
Abstract
A Layered Double Hydroxide (LDH) compound LDH ([Mg2Al(OH)6]+ x 2 H2O) intercalated with a redox active organic anion, Anthraquinone-2-sulfonate (AQS), has been envisioned as an electrode material for high power aqueous based battery. The purpose is to use this interlayer redox active molecule for the enhancement of the specific capacity at the LDH composite electrode, which should allow fast charge transfer at the negative electrode for high power storage applications. This is achieved by the reduction of AQS in charge and oxidation in discharge within a redox inactive LDH matrix. The first charge of this new material [Mg2Al(OH)6]+[AQSO3]− x 2 H2O leads to a capacity of 100 mAh g−1 at − 0.78 V vs Ag/AgCl (based on the weight of the active material) when operated in aqueous 1 M sodium acetate electrolyte. However, low cycling stability was observed, since a drastic loss in specific capacity occurs after the first charge. This study focuses at elucidating the mechanism behind this phenomenon via in situ UV/vis experiments. Subsequently, the dissolution of charged AQS anions into the electrolyte during the first charge of the anode has been identified and quantified. Such understanding of fading mechanism might lead to the design of improved LDH-based electrodes, which utilize redox active anions working in the positive potential range with enhanced cycling ability.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution Non-Commercial No Derivatives 4.0 License (CC BY-NC-ND, http://creativecommons.org/licenses/by-nc-nd/4.0/), which permits non-commercial reuse, distribution, and reproduction in any medium, provided the original work is not changed in any way and is properly cited. For permission for commercial reuse, please email: permissions@ioppublishing.org.
Layered Double Hydroxides (LDHs), also known as hydrotalcite-like compounds, are a versatile class of 2D intercalation clay compounds described by the general formula: [M2+ 1−xM3+ x(OH)2]x+(An−) x/n.yH2O, where M2+ and M3+ are divalent and trivalent metals cations, respectively, and An− represents an n-valent anion. 1,2 The most popular example showing this characteristic structure is hydrotalcite Mg6Al2(OH)16CO3 4H2O. 3 The cation couple (Mg/Al) has been widely studied in this context but other divalent (Zn, Cu, Mn, etc.) and trivalent (Fe, Cr, Co, etc.) cations can be found and are used in LDHs as well. 1,2,4–12 The structure of a single LDH layer is similar to brucite Mg(OH)2, with edge-sharing M(OH)6 octahedra in which the partial substitution of M3+ for M2+ introduces a net positive charge to the layers balanced by intercalated exchangeable anions (see Fig. 1).

Figure 1. Atomic structure of Hydrotalcite mineral Mg6Al2(OH)16[CO3] x 4H2O as representative of layered double hydroxide family. 129
Download figure:
Standard image High-resolution imageBy modifying the substitution ratio and/or the intercalated anions, it is possible to obtain a large variety of synthesized phases, which enables a high diversity in physical and chemical properties. Different intercalated organic and inorganic anions enable the design of LDHs for many different applications such as drug delivery, 13–16 bio-imaging, 17–19 water purification, 20–23 catalysis, 24–38 sensors, 39–44 additives in polymers, 45–47 anti-corrosion films 48–51 and ion exchange. 52–54
Furthermore, since the 1990s, there have also been investigations on LDH based electrodes in order to clarify their electrochemical characteristics. 55–60 Specifically, LDHs with the possibility to use electroactive cations within the layers are promising in this field. Several studies have demonstrated the electrochemical behavior of these layers, such as Vialat et al. with the cobalt LDHs Co(II)Co(III)-CO3 and CoNiAl-CO3. 61,62 Thin film electrodes composed of an electroactive LDH matrix deposited on a carbon conductor exhibit specific capacities ranging from 14 up to 30 mAh g−1. However, changes of the cation oxidation state usually lead to the destabilization of the layers and to their subsequent delamination. 63,64
These efforts have led to many investigations with the aim to use LDH electrodes (with redox active cations, mainly Ni and Co) as energy storage materials in energy storage devices. 65–98 The literature in this regard, however, must be read carefully, since researchers often report on very high values for specific capacitances C in F g−1 for single electrodes, which are unfortunately misleading and do not represent the actual charge storage process of the active material. 71,73,85,86 Indeed, classical supercapacitor calculations are mistakenly applied to these electrodes although faradic redox behavior is often clearly observed (non-constant current in CV and plateaus in galvanic charge/discharge tests). 99
LDHs have been investigated in various battery-based systems 100–107 e.g. as Li-ion battery electrodes, 108,109 in Li-Sulfur batteries 110–112 and so-called anion, halide or double ion batteries. 113–116 A good example for the application of LDHs in batteries was presented by Yang et al. in 2013. 104 In this study, a ZnAl-LDH carbon nanotube composite was used as an anode material for a Nickel-Zinc-Battery and displayed an improved discharge capacity of 390 mAh g−1 (based on the weight of the anode active material) with high cycling stability compared to classical ZnO, but at the expense of the collapsing of the LDH structure upon cycling.
So far, many energy storage systems from the literature use LDHs with redox active cations within the layers. In this work, however, we specifically target the investigation of the redox activity of the anions intercalated in between these layers to enhance fast charge storage capability. This could increase the energy density of advanced energy storage devices using LDH electrodes, while keeping the LDH backbone all over the cycle life of the electrodes. In order to realize this approach, redox active anionic groups can be intercalated during the synthesis, in between the layers of LDHs with non-redox active cations such as MgAl or CaAl to replace inactive anions like CO3 2−, NO3 −, Cl−, or OH−. 2 Mousty et al. studied this concept in 1994 and successfully intercalated m-Nitrobenzene sulfonate and Anthraquinone sulfonates in a ZnCr-LDH matrix. 117 Furthermore, they were able to show the electrochemical activity of single electrodes derived from these materials but with very low amount of electrode material (few μg) with the purpose of designing a pH sensor. Indeed, such strategy has already been pioneered by our team to enhance the capacity (originated from double layer capacitance) of different types of carbons. 118–120 In addition our team presented a similar approach for the redox activity of riboflavin- and ferrocene based anions in LDH structures in a very recent study. 121 However, besides this work, to the best of our knowledge there is no study of the use of LDHs as matrices for energy storage application. The higher density of LDHs compared to carbons and the one pot synthesis of AQS-intercalated LDHs (instead of multiple steps for functionalized carbons) are two advantages over the use of AQS-grafted carbons.
Specifically, in this work we intercalated the redox active Anthraquinone-2-sulfonate (AQS) between the layers of the redox inactive LDH [Mg2Al(OH)6]+ x 2 H2O to yield the active material [Mg2Al(OH)6]+[AQSO3]− x 2 H2O (MgAlAQS). We decided to use the redox active group of Anthraquinone (AQ), since it displays high specific capacity and a stable redox potential in organic battery applications. 122 The idealized structure of MgAlAQS is shown in Fig. 2A.

Figure 2. (A) Schematic structure LDHs with inserted redox active moieties example AQS, B) Exploited redox reaction of AQS.
Download figure:
Standard image High-resolution imageThe desired reduction process of [AQSO3]− to [AQSO3]3− during the charging process is shown in Fig. 2B. The theoretical capacity Qth in mAh g−1 (based on the mass of the active material) of this reaction was calculated using Eq. 1
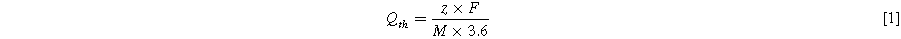
where z is the number of electrons involved in the redox reaction (z = 2), F is the Faraday constant (F = 9.65 × 10−4 C mol−1) and M is the molar mass of MgAlAQS (M = 501 g mol−1). For the calculation, 1 mol of [AQSO3]− was assumed per mol of [Mg2Al(OH)6]+ x 2 H2O. This results in a theoretical capacity of 107 mAh g−1 for MgAlAQS. The reduction potential of Anthraquinone as well as AQS is found to be at −0.68 V vs SHE, which makes MgAlAQS an anode material to be used at the negative electrode of an energy storage device operated in aqueous electrolyte. 123
Therewith, the intercalation of [AQSO3]− in MgAlAQS electrodes results in similar capacities compared to Anthraquinone in organic batteries. 123–125 Meanwhile, the capacities of MgAlAQS electrodes are lower than those obtained in LDHs which use the reaction of the metal cations composing the layers. 126 However, in the latter case the LDH structure is often destroyed, which is not likely to happen for the structure of AQS intercalated in [Mg2Al(OH)6]+.
The resulting schematic setup, which was used in this work, is shown in Fig. 3 and displays the MgAlAQS composite working electrode combined with a counter electrode (of any kind) submerged in the electrolyte during the charging process. Note that the reference electrode, which was also utilized in all electrochemical experiments, is not drawn. Details about this setup are given in the experimental part.

Figure 3. Concept of MgAlAQS electrode in electrochemical energy storage cell.
Download figure:
Standard image High-resolution imageWith this experimental design, the aim of this work is to investigate the redox activity of the [AQSO3]− anion, which is intercalated between the layers of [Mg2Al(OH)6]+ x 2 H2O and therewith to prove the concept of our newly designed approach for the enhancement of the specific capacity of LDH electrodes in energy storage applications.
Experimental
A layered double hydroxide of the MgAl type was prepared using a coprecipitation method similar to that described by the usual adapted procedure. 121 100 ml of deionized and decarbonated water was used and the preparation was carried out in a 250 ml reactor under a nitrogen atmosphere. Anthraquinone-2-sulfonate was dissolved in this media at 80 °C and the pH was adjusted to 10 with 0.1 M NaOH solution following the mole ratio between AQS and Al3+ cations of nAQS = 4nAl. 25 ml of the aqueous metal chlorides solution (0.333 mol l−1 in MgCl2 and 0.167 mol l−1 in AlCl3) was added dropwise to the reactor at 80 °C and under magnetic stirring for 3 h. The pH of the reaction mixture was kept constant at 9.5 by adding a 1 M NaOH solution. After complete addition of the metal salts, the reaction mixture was left to age at 80 °C for 24 h. The precipitate was then centrifuged and washed three times with hot water. The solid was further dried in an oven for 24 h at 40 °C. The X-ray diffraction spectra of the obtained solid are displayed in S1.
X-ray diffraction analyses were performed using a theta−theta PANalytical X'Pert Pro diffractometer equipped with a Cu anticathode (λKα1 = 1.540598 Å, λKα2 = 1.544426 Å) and a X'Celeretor detector. For the phase identification and refinement of the unit cell parameters of the series samples, patterns were recorded in the range of 2−80°/2θ with a step size of 0.325°/min three times.
Scanning electron microscopy (SEM) images were recorded using a JSM-7500F field-emission scanning electron microscope operating at an acceleration voltage of 3 kV and magnifications of ×1 K, ×20 K, and ×50 K or using a Zeiss MERLIN scanning electron microscope with a magnification of ×10 K and an acceleration voltage of 20 kV. Samples to be imaged were mounted on conductive carbon adhesive tabs and coated with a gold thin layer.
The composite working electrodes used in this study have been prepared as described in Ref. 121 and consisted of 60% active material (LDH, MgAlAQS), 30% conductive additive carbon black (Super Graphite, Superior Graphite Co.) and 10% binder (PTFE, Sigma Aldrich, 60% solution in water). The prepared electrodes resulted in an area of 0.5 cm2 with a thickness of around 100 μm and an average mass loading of 9.2 mg cm−2. They were pressed into a stainless steel current collector in the form of a grid under 500 MPa. For the UV/vis experiments, smaller electrodes with an average area of 0.04 cm2 and an average mass loading of 18 mg cm−2 have been used.
The investigated electrochemical cells were assembled in a 3-electrode beaker type setup, where the working electrode was the LDH composite electrode, the counter electrode was a platinum wire and an Ag/AgCl (3 M NaCl) electrode was used as the reference electrode. All electrodes were submerged into 10 ml of 1 M sodium acetate (NaCH3COO) aqueous electrolyte.
Electrochemical tests were performed using a multichannel potentiostatic-galvanostatic workstation (BioLogic Science Instruments, VMP3, operated under ECLab software) at room temperature. Before all electrochemical experiments, the cells were set to equilibrium by leaving at open circuit for 1 h. Cyclic voltammetry (CV) measurements were recorded between 0 and –1.2 V vs Ag/AgCl at a scan rate of 1 or 2 mV s−1. Constant current galvanostatic charge/discharge experiments were performed between 0 to—1.2 V vs Ag/AgCl (3 M NaCl) with a current of 0.43 A g−1 (4 C).
Electrochemical CV tests for UV/vis analysis were performed in the same conditions using a single channel potentiostat-galvanostat (PalmSens, PalmSens 4).
UV/vis analysis was carried out using a UV/Vis/NIR spectrophotometer (PerkinElmer LAMBDA 1050) in transmission mode. The absorption maximum of AQS in the electrolyte was located at 330 nm, and absorption spectra were measured in the range between 300–360 nm.
Results and Discussion
The electrochemical behavior of MgAlAQS electrodes in 1 M NaCH3COOH aqueous electrolyte during cyclic voltammogram (CV) measurements at a scan rate of 2 mV s−1 in the potential range from 0 to −1.2 V vs Ag/AgCl is shown in Fig. 4. The evolution between the first and second CV are shown in Fig. 4A, since these are of particular interest in this experimental series. The highest current density in this graph is measured for the first reduction of [AQSO3]− to [AQSO3]3− at - 1.03 V vs Ag/AgCl with a value of 2.01 A g−1. The related charge determined for this first reduction process is 99 mAh g−1, which is very close to the theoretical capacity of MgAlAQS of 107 mAh g−1 (93%). The corresponding oxidation peak of the first CV cycle is obtained at −0.48 V vs Ag/AgCl with a significant loss in maximum current density and charge at 1.54 A g−1 and 52 mAh g−1. The coulombic efficiency of this first reduction (charge) and oxidation (discharge) couple is close to 50%. This indicates a large hysteresis phenomenon occurring at the working electrode already in the first cycle. In the second cycle, a further decrease in the charge related to both redox peaks, specifically for the reduction process, is also observed. The related capacities are 48 mAh g−1 and 37 mAh g−1 for reduction and oxidation processes respectively, with a coulombic efficiency of 72%.

Figure 4. CVs of MgAlAQS electrodes from 0 to—1.2 V vs Ag/AgCl at 2 mV s−1 in 1 M NaCH3COOH in H2O at the cycles 1, 2, 10, 50, 100 and 200.
Download figure:
Standard image High-resolution imageThe loss in capacity with increasing number of cycles is further illustrated in Fig. 4B. After 200 cycles, the degradation results in a charge of 7 mAh g−1 for the reduction and 3 mAh g−1 for the oxidation (42% coulombic efficiency). The shape of the CV curves towards the more negative potentials suggests an increased contribution from the hydrogen evolution reaction (HER) with increasing cycle number, which could explain the drop in coulombic efficiency.
The results shown in Fig. 4, present a continuous decrease in capacity, with a pronounced drop on the first cycle. Since nearly all the electroactive AQS molecules in the electrode respond at the first reduction (close to 93% of the theoretical capacity), it suggests that the successive reduction reactions involve AQS molecules which remain electrochemically active, demonstrating that the redox reaction in Fig. 2B is reversible. However, it also appears that a second electrochemical reaction, the hydrogen evolution reaction progressively increases with the number of cycles, probably due to an electro catalytic activation of the stainless steel current collector. The onset of the HER on stainless steel in alkaline solution has been reported to be around −0.28 V vs RHE (i.e. around −0.48 V vs Ag/AgCl/KClsat). 127,128
The rapid degradation of the MgAlAQS electrode in terms of specific capacity for the charge/discharge process at a current density of 0.43 A g−1, which corresponds to a 4 C rate (full charge or discharge of the electrode within 15 min if the full capacity is utilized), is clearly depicted in Fig. 5. A specific capacity of 100 mAh g−1 is obtained for the first reduction (charge) of the LDH composite electrode, with a potential plateau at −0.78 V vs Ag/AgCl (Fig. 5A), which matches perfectly with the results recorded in the CVs depicted in Fig. 4. However, upon the first re-oxidation (discharge), the active material delivers a specific capacity of only 44 mAh g−1 (ƞc = 44%) at −0.6 V vs Ag/AgCl. Therewith, it is obvious that also in the charge/discharge process of MgAlAQS a pronounced decrease in specific capacity occurred right after the first reduction of the material and indicates again a strong loss of redox active sites in the working electrode. In the second charge, a specific capacity of 46 mAh g−1 is measured which is close to the previous discharge capacity. This confirms that the AQS activity lost during the first charge is no longer available for the following cycles. The second discharge delivers 37 mAh g−1 (ƞc = 80%) with a loss which is proportionally lower, suggesting that the remaining redox active sites are slightly more stable in the structure of the LDH during cycling.

Figure 5. (A) Charge/discharge profile of MgAlAQS electrodes for the first and second cycle and (B) cycling stability at 0.43 A g−1/4 C in 1 M NaCH3COOH aqueous electrolyte for 200 cycles.
Download figure:
Standard image High-resolution imageThe further cycling behavior of the MgAlAQS electrodes in 1 M NaCH3COOH in H2O is shown in Fig. 5B for 200 cycles at the same current density (0.43 A g−1, 4 C). After the significant decrease in specific capacity during the first charge, a continuous decrease is measured. After only 20 cycles, the specific capacity of the working electrode is reduced to 21 mAh g−1 and further decreases to very low last cycle capacities for charge and discharge processes of 6 and 5 mAh g−1 after 200 cycles, respectively. Apart from the first cycle, the coulombic efficiency ranges from 80% to 90%. The values depicted in Fig. 5B are shown more detailed in S2.
Both Figs. 4 and 5 show a strong loss of the redox capacity in CV and constant current cycling especially in the first reduction. In order to investigate this electrode degradation, the surface of pristine and cycled MgAlAQS composite electrodes have been observed by SEM to identify possible changes of the electrode, which could have caused a decline in specific capacity. However, the SEM images of pristine and cycled MgAlAQS electrodes in Figs. 6A and 6B do not reveal any degradation during cycling, since no difference in the composite material can be depicted.

Figure 6. MgAlAQS electrodes (A) pristine and (B) after 200 cycles of charge/discharge in 1 M NaCH3COOH in H2O with a magnification of ×10 k, the scale at the bottom left corner is 1 μm.
Download figure:
Standard image High-resolution imageThis together with the low coulombic efficiencies obtained of the electrodes indicate that the absence of structural electrode film failure, and instead that unwanted side processes during the cycling are most likely responsible for the degradation of the active material. Furthermore, since neither an apparent change of the electrodes nor additional peaks or plateaus in the cycling experiments were observed, a possible explanation is the dissolution of electro-active AQS anions into the electrolyte. To understand this process and to find evidence for such hypothesis, in situ UV/vis measurements were carried out to identify changes in the electrolyte during cycling.
The dissolution of AQS shown by an UV/vis experiment in an in situ beaker type glass cell with a MgAlAQS working electrode after 40 min of CV at 1 mV s−1 (one forward sweep = 20 min, 40 min for a full cycle) between 0 V and −1.2 V vs Ag/AgCl (3 M NaCl) and another MgAlAQS electrode in the same setup during 40 min at open circuit potential (OCP) are compared in Fig. 7A. The quantity of AQS in the aqueous electrolyte was calculated from the increase in the absorption maximum of anthraquinone at 330 nm. The quantity in % refers to the amount of AQS remaining in the LDH host structure vs the initial amount of AQS present in the WE. In the first 10 min, no significant dissolution is observed neither during OCP nor CV measurement. After 10 min, once the potential reaches a critical value on the CV experiment, the evolution of the two curves differs significantly. After 40 min at OCP no significant amount of AQS was found in the electrolyte (100% remaining in the electrode), but a strong dissolution was observed after 10 min of the CV reduction sweep. At this point, a steep decrease of the AQS in the WE is calculated until a plateau is reached at 25% after 25 min of CV, i.e. a strong increase of AQS in the electrolyte is observed up to 75% of the initial amount intercalated in the electrode (Fig. 7A). At the end of the CV, after 40 min, the quantity of AQS in the electrode does not change significantly in the last 15 min of the experiment (i.e. during the oxidation sweep), and stays at 25%.

Figure 7. (A) In-situ UV–vis measurements of MgAlAQS electrodes in cuvette, (B) UV–vis comparison of MgAlAQS electrode in situ after 19 d of OCP and 40 min CV.
Download figure:
Standard image High-resolution imageIt is therefore clear that during a CV measurement much more AQS is dissolved in the electrolyte in a very short time in comparison to the amount observed during the OCP test. Figure 7B emphasizes this fact, since here even after 19 d at OCP, there is still 95% of residual AQS in the electrode material, which is much more AQS left in the electrode compared to the CV test.
Based on these results, it is obvious that due to the potential changes during the CV experiment, the majority of the redox active AQS is released from the electrode in the time range between the 10th and the 25th minute. To elucidate this process, Fig. 8 overlaps the potential at the electrode, the time and the current (I vs t) from 0 V to −1.2 V vs Ag/AgCl at a scan rate of 1 mV s−1 with the evolution of the remaining AQS in the working electrode.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Loss of AQS in MgAlAQS electrodes at 1 mV s−1 overlaid with unfolded CV current I vs time t.
Download figure:
Standard image High-resolution image{kind=link}
The monitored loss of active sites vs the potential at the MgAlAQS based electrode is shown in Fig. 8. The two plots highlight that the dissolution of redox active AQS is caused by the reduction of [AQSO3]− to [AQSO3]3− at the potential range between −0.6 V and −0.9 V vs Ag/AgCl (between minute 8 and 15). The appearance of reduced [AQSO3]3− in the electrolyte starts within a short delay after the reduction of the [AQSO3]− anions at 11 min and continues until a plateau is reached at 25 min, which corresponds to the end of the reduction sweep. From there, the following oxidation sweep does not lead to an increase in AQS in the electrode. The final amount of residual AQS in the WE amounts to 25% after only one CV cycle. This means 75% is dissolved in the electrolyte and no longer accessible for efficient charge storage for the subsequent cycles. It also shows that the remaining 25% reduced AQS that are still in the electrode can be re-oxidized as indicated from the oxidation peak around −0.65 V vs Ag/AgCl. The redox reaction displayed in the unfolded CV of Fig. 8, results in a coulombic efficiency of 23%, which nicely matches the displayed loss in AQS anions after the first reduction. Therefore, it appears that none of the dissolved AQS reacts at the surface of the electrode. In repeated experiments with the same conditions, other values for the remaining percentage of AQS have been obtained (S3). However, in all tests the same trend and the same potential for the release of AQS anions was observed.
The described loss of active sites in the WE is caused by a charge imbalance after the reduction (charging) of the active material. In the discharged state of [Mg2Al(OH)6]+[AQSO3]− x 2 H2O, [AQSO3]− anions compensate the positive charge of the [Mg2Al(OH)6]+ layers. In the charged electrode, however, this equilibrium is no more valid due to the presence of [AQSO3]3−. This specie demands three times more positive charge than [AQSO3]−, which can, however, not be provided by the LDH layers. Therefore, to restore the charge equilibrium, [AQSO3]3− anions need to leave the LDH structure. In theory, this process should result to the release of 66% of the initial quantity of AQS anions out of the electrode to yield to the charged LDH active material [Mg2Al(OH)6]+([AQSO3]3−)0.33. This means 33% of AQS is in the LDH electrode after reduction and 66% is dissolved in the electrolyte. In reality, release percentages between 60 and 90% have been measured after 1 CV cycle (40 min) in repetitive experiments (S3). This deviation is possibly explainable by the interaction of the WE with acetate anions from the electrolyte, which are present in abundance, and which can be exchanged with [AQSO3]3− upon cycling the electrode. Additionally, during oxidation and the corresponding polarization towards more positive potentials, the re-intercalation of [AQSO3]3− is statistically unlikely, since vastly more acetate anions are available in the electrolyte for this process. This release of AQS anions out of the electrode into the electrolyte explains the evident decrease in specific capacity measured after the first reduction of the electrode.
This means that, although we demonstrated the redox activity of AQS in the structure of MgAlAQS, the system is by nature not stable during charging and more efforts must be dedicated to the stabilization of the capacity upon cycling, mainly by preventing electroactive molecules to escape from the LDH interlayer space.
Conclusions
In this study, we proved the usability of redox active anions intercalated in between LDH layers for energy storage application. In these experiments, the specific capacity of the first reduction/charge of AQS anions in MgAlAQS was measured at the potential of −0.78 V vs Ag/AgCl with a total charge capacity of 100 mAh g−1, which is close to the theoretic capacity and therewith highlights the complete electrochemical activity of the anions immobilized in the LDH interlayer spacing.
However, during all electrochemical experiments, insufficient cycling stability of MgAlAQS was observed, which is caused by the dissolution of charged [AQSO3]3− into the electrolyte after the first reduction, which has been evidenced by in situ UV/vis experiments. In this process, the charged anion [AQSO3]3−, unlike the discharged [AQSO3]−, causes a charge imbalance in the LDH structure and therewith is forced out of the interlayers of the LDH working electrode. With the release of [AQSO3]3− for charge compensation, these active sites are not accessible for charge storage during subsequent cycles. In order to exploit the redox activity of AQS anions, however, this species has to be reduced, which will in any case cause the described intrinsically low cycling stability of the presented active material [Mg2Al(OH)6]+[AQSO3]− x 2 H2O.
Therefore, it is necessary to search for other intercalated LDHs, with redox active anions, which show less dissolution during charging or ways to prevent the dissolution of the active species into the electrolyte. We will investigate this approach in future studies. Nevertheless, with the findings of this work a vast variety of possible combinations of LDH structure and redox active groups is opened to be investigated for energy storage application.
Acknowledgments
This research was funded by the French Research Agency ANR AAPG2020 "LaDHy," ANR-20-CE05–0024–01. Labex STORE-EX (ANR-10-LABX-76–01) is also acknowledged for financial support.
Supplementary data (0.6 MB DOCX)