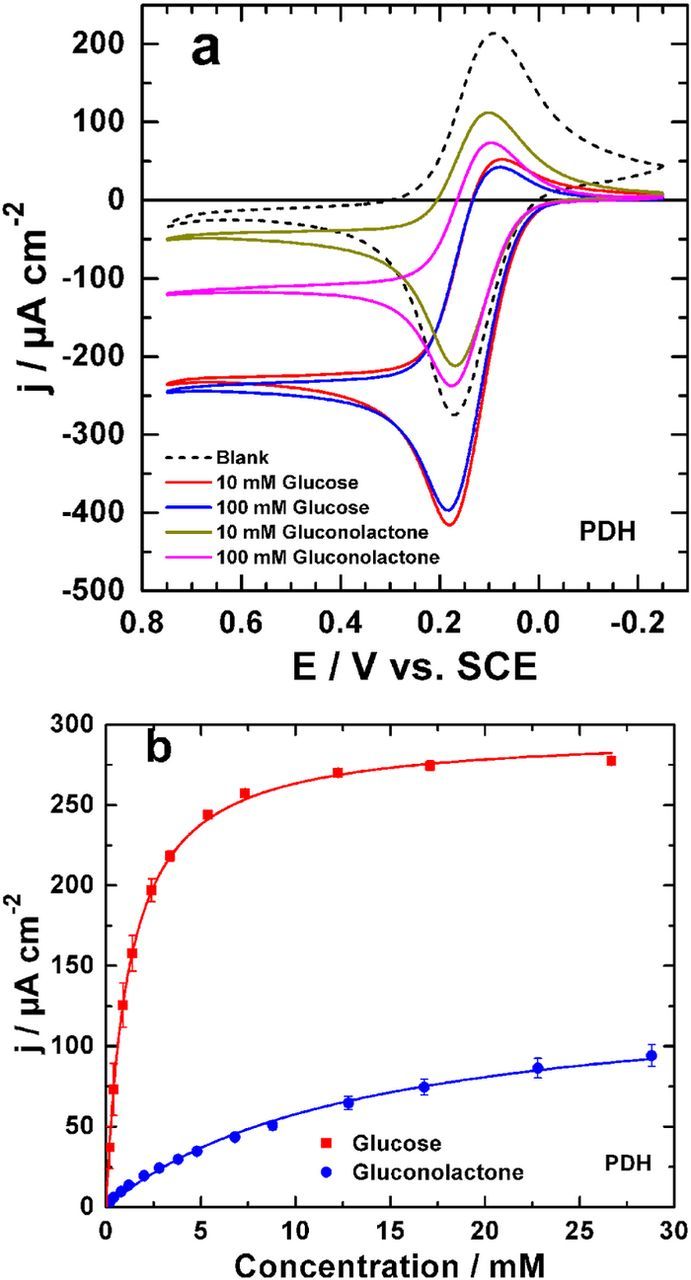
Figure 1. (a) Cyclic voltammograms of PDH bioelectrodes recorded at a scan rate of 10 mV s−1 in citrate/phosphate buffer (0.2 M, pH 6.5) containing glucose or gluconolactone (solid lines). The dashed line represents the blank experiment performed in the absence of substrate. (b) The corresponding apparent steady-state Michaelis-Menten kinetics of PDH bioelectrodes, determined using amperometric i-t analysis in a slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) at an applied potential of +0.23 V vs. SCE (+50 mV of the oxidation peak potential of Fc+/Fc). Error bars represent one standard deviation (n = 3). The kinetics were determined by non-linear regression of the Michaelis-Menten model.
Cyclic voltammetry (CV)
All CV tests were conducted in a conventional three-electrode cell using a CHI 611 C potentiostat (CH Instruments, Inc., U. S. A.). The reference electrode was a saturated calomel electrode (SCE) and a Pt mesh was used as a counter electrode. The working electrode for CV consists of 10 μL bioanode catalytic ink deposited onto Toray carbon paper electrode with a geometric surface area of 0.25 cm2. Current densities (j) are given as a function of the geometric surface area of the respective bioelectrode.
Enzymatic fuel cell (EFC) tests
All EFC experiments were performed in a slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) used as electrolyte solution in a non-compartmentalized cell and without any external addition of O2. After preliminary experiments, the exposed electrode areas of the bioanode and cathode were fixed to 0.25 and 1 cm2, respectively. EFC characterization was performed by recording: (i) the open circuit voltage (OCV) for 10 min and (ii) polarization curves by the quasi-steady state galvanostatic measurement from the OCV to short circuit (short circuit current density, jsc) at a current ramp of 0.1-1.2 μA s−1 (see Enzymatic fuel cells: method optimization and limiting component determination section). The current density (j) and power densities (P) are given as a function of the geometric surface area of the current-limiting electrode that is determined to be the bioanode (see Enzymatic fuel cells: method optimization and limiting component determination section). EFC tests were conducted using a CHI 660 E potentiostat (CH Instruments, Inc., U.S.A.). Other details will be provided in Results and discussion section.
Long-term electrolysis and analytical analysis
Electrolysis was performed in a non-compartmentalized cell in conventional three-electrode configuration using a Pt mesh and a SCE as the auxiliary electrode and reference electrode, respectively. The working electrode consists of a Toray carbon paper piece that was cut into an L-shape and coated with paraffin wax at the connecting ends resulting in a 1 cm2 square plate as the exposed electrode area. The long-term electrolysis was performed at fixed electrode potential of 0.3 V vs. SCE for 4 h. The cell was filled with 10 mL of solution. To avoid the presence of citrate (carbon source) in the final product distribution, the electrolytic solution for electrolysis was a simple sodium phosphate buffer (0.2 M, pH 6.5). Finally, the electrolyses were carried out using a CHI 611 C potentiostat (CH Instruments, Inc., U.S.A.).
Results and Discussion
Electrochemical characterization by cyclic voltammetry and amperometric i-t analysis
Before coupling bGOx to PDH, we initially evaluated the ability of the fabricated PDH bioelectrodes to oxidize glucose and gluconolactone (the inevitable intermediate formed during glucose oxidation at the anomeric carbon, by GOx), both employed as models fuels. Figure 1a presents typical CVs obtained for PDH bioelectrodes operating in the presence of glucose or gluconolactone. The blank electrode (in the absence of substrates) shows a pair of redox peaks, with an approximate E0 of +0.195 V vs. SCE, which is attributed to the Fc/Fc+ redox couple of the FcMe2-C3-LPEI redox polymer.37 Following the addition of each substrate, an oxidative catalytic wave is observed with an approximate onset potential of 0 V vs. SCE, which was attributed to the mediated bioelectrocatalytic oxidation of glucose or gluconolactone, as supported by the control experiments (Figure S1). For gluconolactone, the increase of the concentration is accompanied by a significant current increase in the limited region (E ≥ 0.4 V vs. SCE). The current density for glucose oxidation does not significantly change when increasing glucose concentration from 10 to 100 mM, implying that 10 mM glucose is enough to saturate the bioelectrode. Saturation of glucose bioelectrodes at low glucose concentrations is desirable (when coupled to large current densities), since minimal fuel is required for EFC operation and the EFC could effectively operate under physiological glucose concentrations (ca. 5 mM).40 These findings suggest that the apparent affinity (KMapp) is relatively small for glucose compared to gluconolactone. To probe the hypothesis of fast glucose saturation at a PDH bioelectrode, we determined the apparent Michaelis-Menten kinetics for the PDH bioelectrodes for glucose and gluconolactone by performing amperometric i-t evaluation at a fixed applied potential of 0.23 V vs. SCE. The resulting kinetic plots and associated non-linear regressions (to the Michaelis-Menten model) are reported in Figure 1b. The KMapp for glucose was determined to be 1.2 ± 0.0 mM (jmax = 298 ± 2 μA cm−2) compared to gluconolactone with a KMapp = 13.4 ± 0.6 mM (jmax = 145 ± 2 μA cm−2). Under our experimental conditions, i.e. pH = 6.5, the reduced performance of gluconolactone is likely a function of its expected hydrolysis into a non-pyranosic structure (gluconate), since the pKa of the gluconic acid is 3.76.45 It is possible, however, that substrate recognition of gluconolactone by PDH is diminished in its non-pyranosic form. Overall, these results clearly demonstrate that the FcMe2-C3-LPEI redox polymer can be used to facilitate mediated electron transfer (MET) from PDH.
We then proceeded by analyzing the compatibility of bGOx and PDH within the same bioelectrode architecture. As previously demonstrated, bGOx is able to oxidize a wide range carbohydrates at their anomeric position (when the C1 is not involved in a glycosidic linkage).37 Due to the ability of PDH to facilitate oxidations at alternative C positions to bGOx for many substrates (with multiple oxidations possible for many substrates, i.e. C2 + C3), the combination of bGOx and PDH within the same electrode architecture could yield a bioelectrode that is able to extract up to 6e− from a single molecule of glucose. We envisaged that the oxidative product of bGOx could still undergo oxidation of PDH, or vice versa. The real suitability of this approach, however, could be experimentally hampered by the inability of the product of either bGOx or PDH to undergo further oxidation by its counterpart. To evaluate the activity of the individual enzymes (where control bioelectrodes were prepared using denatured counterpart enzymes, PDH-dbGOx and dPDH-bGOx) as well as the final complete PDH-bGOx bi-enzymatic bioelectrodes, differing saccharides were used to perform a series of CV experiments in citrate/phosphate buffer (0.2 M, pH 6.5). Figure 2 presents the bioelectrocatalytic oxidation of glucose, galactose, lactose, maltose, cellobiose and xylose by the individual enzymes and the bi-enzymatic bioelectrode. Additional CVs for the oxidation of ribose, sucrose, mannose, 2-deoxy-D-glucose, fructose and gluconolactone (all of which were previously demonstrated to be silent for oxidation by bGOx) are reported within the Supporting Information (Figure S2).37 For blank electrodes (in the absence of substrates), the same pair of redox peaks was observed with an approximate E0 of +0.195 V vs. SCE, assigned to the Fc/Fc+ redox couple of the FcMe2-C3-LPEI redox polymer.37,46 Following the addition of each substrate, an oxidative catalytic wave is observed with an approximate onset potential of 0 V vs. SCE. The chosen concentration of each substrate in these CV experiments was based on the KMapp values (see below) and based on several preliminary experiments. With the exception of cellobiose, the bi-enzymatic bioelectrode consisting of PDH and bGOx exhibits larger current densities in the limited region (E ≥ 0.2 V vs. SCE) of each CV, indicating that the products of either bGOx or PDH are still active on the electrode (for all substrates). It is also likely that the response observed for cellobiose is simply a function of the experimental conditions (i.e., hydrostatic, fast scan rate), so steady-state experiments were performed and are detailed below and within the Supporting Information. Herein, PDH and bGOx are randomly distributed within the electrode architecture. However, the promiscuous nature of each enzyme eliminates the necessity of the reaction intermediates to diffuse between enzymes in a specific reaction sequence. These cyclic voltammograms suggest that both enzymes are compatible within the same bioelectrode architecture, although these experiments were only compared at a single substrate concentration that may or may not be a saturating concentration for either/both enzyme(s).
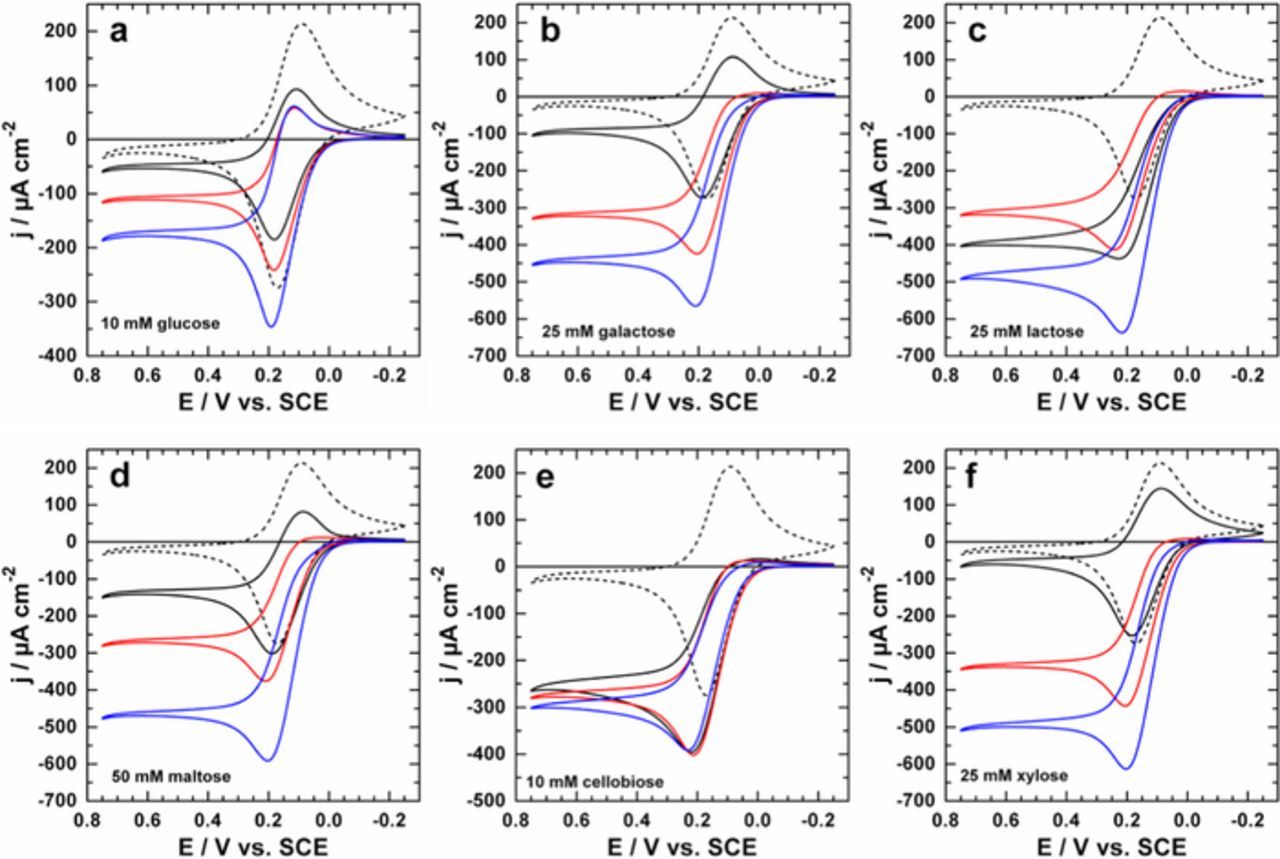
Figure 2. (a-f) Representative cyclic voltammograms of the mono- and bi-enzymatic bioelectrodes in citrate/phosphate buffer (0.2 M, pH 6.5) containing different (oligo)saccharides (solid lines). Dashed lines represent blank experiments performed in the absence of substrates. All bioanodes were prepared on Toray carbon paper electrodes: dPDH-bGOx (black line), PDH-dbGOx (red line) and PDH-bGOx (blue line). Experiments were performed at a scan rate of 10 mV s−1.
We next evaluated the apparent Michaelis-Menten kinetics of each substrate under quasi-steady state conditions, to compare the KMapp and jmax of each substrate at the bioelectrodes by continuously increasing the concentration of each individual substrate. The solution was slightly stirred during the entire experiment with an applied potential of 50 mV more positive than the oxidative peak potential, typically Eapplied = +0.23 V vs. SCE. The obtained amperometric response resulting from the addition of each saccharide is reported within the Supporting Information (Figures S3-4). The Michaelis-Menten model was applied by non-linear regression and used to evaluate the apparent kinetics of the bioelectrodes for each substrate. The determined KMapp and jmax values are reported in Table I. Kinetic data for PDH and bGOx in homogeneous solutions (not immobilized at an electrode surface) have been previously reported.47–49 Among the investigated substrates and with the exception of lactose, PDH exhibits lower KMapp values than bGOx (and thus, improved affinity). Subsequently, when both enzymes are active within the bi-enzymatic bioelectrode, the value of KMapp is situated between those of the individual enzymes. For the bi-enzymatic PDH-bGOx bioelectrodes, the lowest value of KMapp is observed for xylose (1.7 ± 0.1 mM), galactose (2.1 ± 0.1 mM) and glucose (2.6 ± 0.4 mM). However, jmax increases in the following order: galactose (306 ± 4 μA cm−2) > glucose (265 ± 7 μA cm−2) > xylose (236 ± 3 μA cm−2). Cellobiose yields the largest maximum current density (jmax = 353 ± 2 μA cm−2) but has a relatively higher KMapp (3.0 ± 0.1 mM) that is ca. 2-fold larger than that of xylose. By taking into account the values of KMapp and jmax, EFCs operating on glucose, xylose, galactose and cellobiose are expected to deliver improved performances over other substrates.
Table I. Apparent Michaelis-Menten kinetics of PDH-bGOx bioelectrodes for differing saccharides determined in a slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) at an applied potential of +0.23 V vs. SCE. Note: Values are reported as mean ± one standard deviation (n = 3). The kinetics were determined by non-linear regression of the Michaelis-Menten model. n.d. = not determined.
| KMapp (mM) | jmax (μA cm−2) | |||||
|---|---|---|---|---|---|---|
| Substrates | dPDH-bGOx | PDH-dbGOx | PDH-bGOx | dPDH-bGOx | PDH-dbGOx | PDH-bGOx |
| Glucose | 54.3 ± 1.5 | 1.2 ± 0.0 | 2.6 ± 0.4 | 324 ± 3 | 298 ± 2 | 265 ± 7 |
| Galactose | 214 ± 4 | 2.1 ± 0.0 | 2.1 ± 0.1 | 313 ± 2 | 204 ± 1 | 306 ± 4 |
| Lactose | 2.4 ± 0.1 | 42.0 ± 2.8 | 8.5 ± 0.8 | 289 ± 5 | 225 ± 4 | 249 ± 6 |
| Maltose | 117 ± 3 | 6.0 ± 0.2 | 14.5 ± 1.3 | 193 ± 1 | 157 ± 1 | 187 ± 4 |
| Cellobiose | 3.8 ± 0.3 | 3.5 ± 0.1 | 3.0 ± 0.1 | 367 ± 9 | 226 ± 1 | 353 ± 2 |
| Xylose | 43.8 ± 2.9 | 1.7 ± 0.1 | 1.7 ± 0.1 | 112 ± 2 | 205 ± 2 | 236 ± 3 |
| Gluconolactone | n.d. | 13.4 ± 0.6 | 13.2 ± 0.5 | n.d. | 145 ± 2 | 146 ± 2 |
| 2-Dexoy-D-glucose | n.d. | n.d. | 20.9 ± 1.1 | n.d. | n.d. | 262 ± 4 |
While CV and amperometric/Michaelis-Menten data suggest some substrates may be good candidates to undergo multiple/sequential oxidations at these bi-enzymatic bioelectrodes, further evidence is required to confirm such a mechanism; Determination of the depth of saccharide oxidation section below details substrate analysis and charge quantification experiments.
Enzymatic fuel cells: method optimization and limiting component determination
Based on the aforementioned CV results, we used the bi-enzymatic PDH-bGOx bioelectrode for optimization of the EFC tests. Among the multitude of methods for EFC testing available and based on our previous preliminary tests, we chose a quasi-steady-state galvanostatic method, where the rate depends on the nature of the fuel cell. The EFC performances were compared in terms of open circuit voltage (OCV), maximum power density (Pmax) and maximum current density that can be drawn also known as the short circuit current (jsc). We first examined whether the rate of the current ramp influences the performance of the EFCs. Figure S5 depicts the obtained fuel cell polarization curves where the current ramp rate varies between 0.1 to 1.2 μA s−1, employing 40 mM glucose as the fuel. By increasing the rate, the Pmax increases from 144 μW cm−2 (0.1 μA s−1) and reaches a maximum around 159 μW cm−2 (1.2 μA s−1). It should be pointed out that a rapid current ramp rate does not enable the EFC to reach equilibrium. This could consequently lead to the overestimation of EFC performances. As a compromise between EFC under/over estimation and practicality, a current ramp rate of 0.2 μA s−1 (Pmax = 125 μW cm−2, jsc = 627 μA cm−2) was selected and used for all remaining EFC characterization throughout this study.
Furthermore, the changes in the anodic (EA) and cathodic (EC) electrode potentials when the fuel cell delivers current were evaluated using 40 mM glucose as the model fuel. As reported in Figure 3, when the anode electrode size is 0.25 cm2, EA undergoes a prominent increase with a ΔEA of 0.48 V from the Ecell of 0.62 V (OCV) to 0.09 V while the ΔEC remains at a small value of 0.08 V. If the size of both electrodes is 1 cm2, the ΔEA is 0.40 V and the ΔEC is 0.20 V. In other words, the anode is the limiting-electrode. Such results clearly demonstrate that the EFC performances are limited by the anodic reaction, which is desired in order to compare the performances of each EFC as a function of the ability of the bi-enzymatic bioanode to oxidize multiple substrates. Consequently, we chose the electrode sizes of 0.25 cm2 for the bioanode and 1 cm2 for the biocathode, so that the EFC performances will be accurately compared on the only basis of each (oligo)saccharide.
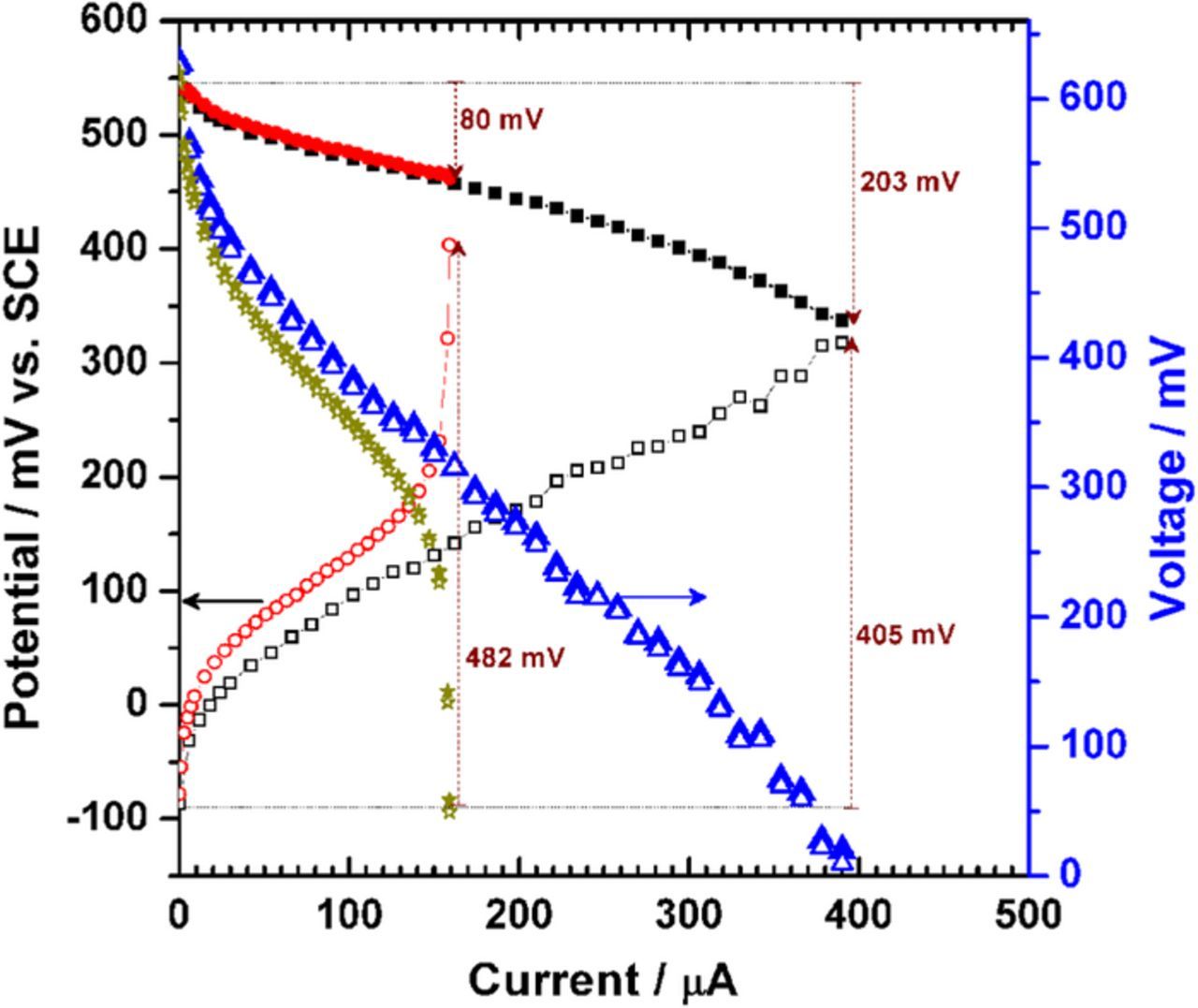
Figure 3. Left y-axis: Representative behavior of the anode ( ) and cathode (
) and cathode ( ) potentials vs. SCE, recorded during the polarization curve measurements (cell voltage E: right y-axis) for an anode electrode size of 0.25 cm2 (
) potentials vs. SCE, recorded during the polarization curve measurements (cell voltage E: right y-axis) for an anode electrode size of 0.25 cm2 ( ) and 1 cm2 (
) and 1 cm2 ( ). The bi-enzymatic bioanode (PDH-bGOx) and cathode (BOx) were prepared on Toray carbon paper electrodes. The polarization curves are recorded by sweeping the current at 0.2 μA s−1 from zero until short circuit. All experiments were performed in a slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) used as the electrolyte solution containing 40 mM glucose in a non-compartmentalized cell and without any external addition of O2.
). The bi-enzymatic bioanode (PDH-bGOx) and cathode (BOx) were prepared on Toray carbon paper electrodes. The polarization curves are recorded by sweeping the current at 0.2 μA s−1 from zero until short circuit. All experiments were performed in a slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) used as the electrolyte solution containing 40 mM glucose in a non-compartmentalized cell and without any external addition of O2.
Multi-substrate enzymatic fuel cells
As evaluated above, CVs of the bi-enzymatic bioelectrodes suggest that both enzymes are compatible. Additionally, amperometric i-t analysis and its application to the Michaelis-Menten model suggest that multiple oxidative reactions are possible (especially in cases where the jmax for the bi-enzymatic bio-electrodes exceeds the total jmax for the combined individual bioelectrodes). To demonstrate the versatility of this EFC configuration based on the bi-enzymatic PDH-bGOx bioanode, we examined a range of substrates as fuels (50 mM). The EFCs were operated in a single compartment (membrane-less) using a BOx direct electron transfer-type (DET) biocathode and operated at pH 6.5 and room temperature; O2 or additional air purging was not performed and the bioanodes were the limiting components as demonstrated previously. Figure S6 depicts the recorded OCV as a function of time, where a steady-state was typically reached after a few minutes. Table II reports the recorded OCV in the presence of different substrates varying between 0.56 V (fructose) to 0.64 V (cellobiose). The low value of OCV for fructose is undoubtedly due to the fact that fructose shows the poorest catalytic activity (see Figure S2e).
Table II. Fuel cell performances with different substrates: open circuit voltage (OCV), short-circuit current density (jsc) and maximum power density (Pmax) of PDH-bGOx/BOx EFCs operating in slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) containing different saccharides (50 mM).
| Substrate | OCV (V) | Pmax (μW cm−2) | jsc (μA cm−2) |
|---|---|---|---|
| Glucose | 0.62 ± 0.00 | 167 ± 1 | 705 ± 5 |
| Gluconolactone | 0.58 ± 0.01 | 54 ± 1 | 195 ± 5 |
| Galactose | 0.61 ± 0.00 | 118 ± 3 | 540 ± 7 |
| Fructose | 0.56 ± 0.01 | 34 ± 0 | 125 ± 1 |
| Ribose | 0.58 ± 0.00 | 66 ± 1 | 253 ± 2 |
| Xylose | 0.59 ± 0.01 | 130 ± 1 | 586 ± 3 |
| Maltose | 0.62 ± 0.00 | 109 ± 1 | 429 ± 6 |
| Mannose | 0.56 ± 0.01 | 45 ± 1 | 188 ± 3 |
| Sucrose | 0.58 ± 0.00 | 73 ± 0 | 174 ± 1 |
| Lactose | 0.62 ± 0.01 | 55 ± 1 | 225 ± 3 |
| Cellobiose | 0.64 ± 0.00 | 104 ± 5 | 490 ± 6 |
| 2-Deoxy-D-glucose | 0.59 ± 0.00 | 97 ± 2 | 364 ± 5 |
Figure 4 presents the polarization and resulting power cures for different fuels. The extracted parameters of interest, i.e., Pmax and jsc are reported within Table II. The best performances are obtained when glucose is used as the fuel (Pmax = 167 ± 1 μW cm−2, jsc = 705 ± 5 μA cm−2). Using 50 mM glucose, the obtained OCV of 0.6 V and Pmax of 167 μW cm−2 surpass the only reported hybrid EFC (using PDH-CDH bioanode and Pt cathode), that yielded an OCV of 0.5 V and Pmax of 135 μW cm−2 operating in 100 mM glucose.33 The origin of this enhanced performance is attributed to different reasons including: (i) the performance of the engineered glucose oxidase compared to CDH, (ii) the FcMe2-C3-LPEI redox polymer used to facilitate MET from PDH compared to the Os redox polymer hydrogel employed in the reference and (iii) the use of bilirubin oxidase at the biocathode that enables the a 4e− reduction of O2 to H2O at a high potential compared to a Pt abiotic cathode.
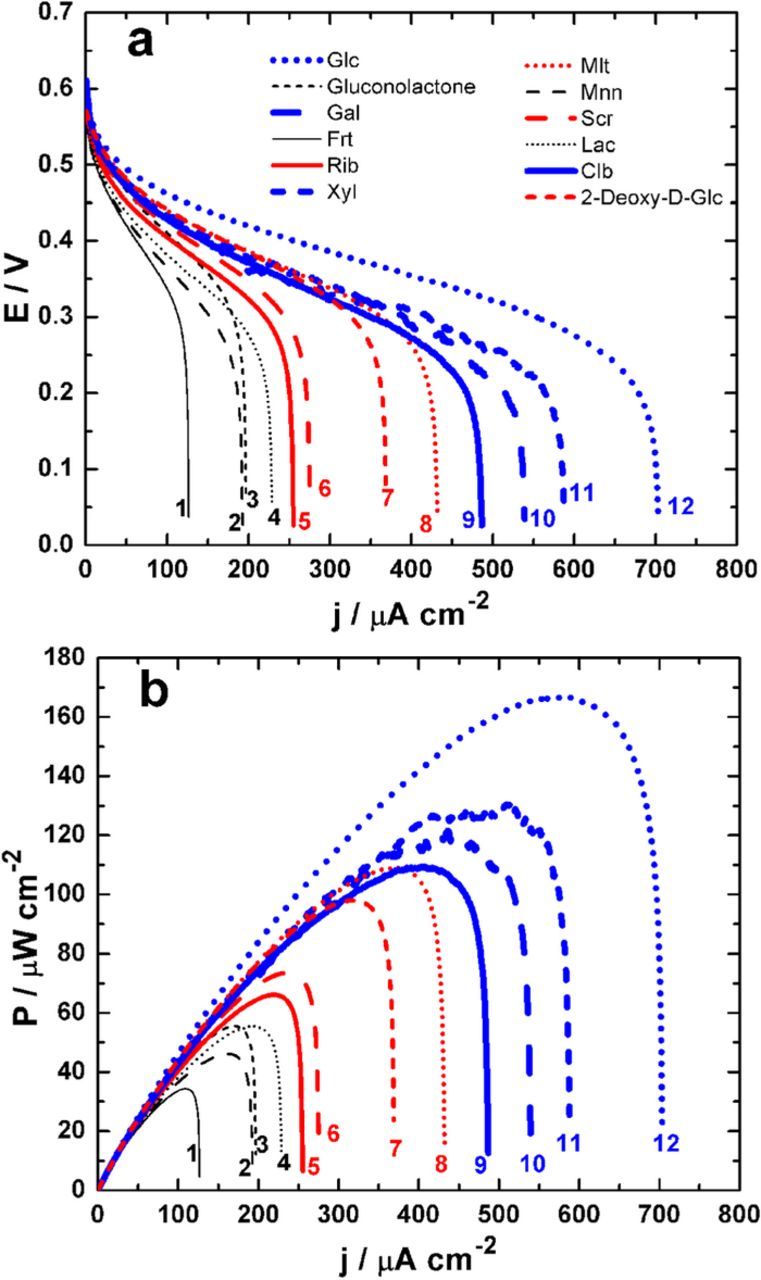
Figure 4. Representative EFCs performances recorded in a slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) containing different saccharides (50 mM). Fuel cell polarizations in terms of: (a) cell voltage E and (b) power density P. The polarization curves are recorded by sweeping the current at 0.2 μA s−1 from zero until short circuit (E = 0), in a non-compartmentalized cell and without any external addition of O2. Note: 1 = Frt (fructose), 2 = Mnn (mannose), 3 = gluconolactone, 4 = Lac (lactose), 5 = Rib (ribose), 6 = Scr (sucrose), 7 = 2-Deoxy-D-glc (2-deoxy-D-glucose), 8 = Mlt (maltose), 9 = Clb (cellobiose), 10 = Gal (galactose), 11 = Xyl (xylose), 12 = Glc (glucose).
Even though xylose is expected to offer improved performance over glucose at PDH bioelectrodes,32 the kinetics of bGOx toward xylose oxidation is very low (KMapp = 44 ± 3 μA cm−2 vs. KMapp = 1.8 ± 0.1 μA cm−2 for PDH) which leads to lower performances (Pmax = 130 ± 1 μW cm−2, jsc = 586 ± 3 μA cm−2). Furthermore, the EFC tests show that deeper glucose oxidation can effectively be facilitated by the EFC since gluconolactone leads to an OCV of 0.58 ± 0.01 V, a Pmax of 54 ± 1 μW cm−2 and a jsc of 95 ± 5 μA cm−2. To further demonstrate the suitability of this PDH-bGOx bioanode in EFCs fueled by alternative substrates to glucose, EFC tests were performed with other sugars such as galactose, lactose, cellobiose, maltose, mannose, sucrose, ribose and 2-deoxy-D-glucose. Figure 4 and Table II demonstrate that galactose, cellobiose and maltose can be potential substrates for this EFC configuration. Since the performances of disaccharides are lower than that of glucose, we hypothesize that hydrolysis does not occur and that oxidation takes place at complete disaccharides.
Evidence of multiple oxidative steps in EFCs
With the aim to determine exactly the role of each enzyme during the EFC operation, we performed three series of tests using mono- or bi-enzymatic bioanodes: PDH + bGOx, PDH + dbGOx and dPDH + bGOx, where 50 mM xylose was used as the model fuel. As displayed in Figure S7, the recorded OCV strongly depends on the nature of the bioanode. When only PDH is active, the OCV is 0.61 ± 0.00 V (PDH + bGOx). This value decreases to 0.56 ± 0.01 V (dPDH + bGOx) in the case of bGOx. Interestingly, the presence of both enzymes leads to an intermediate OCV of 0.59 ± 0.01 V (PDH + bGOx). Consequently, we postulated that the first step on the multiple oxidations might take place at the active site of bGOx and the kinetics of the "hybrid biocatalyst" is limited by the diffusion of the intermediate (resulting from the carbon C1-position oxidation) toward the PDH active site where it is further oxidized at the C2 and/or C3 position. The recorded fuel cell polarization curves (as shown in Figure 5) substantiate the above hypothesis. PDH + bGOx facilitates a Pmax = 130 ± 1 μW cm−2 and a jsc of 586 ± 3 μA cm−2. For PDH + dbGOx the Pmax and jsc decrease to 107 ± 1 μW cm−2 and 440 ± 4 μA cm−2. Because of the high KMapp of bGOx, the performance of dPDH + bGOx is limited to a Pmax of 31 ± 1 μW cm−2 and a jsc of 120 ± 1 μA cm−2. It is important to note that these sets of measurements were also performed using 10 mM glucose (plots not shown herein), resulting in the same conclusions. Overall, the ability for the bGOx to be coupled to an additional enzyme such as PDH provides wider practical applicability using various (oligo)saccharides, with additional deeper fuel oxidation in multiple oxidative steps. We are highly confident that upcoming improvements in cell design, namely the size and flow conditions in a suitable operating temperature, will lead to current and power increases for a wide range of applications.
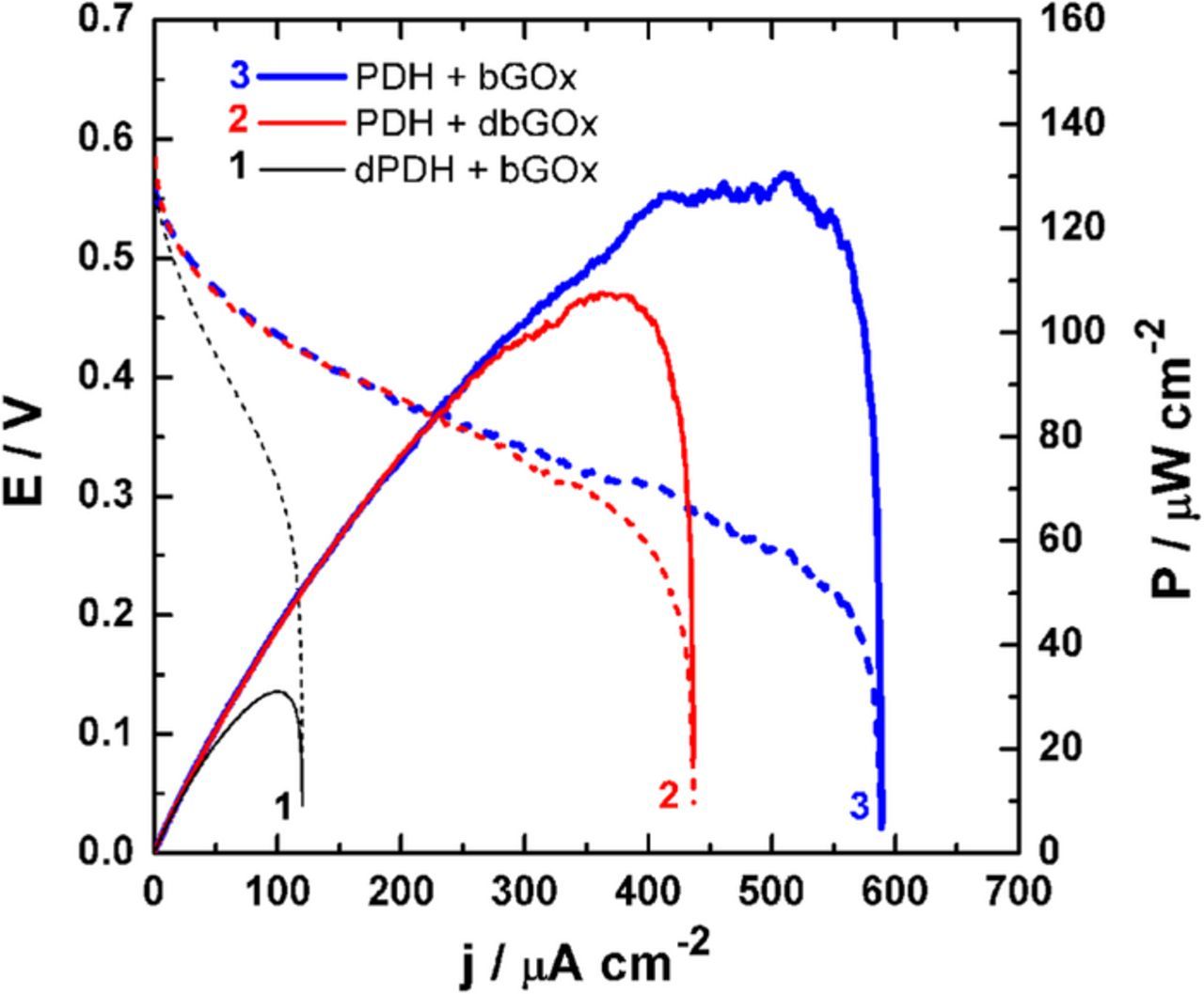
Figure 5. EFC tests performed in 50 mM xylose using mono- or bi-enzymatic bioanodes (see inset). Fuel cell polarizations are presented in terms of cell voltage E (dashed lines: left y-axis) and power density P (solid lines: right y-axis). Experiments were performed in a slightly stirred citrate/phosphate buffer (0.2 M, pH 6.5) used as the electrolyte solution. The cathode is BOx and polarization curves are recorded by sweeping the current at 0.2 μA s−1 from zero until short-circuit (E = 0), in a non-compartmentalized cell and without any external addition of O2.
Durability of the enzymatic fuel cells
Although EFCs are able to produce high power densities from a range of fuels under mild conditions, unsatisfactory long-term stability EFCs is commonly a significant drawback. To this end, we initially investigated the durability of the final EFC configuration by recording several successive polarization curves without refreshing the bioelectrodes. The obtained results are presented in Figure 6a and the corresponding parameters of interest, i.e., the Pmax and jsc are reported in Figure S8a. After 10 rounds of discharge, the remaining Pmax and jsc values are 89% and 92% of their initial values. To further demonstrate the industrial applicability of our system, we performed a continuous 12-hour durability measurement by recording the current that can be produced at a cell voltage of 0.4 V. This long-term evolution of the current density (left y-axis) and output power density (right y-axis) is displayed in Figure S8b. The first hour of the EFC operation (Figure 6b) does not show any significant performance losses. However, the progressive decrease of the current and power after 2 hours may be attributed reasonably to glucose consumption since there is no continuous renewal of the substrate. Furthermore, even if the solution is slightly stirred during the 12 hours, the performances will be limited by the mass-transport phenomenon at the anodic reaction as expected, as the designed EFC operates under batch conditions without fuel renewal and the prompt removal of reaction products from electrodes.
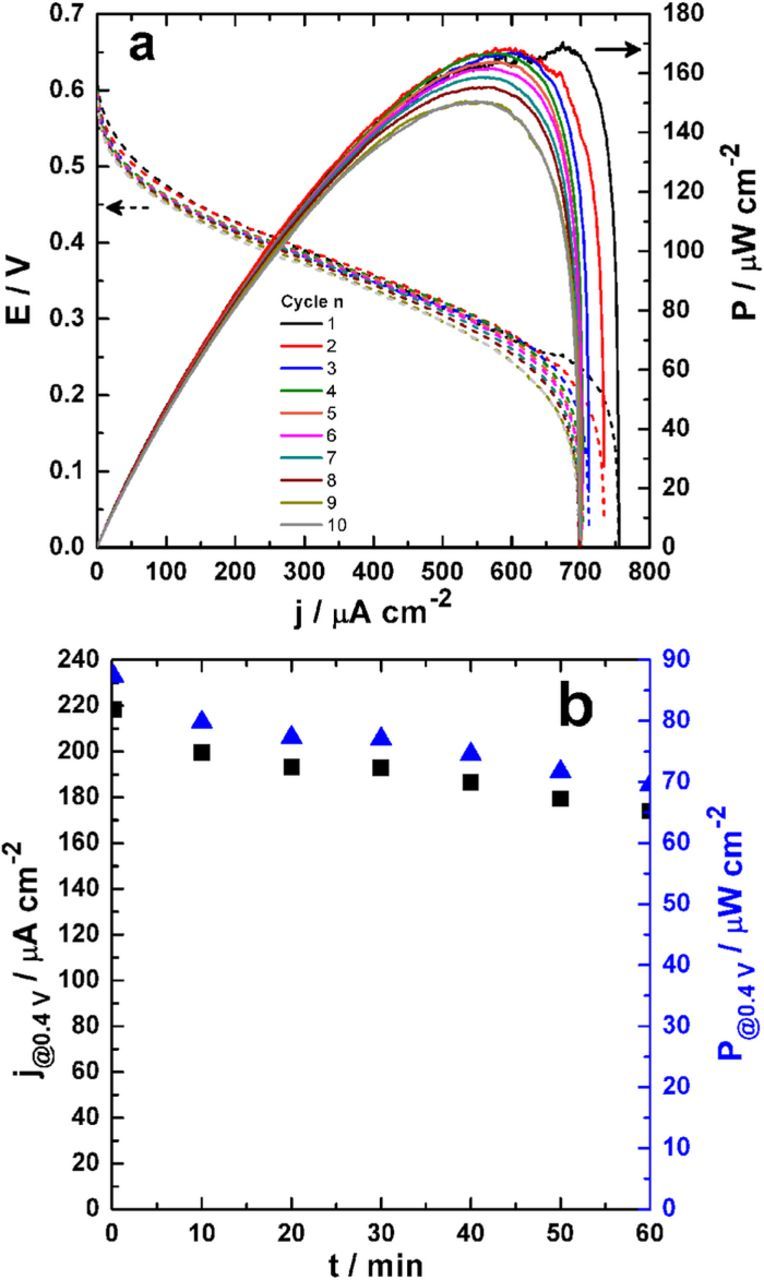
Figure 6. EFC durability tests performed in 50 mM glucose. (a) Successive polarization curves for different cycles in terms of cell voltage E (dashed lines: left y-axis) and power density P (solid lines: right y-axis). (b) The first hour of long-term evolution of the current density (j: left y-axis) and power density (P: right y-axis) at a cell voltage of 0.4 V. Experiments were performed in a slightly stirred citrate/phosphate buffer (pH 6.5, 0.2 M) used as electrolyte solution. The anode is PDH-bGOx and cathode is BOx. For panel (a), the polarization curves are recorded by sweeping current at 0.2 μA s−1 from zero until short circuit (E = 0), in a non-compartmentalized cell and without any external addition of O2.
Determination of the depth of saccharide oxidation
As investigated in Electrochemical characterization by cyclic voltammetry and amperometric i-t analysis sections and Evidence of multiple oxidative steps in EFCs above, current responses in the presence of differing substrates can be used to provide preliminary evaluation of the depth of substrate oxidation; however, this remains only a preliminary evaluation in the absence of chemical analysis due to potential kinetic/enzymatic complications (such as substrate/product inhibition, hydrolysis, etc.). Thus, we next sought to determine the depth of substrate oxidation by performing electrolysis experiments and measuring the concentration of remaining (unreacted) substrate within the electrochemical cell. Following an extended period of potentiostatic electrolysis, the amperometric trace was integrated to determine the charge passed (coulometry) during the experiment, which was combined with chemical analysis to determine the concentration of unreacted substrate within the electrochemical cell. This enables the determination of the number of electrons that were obtained from the oxidation of the substrate (i.e., the depth of substrate oxidation).
Long-term electrolysis
To confirm the extent of glucose oxidation by bGOx and PDH, we performed long-term electrolysis followed by substrate analysis. We hypothesize that the biocatalytic oxidation of glucose using bGOx bioelectrode concerns only the carbon at C1 position, thus involving an exchanged number of electrons (nex) of 2. According to Peterbauer and Volc,32 PDH has the ability to oxidize galactose exclusively at the C2 position (nex = 2), while the oxidation may happen at carbons C2 and C3 separately (nex = 2) as well as simultaneously (nex = 2 + 2 = 4) in the case of glucose or gluconolactone. Electrolyses were performed for glucose, galactose and gluconolactone independently at a concentration of 50 mM using PDH bioelectrodes. Following this, bGOx and PDH + bGOx were used for glucose electrolysis. Figure 7a compares the oxidation current Iox (left y-axis) to the quantity of electricity (Qox, right y-axis) resulting from the oxidation of each substrate. The fast current decrease during the first 5 min is attributed to the leaching of partially unbound enzyme or redox mediator into solution. However, after a relatively long time of electrolysis, the Iox is expected to decrease because of substrate consumption and/or catalyst deactivation. It should be pointed out that the overall Iox vs. time behavior during such experiments strongly depends on the diffusion coefficient of each involved reactant and its reaction intermediate(s) and/or product(s). After 4 h of glucose electrolysis, Qox was determined to be 5.6, 8.5 and 9.5 C for bGOx, PDH + bGOx and PDH, respectively. For the electrolysis of galactose and gluconolactone by PDH, Qox was found to be 4.8 C and 1.9 C, respectively. For a theoretical nex = 2, the theoretical quantity of electricity (Qth) is 2 × 0.05 × 0.01 × 96485 = 96.5 C for full conversion of 50 mM substrate in 10 mL of buffer, where the substrate concentration was 50 mM, the volume of the electrolyte was 10 mL and Faraday's constant is 96,485 C mol−1. Subsequently, a Qth of 192.9 and 289.5 C for nex = 4 and nex = 6 was calculated, respectively. After 4 h, the conversion of galactose at the PDH bioelectrode is estimated to be ca. 5%, Figure S9 reports the different scenarios depending on the value of nex. The electrolysis of gluconolactone on the PDH bioelectrode gives the lowest oxidation and Qox. This is in agreement with the previous results obtained by CV and EFC. This very poor performance compared to glucose can be explained by the pH (6.5) of the medium, in which the gluconolactone (cyclic) is swiftly hydrolyzed into gluconate (linear). This hydrolyzed "opened structure" exhibits decreased activity at PDH than its corresponding pyranosic (cyclic) counterpart. Gluconate is expected at pH = 6.5, since the pKa for gluconic acid is 3.76.
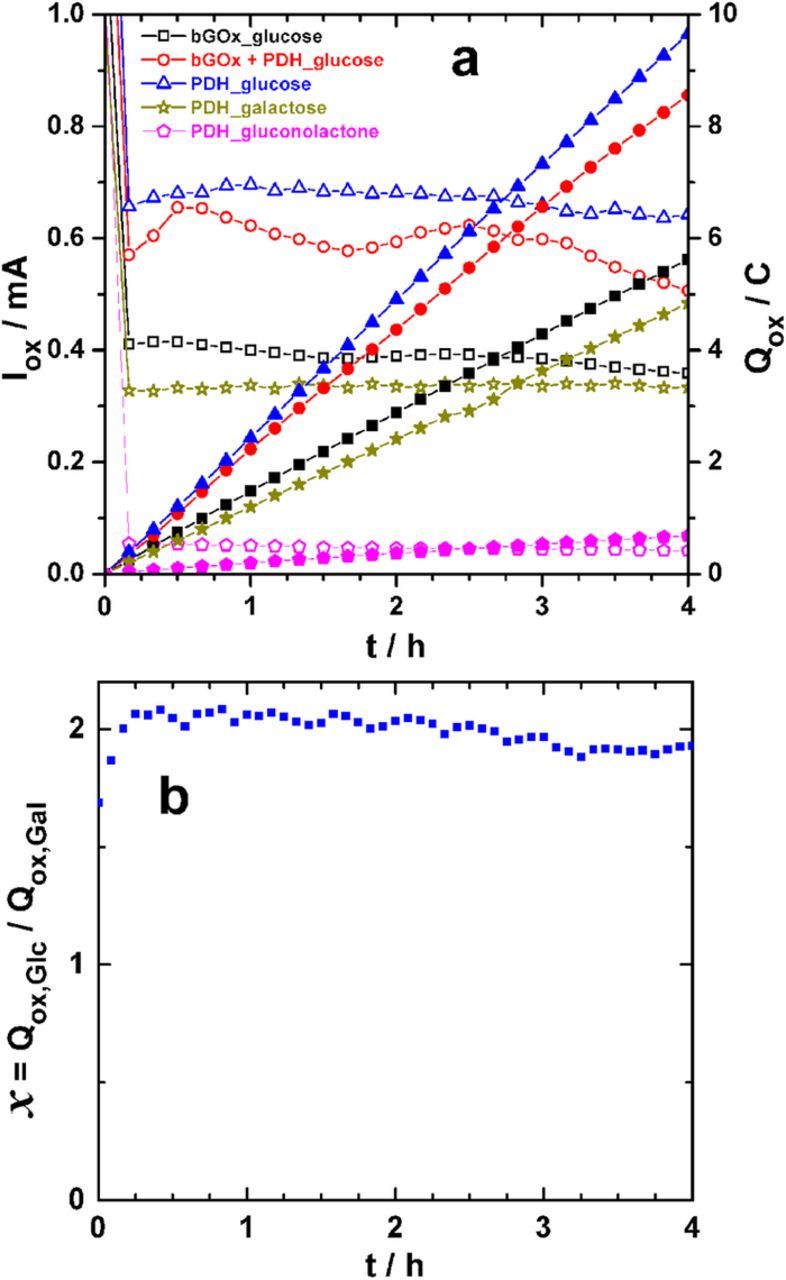
Figure 7. (a) Evolution of the oxidation current (Iox, left y-axis: opened symbols) and quantity of electricity (Qox, right y-axis: filled symbols) during the long-term electrolyses of glucose, galactose or gluconolactone (50 mM) at 0.3 V vs. SCE on different biocatalysts. (b) Time-dependent variation of the ratio between Qox from glucose (Qox,Glc) and that from galactose (Qox,Glt). Experiments were performed in a slightly stirred sodium phosphate buffer (0.2 M, pH 6.5) used as electrolyte solution.
For the simultaneous glucose oxidation at carbons C2 and C3, nex = 4 is expected for PDH bioelectrodes. In the case of galactose, we expect a total of 2e− (exclusive oxidation at the C2 position). Then, by assuming the same conversion of glucose and galactose when the PDH bioelectrode is employed (same enzyme loading and electrode size), the ratio between the quantity of electricity from glucose and galactose electrolyses is expected to be x = Qox,Glc/Qox,Gal = 4/2 = 2. Figure 7b shows the evolution of this ratio during the entire electrolytic experiment. Our finding is in perfect agreement with the theory, thus underpinning the conclusion that galactose and glucose are oxidized in 2-electron and 4-electron processes at the PDH bioelectrode, respectively. The goal of this exploratory work is not to perform complete substrate conversion, but rather to provide an overview of the degree of substrate oxidation that can be obtained by PDH, and later, the PDH/bGOx bi-enzymatic bioelectrode. In the case of glucose electrolysis by PDH, we determined the glucose concentration remaining following the electrolytic experiment using a TRUE-2-Go commercial Blood Glucose Meter (MeterTRUE2go, NIPRO Diagnostics; U.S.A) that employs a pyrroloquinoline quinone-dependent GDH. Following electrolysis, the remaining glucose was determined to be 8.53 mg mL−1 (ca. 47.4 mM). For a Qox of 9.5 C, the mean experimental exchanged number of electrons (nexp) was evaluated by Eq. 1 to be 3.8, supporting the ability of PDH to be able to facilitate multi glucose oxidative reactions when mediated by FcMe2-C3-LPEI and immobilized at an electrode surface. Based on Eq. 2 and nex = 4, a faradaic yield of 95% is reached for the oxidation of glucose at the C2 and C3 positions when assuming a 4e− process. Furthermore, the conversion rate can be readily manipulated by increasing the electrode area and enzyme loading on the electrode.
![Equation ([1])](https://content.cld.iop.org/journals/1945-7111/164/3/H3073/revision1/d0001.gif)
![Equation ([2])](https://content.cld.iop.org/journals/1945-7111/164/3/H3073/revision1/d0002.gif)
where Qox[C] is the charge, Δc[mol L−1] = c0-cf, which are the initial and final concentrations of glucose, V[L] is the volume of the electrolysis solution (V = 0.01 L; τF is the faradaic yield).
Evidence for deep bioelectrocatalytic oxidation
To validate the ability to perform multiple oxidation reactions of a single saccharide molecule using a bi-enzymatic bioelectrode architecture, extended electrolysis (7 hours in potassium phosphate buffer (0.2 M, pH 6.5)) of glucose and xylose was performed for electroanalytical investigation. Figure S10 shows the obtained data, highlighting the excellent stability of the bioelectrode. The results in terms of Iox and Qox are in agreement with the previous kinetic studies where KMapp values of 43.8 ± 2.9, 1.7 ± 0.1 and 1.7 ± 0.1 mM for bGOx, PDH and PDH + bGOx bioelectrodes were obtained (in the case of xylose), respectively. These results suggest that the kinetics at the bi-enzymatic bioelectrode PDH + bGOx could be limited by the first oxidative step that presumably takes place at bGOx. Initially, we employed the previous TRUE-2-Go Blood Glucose test, to estimate the remaining glucose concentration and then evaluated nexp (from Eq. 1) for bGOx, PDH and PDH + bGOx bioelectrodes separately. It should be noted that the amount of the enzyme is kept constant in the bi-enzymatic electrode (for better comparison). A custom electroanalytical method was employed to determine the remaining concentration of xylose that will enable the assessment of nexp, since the commercial glucose sensor is not able to detect xylose; we turned to our previously-reported FAD-GDH bioelectrode for xylose detection.40 Figure 8a presents the typical amperometric response of the FAD-GDH bioelectrode upon the addition of xylose, with an applied potential of 0.25 V vs. SCE (MET of FAD-GDH is facilitated by the FcMe2-C3-LPEI redox polymer). The Michaelis-Menten kinetic model (Figure S11) was applied by nonlinear regression, demonstrating a linear range at low substrate concentrations (c ≪ KMapp, inset within Figure 8a). Figure 8b presents the ability of the FAD-GDH bioelectrode to quantify the remaining xylose concentration in an electrolytically-treated sample, whereby small aliquots of electrolyte (containing remaining xylose) obtained following electrolysis by mono- and bi-enzymatic PDH/bGOx bioelectrodes is injected into an electrolyte absent of xylose (in which the FAD-GDH bioelectrode is operating).
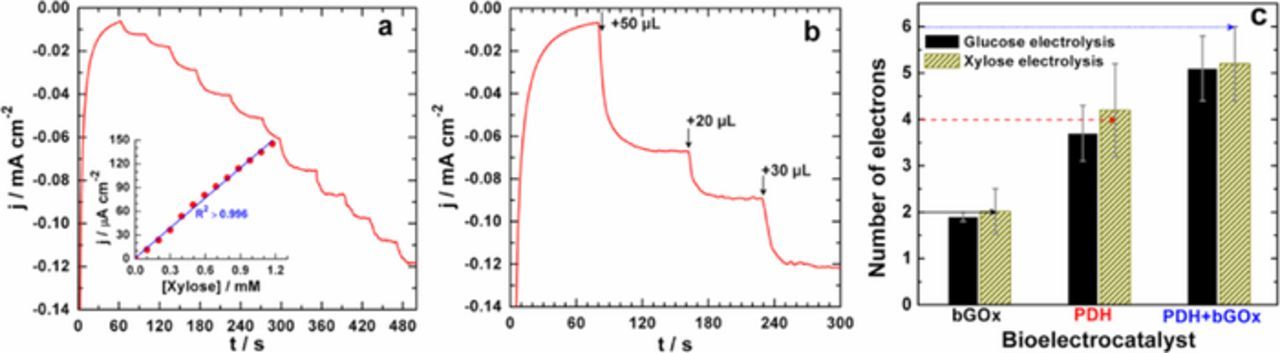
Figure 8. (a) Amperometric response of the FAD-GDH bioelectrode following successive additions of xylose. Inset: Data fitted using linear regression (least squares method) at low concentrations of xylose (Error bars represent one standard deviation, n = 3). (b) Amperometric response of the FAD-GDH bioelectrode following the addition of the electrolytic solution products of xylose at a PDH-bGOx bioelectrode. (c) The determined experimental exchanged number of electrons (nexp) per substrate molecule based on the electrolysis of glucose and xylose (Error bars represent one standard deviation, n ≥ 4): the horizontal lines represent the theoretical maximum exchanged number of electrons per molecule of substrate (nex). Amperometric experiments (a, b) were performed with the applied potential at 0.25 V vs. SCE in a slightly stirred potassium phosphate buffer (0.2 M, pH 6.5) used as electrolyte solution. The FAD-GDH bioelectrode was prepared as previously reported.40
The experimentally determined nexp following the electrolytic experiments is displayed in Figure 8c, for both glucose and xylose oxidations for mono- and bi-enzymatic PDH/bGOx bioelectrodes. For bGOx-only bioelectrodes, the obtained nexp is 1.9 ± 0.1 (glucose) and nexp = 2.0 ± 0.5 (xylose), which are close to the expected theoretical value (nex = 2), providing experimental evidence that glucose and xylose are oxidized in a single 2e− process. Thus, the estimated faradaic efficiency (Eq. 2) on the basis of nex = 2 is nearly τF = 100%. For PDH-only bioelectrodes, nexp was determined to be 3.7 ± 0.6 (glucose) and 4.2 ± 1.0 (xylose), certifying their multiple oxidations at the C2 and C3 positions, in agreement with the literature;32 the oxidation at one carbon (C2 or C3) yields nex = 2 and a twin oxidation (C2 + C3) yields nex = 4. It is important to note that the relatively-large deviation in error may come from the possible interference of the reaction products (C2/C3 oxidized products) with FAD-GDH during the assay. Interestingly, nexp values of 5.1 ± 0.7 (glucose) and 5.2 ± 0.8 (xylose) confirm that the bi-enzymatic bioelectrodes are able to facilitate multiple substrate oxidative reactions (up to 6e−) with an associated faradaic efficiency of ca. 90% toward a 6e− process for both substrates. Since nexp is lower than the theoretical prediction "nex = 2 + 4 = 6", we assume that the reaction may lead to a mixture of C1 + C2/C3 (first step: 2 + 2 = 4 electrons) and C1 + C2 + C3 (second step: 2 + 2 + 2 = 6 electrons) oxidation. In this way, the value of nexp will be between 4 and 6, as obtained experimentally. Future work will focus on the optimization of the experimental conditions for alternative analytical techniques.
In summary of these bulk electrolytic experiments, a commercial selective (by definition) glucose biosensor was used to determine the concentration of glucose remaining in solution following a period of electrolysis (bioelectrocatalytic glucose oxidation at the bioelectrode). By determining the remaining concentration of glucose after electrolysis, we demonstrated that a bioelectrode prepared with bGOx only returned approximately 2 electrons per molecule of oxidized glucose (expected for a single 2 electron oxidation at the C1 position). Using the same approach, PDH-only electrodes returned approximately 3.7 electrons per molecule of oxidized glucose (up to 4 electrons expected for oxidation at the C2 and C3 positions). Remarkably, this value increased to approximately 5.1 electrons per molecule of oxidized glucose (where 6 is the theoretical maximum if operating at 100% efficiency), providing strong evidence for instances of 3 × 2 electron oxidations of single glucose molecules. To provide additional support for this claim, these experiments were repeated using xylose as the substrate (synonymous to glucose for these experiments) and xylose concentrations were determined using a novel xylose biosensor. In summary, the findings presented for glucose are mirrored for xylose oxidation, further confirming the ability to perform multiple substrate oxidations using this bienzymatic approach.
General scheme for the deep oxidation of substrate
Scheme 1 represents the general approach for the ferrocene redox polymer mediated electron transfer (MET) of bGOx and/or PDH, using glucose as a model substrate. Panel (a) displays the MET of the mediated bioelectrocatalytic oxidation by either PDH or bGOx. Panel (b), shows the single enzyme mechanism where bGOx only performs a single 2e− oxidation at the anomeric (C1) position whereas PDH has the capability to perform single or twin oxidations of glucose (at the C2 and/or C3 position) yielding up to 4e−. Then, an intelligent combination of these two enzymes (PDH and bGOx) that have different substrate specificities within the same bi-enzymatic electrode architecture enables performing deep oxidation of a single molecule. Herein, the product of glucose oxidation by bGOx is able to undergo a further round of oxidation PDH (red arrow) or vice versa (blue arrow), yielding to a theoretically-possible 6e− from a single molecule of glucose, that has been used here as model fuel.
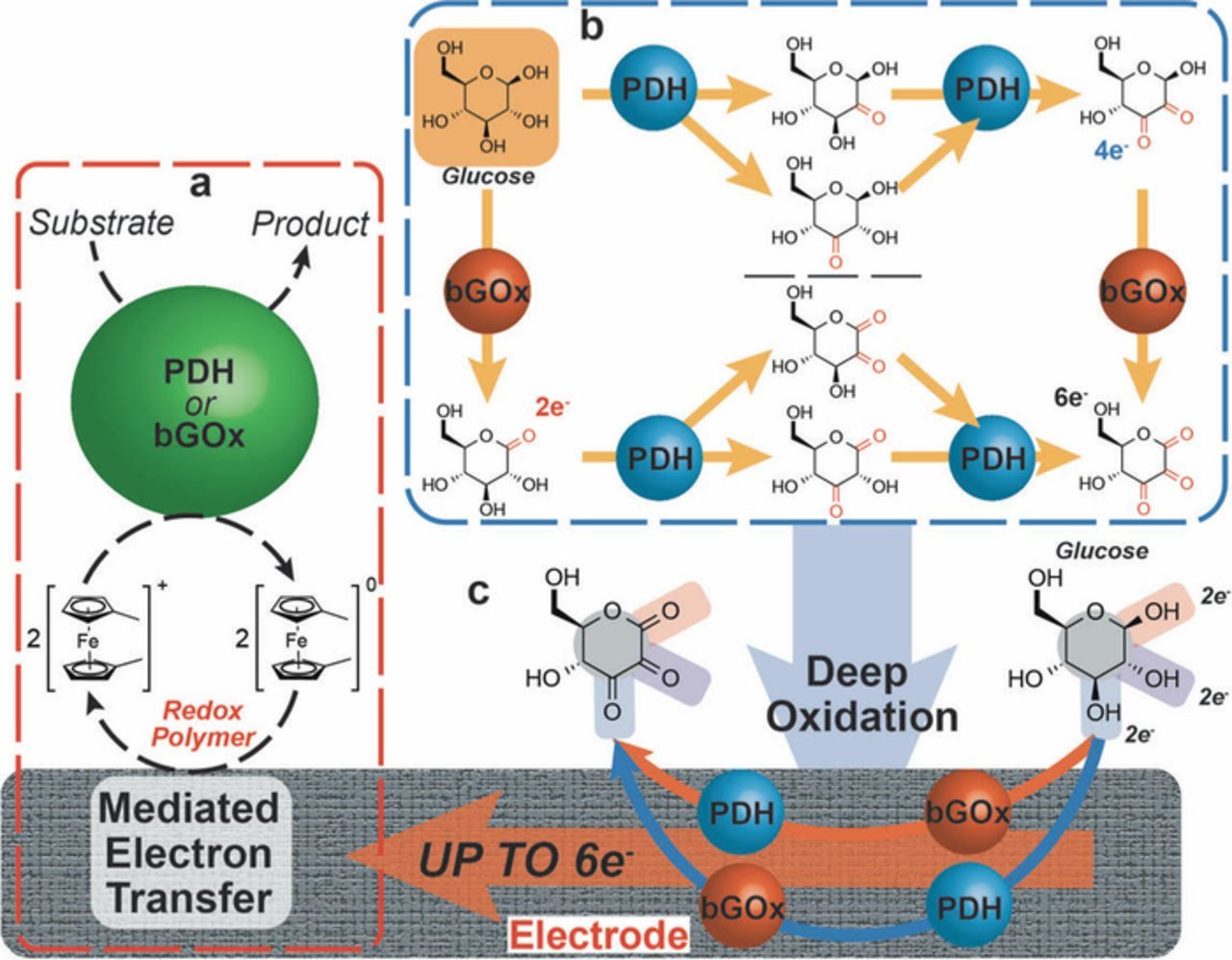
Scheme 1. The rational design of a bi-enzymatic bioelectrode by combining two enzymes that possess diverse substrate specificities; pyranose dehydrogenase (PDH) and an engineered glucose oxidase (bGOx). (a) Ferrocene redox polymer facilitates the mediated bioelectrocatalytic oxidation by either PDH or bGOx. (b) Single enzyme mechanism where bGOx only performs a single 2e− oxidation at the anomeric (C1) position whereas PDH has the capability to perform single or twin oxidations of glucose (at the C2 and/or C3 position) yielding up to 4e−. (c) Deep oxidation of a single molecule of glucose by a bi-enzymatic electrode architecture achieved by combining PDH and bGOx: The product of glucose oxidation by bGOx is able to undergo a further round of oxidation by PDH (red arrow) or vice versa (blue arrow), yielding up to theoretically-possible 6e− from a single molecule of glucose.
Conclusions
This work reports the ability of a FcMe2-C3-LPEI redox polymer to facilitate the mediated bioelectrocatalytic oxidation of multiple (oligo)saccharides by PDH. Additionally, we have demonstrated that the resulting PDH bioelectrodes can perform multiple oxidative reactions (up to 4e−) on a single molecule of glucose and xylose. This suggests that PDH bioelectrodes can be easily employed for EFCs with improved efficiencies, since essentially all existing glucose/O2 EFCs only utilize a single 2e− oxidation of glucose.
We recently reported a bGOx bioelectrode that could oxidize a range of (oligo)saccharides as a 3-electrode configuration or with a EFC configuration (using a typical O2-reducing biocathode).37 We previously hypothesized, however, that bGOx only performs a single 2e− oxidation at the anomeric (C1) position of each substrate (when the C1 is not involved in a glycosidic linkage within oligosaccharides). This led us to hypothesize that a combination of bGOx and PDH within the same electrode architecture (where the FcMe2-C3-LPEI redox polymer can facilitate MET for both enzymes) could yield a versatile bioelectrode that could operate on many substrates while also performing multiple oxidative reactions on single substrate molecules (up to 6e−). Initially, voltammetric and amperometric experiments demonstrated that the presence of both enzymes within the bi-enzymatic bioelectrode does not affect either enzyme. The preparation of an EFC incorporating the bi-enzymatic bioanode resulted in a single EFC configuration that was able to convert chemical energy to electrical energy for a total of 12 different saccharides tested to date.
To probe the ability of the PDH bioelectrodes to perform multiple oxidation reactions and of the PDH/bGOx bi-enzymatic bioelectrodes to oxidize substrates at alternative positions, we used glucose and xylose sensing capabilities to determine the number of electrons, whereby up to 6e− can theoretically be harvested. For xylose and glucose oxidation at a bi-enzymatic bioelectrode, approximately 5 electrons were obtained per molecule of substrate. This evidence confirms that the product of bGOx is able to undergo a further round of oxidation by PDH (or vice versa), demonstrating the ability of the combination of enzymes to facilitate deeper oxidative reactions of substrates. Future work will attempt to evaluate the "depth" of oxidation of alternative substrates for PDH and bGOx combined. Additionally, alternative product analysis techniques will be employed to further investigate the mechanism of substrate oxidation by the bi-enzymatic bioelectrodes.
Acknowledgments
Y. H., M. Y., R. D. M., D. P. H. and S. D. M. thank the National Science Foundation (grant #1158943) and the Army Research Office for funding. R. D. M. acknowledges funding from a Marie Curie-Skłodowska Individual Fellowship (Global) under the EUR Commission's Horizon 2020 Framework (project "Bioelectroammonia", ID: 654836). CKP was supported by grant TRP-218 of the Austrian Science Foundation FWF. The authors thank Amano Enzymes (Japan) for providing bGOx.