
Abstract
Ionic liquids, like bis(fluorosulfonyl)imide (FSI) ionic liquids, are promising solvents for lithium ion batteries due to their high thermal stability and wide electrochemical window. Here, electrolytes based on LiFSI and ionic liquids of different cations; pyrrolidinium (PYR13+), imidazolium (EMI+) and phosphonium (P111i4+), and FSI and bis(trifluoromethanesulfonyl)imide (TFSI) anions, have been cycled together with silicon anodes and LiFePO4 cathodes at 60 °C at rates up to 2 C. The thermal stability was determined through differential scanning calorimetry (DSC). Ionic conductivities were measured in the temperature range −20 °C to 80 °C and the lithium ion mobilities, relative to standard carbonate electrolytes, were determined at room temperature and 60 °C. Significant differences in the temperature dependence of the transport properties were revealed, with a considerable increase in the lithium ion mobility from 20 °C to 60 °C for electrolytes based on EMIFSI and P111i4FSI. The best rate performance at 60 °C was demonstrated for the P111i4FSI based electrolyte, with discharge capacities around 1000 mAh/gSi at a 2 C rate. The improved performance was attributed to a combination of the improved lithium ion mobility and formation of a more conductive solid electrolyte interphase (SEI) for this electrolyte.
Highlights
The thermal stability in the temperature range −20 to 80 °C, ionic conductivity up to 60 °C, and limiting current density at room temperature and 60 °C, was determined for electrolytes based on the ionic liquids, PYR13FSI, PYR13TFSI, EMIFSI, and P111i4FSI with LiFSI salts.
The P111i4FSI based electrolyte showed best conductivity and the highest limiting current density at 60 °C
In combination with Silicon electrodes, the best stability and the best rate performance was demonstrated for the P111i4FSI based electrolyte.
The lowest electrode resistance after cycling at 60 °C was obtained for the P111i4FSI based electrolyte
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Silicon (Si) is heavily researched as an anode material for Li-ion batteries (LIBs) because of its high specific capacity of 3579 mAh g−1 (2194 mAh cm−3) and fairly low delithiation potential with lithium (∼0.4 V vs Li/Li+), 1 when compared to the conventional graphite anode (372 mAh g−1, 837 mAh cm−3, 0.1–0.2 V vs Li/Li+). 2,3 Replacing graphite with silicon could thus increase the energy density of LIBs, and this is already done with small amounts (∼4–5 wt% Si/SiOx) in some commercial graphite-NMC cells. 4–6 Poor electrode stability, caused by the large volume changes in silicon upon (de)lithiation (∼280% for Si→Li15Si4) 7 compared to graphite (13.2% for C → LiC6) limits the amounts of Si used. 8 Loss of active material through isolation (particle cracking, delamination) or insulation (excessive solid electrolyte interphase (SEI) formation) are the main issues produced by the volume change. 9,10 Several methods are proposed to mitigate these issues, generally categorizable as: (i) morphology and dimension control, (ii) composite formation, (iii) grafting/coating/encapsulation and (iv) electrolyte modification. 10,11 A combination of these will likely be needed to realize a stable high silicon content anode.
Electrolyte modification is the focus of this work. The highly reducing conditions of the anode causes electrochemical decomposition of the electrolyte, forming surface passivation layers known as the SEI. 12,13 Although some conventional carbonate electrolytes with additives like fluoroethylene carbonate (FEC) and vinylene carbonate (VC) have been shown to improve the stability of Si anodes through a more stable SEI, 14–17 the performance is still not satisfactory and the additive is continuously consumed. 18 In addition, the high flammability and volatility of these electrolytes are a safety concern, especially in transport applications where LIBs are essential in the ongoing electrification. 19–24 Ionic liquids (ILs) have high thermal and electrochemical stability, high lithium salt solubility, and importantly low volatility and flammability due to a very low vapor pressure. 21,25–28 Hence, ILs in combination with different lithium salts are interesting as potentially safer electrolytes for LIBs. Some ionic liquid electrolytes (ILEs) have over the years shown more and less promising capacities and cycling stability with electrodes made from a silicon thick film, 29 pre-conditioned nanosilicon, 30 graphene-coated nanosilicon, 31 nanostructured micrometric Si, 32 silicon composite, 33 and even microsilicon. 34 Especially ILEs containing FSI− anions in the IL itself or from the added lithium salt have been shown to have good properties like relatively low viscosity, good SEI-forming abilities and Al corrosion inhibition (high purities, low Cl− content), 32–39 although there have also been reports on the benefit to performance of utilizing a mixture of FSI− and TFSI− anions. 40,41
Our previous work demonstrated excellent cycling stability at C/5 rate for two FSI-based ILEs for μMG-Si anodes in combination with oversized LFP cathodes, namely electrolytes based on pyrrolidinium and phosphonium cations with bis(fluorosulfonyl)imide (FSI) anion, and LiFSI salt dissolved. Poor capacity retention was observed for the same cells with imidazolium FSI, as well as LiFSI dissolved in a standard carbonate solvent mixture. Similarly, the pyrrolidinium TFSI failed to deliver any significant capacity. 42 Differences in the electrochemical performance could be attributed to differences in transport properties, as well as the quality of the SEI. The ionic liquids do however in general exhibit lower ionic conductivities than carbonates at equal temperatures, mainly due to higher viscosity, which limits the rate performance of LIBs with these electrolytes. 43,44 The higher thermal stability can enable higher temperature cycling, 44–48 which will also improve the rate performance of LIBs with ILEs. Improved rate performance at temperatures up to 60 °C was demonstrated for NMC half-cells upon cycling in a Pyr13FSI ionic liquid electrolyte, 44 for which carbonate electrolytes showed a significant drop in the rate performance for the same temperatures. Improvements in rate performance was also demonstrated for electrolytes based on P1222FSI and Pyr13FSI, in combination with electrodes fabricated from silicon nanopowders for rates up to 2 C. 32 Similarly, higher discharge capacity has been reported in Li-ion hybrid capacitors with electrolytes based on EMImFSI when increasing from 25 °C to 50 °C. 47
In this study we will evaluate the feasibility of operation of battery cells based on μMG-Si anodes, at a moderately elevated temperature (60 °C), with four selected room temperature ionic liquids as electrolytes. The ionic liquids include pyrrolidinium, imidazolium and phosphonium cations with the bis(fluorosulfonyl)imide (FSI) anion, and pyrrolidinium with bis(trifluoromethanesulfonyl)imide (TFSI)). Electrolytes are tested for their thermal behavior and stability through differential scanning calorimetry, the total conductivity as a function of temperature is determined up to 80 °C and the relative Li+ mobility with respect to a standard 1.0 M LiPF6 in EC:DEC (50:50 v:v, "LP40") electrolyte is reported. The rate performance of the electrolytes in μMG-Si vs LiFePO4 pseudo full cells is compared for the temperatures 20 °C and 60 °C. We are thus able to systematically compare the physical properties (conductivity, glass transition temperature, crystallization temperature and melting point), and the electrochemical performance (limiting current density, galvanostatic cycling and impedance) in laboratory scale battery cells for these ionic liquids at an elevated temperature, for which results are scarce in the open literature.
Experimental
Electrolyte preparation
N-propyl-N-methylpyrrolidinium bis(fluorosulfonyl)imide (PYR13FSI, purity 99.9%), N-propyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide (PYR13TFSI, purity 99.9%) and 1-ethyl-3-methylimidazolium bis(fluorosulfonyl)imide (EMIFSI, purity 99.9%) were acquired from Solvionic (France) and used as received (<20 ppm H2O by Karl Fischer). Trimethyl(isobutyl)phosphonium bis(fluorosulfonyl)imide (P111i4FSI, purity > 98%, < 200 ppm H2O) was acquired from IoLiTec GmbH (Germany) and dried under vacuum at 80 °C for 12 h before use. The structural formula of the ILs are given in Fig. 1. Battery grade ethylene carbonate (EC), dimethyl carbonate (DMC), fluoroethylene carbonate (FEC) and vinylene carbonate (VC) were acquired from Sigma Aldrich and used as received. Lithium bis(fluorosulfonyl)imide (LiFSI, purity >99.9%) was acquired from American Elements (USA) and dried under vacuum at 80 °C for 12 h before use. A 1.0 M LiPF6 in EC:DEC (50:50 v:v, <15 ppm H2O) "LP40" standard battery electrolyte was acquired from Merck KGaA (Sigma Aldrich) and used as received. The electrolytes summarized in Table I were mixed in an argon-filled glove box (<0.1 ppm H2O/O2, MBraun, Germany) using a hot plate and stirring where necessary to dissolve the LiFSI.

Figure 1. Schematic drawing of the structural formula of the four ionic liquids used in this work.
Download figure:
Standard image High-resolution imageTable I. Composition of the electrolytes used in this work.
| Electrolyte | Composition |
|---|---|
| ILE1 | 0.77 m LiFSI in PYR13FSI (22:78 mol%) |
| ILE2 | m LiFSI in PYR13TFSI (26:74 mol%) |
| ILE3 | 0.74 m LiFSI in EMIFSI (20:80 mol%) |
| ILE7 | 0.79 m LiFSI in P111i4FSI (22:78 mol%) |
| STD2 | 0.74 m LiFSI in EC:DMC:FEC:VC (EC:DMC (1:2 w/w), 5 wt% FEC, 1 wt% VC) |
Electrode preparation and cell assembly
Silicon anodes (73.2 wt% Elkem Silgrain® e-Si 400, 11.0 wt% Timcal C65 carbon black, 7.3 wt% Na-CMC binder from Sigma Aldrich and 8.5 wt% leftover buffer chemicals from citric acid and KOH) with a loading of 0.748 ± 0.042 mgSi cm−2 (corresponding to ≈2.68 mAh cm−2 based on 3579 mAh/gSi) 1 screen printed onto dendritic Cu-foil (16 μm thick) were provided by Institute for Energy Technology (IFE, Norway). The electrodes were cut into 12 mm diameter discs (1.13 cm2) and dried at 100 °C for 12 h before inert transfer and storage in an argon-filled glove box (<0.1 ppm H2O/O2, MBraun, Germany).
LiFePO4 (LFP) cathodes (89 wt% active material, ≈3.5 mAh cm−2) acquired from Custom Cells (Germany) were cut into 14 mm discs (2.01 cm2) and dried at 110 °C for 12 h before inert transfer and storage in the glove box mentioned above. The capacity of the LFP cathodes was purposefully oversized by >100% with respect to the Si anodes to assemble Si-LFP pseudo-full cells, as introduced by Wetjen et al. 49 to (i) have a stable reference potential of 3.45 V vs Li/Li+ to monitor the silicon anode potential in a two-electrode configuration, (ii) provide a defined lithium reservoir to be able to exclude capacity loss from depletion of lithium inventory and (iii) minimize side reactions of the electrolyte at the counter electrode (compared to highly reactive Li metal). An additional reason was that the combination of Si-Li half cells and ILEs created issues with stray currents, in our experience. The pseudo-full cells were assembled in either PAT-cells (EL-Cell, Germany) with 316 L stainless steel plungers or pouch (coffee bag) cells in an argon-filled glove box. An Evopor 5E02A (30 μm, Lydall) separator was used, soaked by 50 μl (PAT) or 30 μl (pouch) electrolyte.
Ionic transport properties
The total ionic conductivity of the electrolytes was measured by alternating current (AC) impedance spectroscopy using an Autolab PGSTAT204 (Metrohm, Germany) with a FRA32M Frequency Response Analyser module for the frequency range 500 kHz to 100 Hz (10 mV DC) in 10 °C intervals from 80 °C to −20 °C for the ionic liquid electrolytes, and from −20 °C to 60 °C for the standard carbonate electrolyte. An airtight TSC70 cell (RHD Instruments, Germany) with platinum working and counter electrodes was used to carry out the measurements. The cell was filled with 70 μl of electrolyte in an argon-filled glove box (<0.1 ppm H2O/O2, MBraun, Germany) and closed before being mounted onto a Microcell HC temperature-controlled cell stand (RHD Instruments/Autolab, Germany, accuracy ±0.1 °C). The temperature was ramped up/down at the default instrument speed and kept at each interval temperature for six minutes to stabilize before measurements were performed. The cell constant was determined by measurements on a standard solution of 0.1 M KCl at 20 °C, 30 °C and 40 °C. The cell constant was determined before and after the measurements on the studied electrolytes, and the cell constant used to calculate the ionic conductivity for each electrolyte was determined from linear interpolation between these two values. The resistance (Ω) of the electrolytes was determined from fitted Nyquist plots using a simple RC-circuit.
The limiting current density (ilim) was determined by programmed-current chronopotentiometry using a VMP300 and BSC805 battery testing system (BioLogic Sciences Instruments, France). Symmetrical Li-Li (∅ 14 mm) pouch cells with nickel current collectors were mounted with a GF/A glass fiber separator (260 μm, Whatman) or El-Cell glass fiber separator (1.55 mm), soaked by 80 or 400 μl electrolyte respectively, in an argon-filled glove box. The cells were subjected to a current ramp of 15 μA s−1 from 0 A to 0.1 A with a cut off limit of 10 V at 20 °C and 60 °C. The limited current density was taken as the current value where the voltage increases drastically in the E-I plot (see Figs. S5 and S6).
Cycling performance
Rate test experiments (RT) were performed to measure the capacity retention of the Si-LFP pseudo-full cells at higher currents at 20 °C and 60 °C. Cells were cycled galvanostatically (CC) between 3.40 and 2.40 V for four consecutive cycles at C/20, C/5, C/2, 1 C, 2 C and C/20, with the maximum charge current kept at C/2, where 1 C is taken as 3579 mA gSi −1. The initial cycle (formation) was performed at 20 °C for all cells at C/20, with potential hold steps until the the current reached a value of 50% of the initial, at the end of both charge and discharge. El-Cell® PAT cells were used for the 20 °C rate test while mostly pouch cells (as shown in our previous work 42 ) was used for the 60 °C rate tests. The motivation for the change of cell type was visible signs of corrosion of the stainless-steel plungers in some of the PAT-cells at 60 °C.
Electrochemical impedance spectroscopy
Electrochemical impedance spectra were recorded after galvanostatic cycling at 20 °C and 60 °C, as described above (rate test experiments), for the pseudo full cells in discharged state, i.e. at 2.4 V. The impedance spectra were recorded at a temperature of 20 °C, and in galvanostatic mode, with a current amplitude of 150 μA. The resulting impedance spectra were fitted to an equivalent circuit as depicted in Fig. 2, where Q represents constant phase elements, and W is a finite length Warburg element.

Figure 2. Equivalent circuit used for fitting of the impedance data, with one or two RQ circuits in series with a Randles circuit.
Download figure:
Standard image High-resolution imageThermal behavior of the electrolytes
The thermal behavior of the electrolytes was measured using differential scanning calorimetry (DSC) on a DSC 214 Polyma (Netzsch GmbH, Germany). Electrolyte (≈10 mg) was placed into a dried Al-pan within an Ar-filled glove box (<0.1 ppm H2O/O2, MBraun, Germany) and the pan was sealed by cold pressing an Al-lid onto it. Samples were run through the following program: 20 min of equilibration at 40 °C, dynamic cooling to −120 °C at 2 °C min−1, isothermal hold at −120 °C for 2 min, dynamic heating to 200 °C (100 °C for carbonate) at 10 °C min−1, dynamic cooling to −120 °C at 40 °C min−1, dynamic heating to 200 °C at 40 °C min−1. The program was completed twice for each sample to check reproducibility. Data from the first round of slow cooling and heating are presented. Crystallization (Tc) and melting (Tm) temperatures were taken at the peak of the respective DSC signatures upon cooling and heating, and the glass transition temperature (Tg) was taken at the midpoint of the small heat capacity change recorded upon heating from −120 °C. Peak onset values and additional DSC signature values are presented in Table SI.
Results and Discussion
Thermal phase behavior and stability
DSC was used to investigate the phase transitions of both the neat ILs and the final electrolytes (ILEs) with LiFSI salt. Figure 3 shows the heat flow during cooling and subsequent heating of the neat ILs and the ILEs along with the standard carbonate electrolyte (STD2). Notable signatures such as crystallization temperature ( ), glass transition (
), glass transition ( ) and melting point (
) and melting point ( ) are summarized for the ILEs in Table II and a more complete summary as well as literature values can be found in Tables SI and SII, respectively.
) are summarized for the ILEs in Table II and a more complete summary as well as literature values can be found in Tables SI and SII, respectively.

Figure 3. Differential scanning calorimetry heat flow curves for electrolytes ILE1 (LiFSI:PYR13FSI), ILE2 (LiFSI:PYR13TFSI), ILE3 (LiFSI:EMIFSI), ILE7 (LiFSI:P111i4FSI) and STD2 (LiFSI:EC:DMC:FEC:VC) and the respective pure ionic liquids (faded curves).
Download figure:
Standard image High-resolution imageTable II. Properties of the electrolytes and the μMG-Si∣∣LFP pseudo-full cells studied in the rate performance experiments. Tc and Tm are the temperature values at the peak of the exo- and endothermic DSC signatures, respectively and Tg is the midpoint value. A more complete overview of DSC signatures is given in Table SI. The ± values indicate one standard deviation based on at least two independent measurements, except for the ionic conductivity which is the propagated error of the bulk resistance determined from Nyquist plots.
| Electrolytes | |||||||
|---|---|---|---|---|---|---|---|
| DSC heat flow signatures | Units | ILE1 | ILE2 | ILE3 | ILE7 | STD2 | LP40 |
| Crystallization, Tc | [°C] | — | −14.3 | — | — | −33.2 | — |
| Glass transition, Tg | [°C] | −93.2 | −96.4 | −89.9 | −88.1 | −88.5 | — |
| Melting, Tm | [°C] | — | 10.2 | — | — | −23.4 | — |
| Limiting current density | |||||||
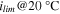 b)
b)
| [mA cm−2] | 8.7 ± 0.9 | 2.1 ± 0.2 | 10.1 ± 0.1 | 6.9 ± 0.1 | 28 c) | 15.3 ± 1.4 |
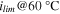 b)
b)
| [mA cm−2] | 9.7 ± 0.2 | 4.7 ± 0.5 | 15.8 ± 1.2 | 10.4 ± 1.8 | — | — |
a)Parameters from fitting the logarithmic VFT equation to conductivity data in an Arrhenius plot, see Section S3. b)See Supplementary Information section S3 for details. c)Only one cell reached ilim. before shortcircuiting, see Fig. S3e) LP40: 1 M LiPF6 in EC:DEC (1:1 v/v)
For the neat IL PYR13FSI, the  is in good agreement with the value reported by Noor et al.,
50
while the
is in good agreement with the value reported by Noor et al.,
50
while the  is between 2 °C to 6 °C higher than reported in other works.
41,50–52
The solid-solid phase transitions at ∼−80 °C are within a few degrees of reported values,
50,52
while the endothermic peak at −16 °C is well in line with literature values.
41,50
Upon addition of 0.77 m LiFSI to PYR13FSI (ILE1), all DSC signatures from the neat IL disappear as the liquid becomes more glass-like, with a
is between 2 °C to 6 °C higher than reported in other works.
41,50–52
The solid-solid phase transitions at ∼−80 °C are within a few degrees of reported values,
50,52
while the endothermic peak at −16 °C is well in line with literature values.
41,50
Upon addition of 0.77 m LiFSI to PYR13FSI (ILE1), all DSC signatures from the neat IL disappear as the liquid becomes more glass-like, with a  of −93.2 °C. No literature values were found for the same composition (0.77 m LiFSI), but Huang and Hollenkamp
52
reported a
of −93.2 °C. No literature values were found for the same composition (0.77 m LiFSI), but Huang and Hollenkamp
52
reported a  of −100 °C for a similar 0.5 m LiFSI in PYR13FSI electrolyte and Paillard et al.
53
a value of −94 °C for a Li0.2(PYR14)0.8FSI. The
of −100 °C for a similar 0.5 m LiFSI in PYR13FSI electrolyte and Paillard et al.
53
a value of −94 °C for a Li0.2(PYR14)0.8FSI. The  is expected to increase with increasing lithium (or other alkaline cation) salt concentration, as has been shown by several authors,
40,53–55
because of increased ion-ion interactions.
is expected to increase with increasing lithium (or other alkaline cation) salt concentration, as has been shown by several authors,
40,53–55
because of increased ion-ion interactions.
The neat PYR13TFSI crystallizes at −17.6 °C upon cooling and no other heat flows are observed before the melting peak upon heating at 14.8 °C. No reports were found on the  during cooling, but a
during cooling, but a  has previously been reported at around −90 °C.
56
Previously reported melting temperatures range from 6 °C to 12 °C,
41,56–58
most of them at the higher temperatures, but still lower than observed in this work. The high purity of the IL in this work could be an explanation. Upon addition of 0.74 m LiFSI to PYR13TFSI (ILE2), the
has previously been reported at around −90 °C.
56
Previously reported melting temperatures range from 6 °C to 12 °C,
41,56–58
most of them at the higher temperatures, but still lower than observed in this work. The high purity of the IL in this work could be an explanation. Upon addition of 0.74 m LiFSI to PYR13TFSI (ILE2), the  shifts to a higher temperature of −14.3 °C, a weak glass transition appears at −96.4 °C and the melting point is lowered to 10.2 °C. In addition, cold-crystallization and melting peaks appear at −42 °C and −25 °C, respectively. No literature values were found for similar compositions to this binary electrolyte. It is peculiar that the crystallization temperature increase upon addition of LiFSI to PYR13TFSI, and that a glass transition temperature appears at lower temperatures than what is reported for the neat IL. The opposite is generally found.
40
shifts to a higher temperature of −14.3 °C, a weak glass transition appears at −96.4 °C and the melting point is lowered to 10.2 °C. In addition, cold-crystallization and melting peaks appear at −42 °C and −25 °C, respectively. No literature values were found for similar compositions to this binary electrolyte. It is peculiar that the crystallization temperature increase upon addition of LiFSI to PYR13TFSI, and that a glass transition temperature appears at lower temperatures than what is reported for the neat IL. The opposite is generally found.
40
Neat EMIFSI crystallizes at −34.4 °C upon cooling and does not show any heat flow before melting is observed at −8.5 °C, in line with results reported by Kerner et al.
40
and Matsumoto et al.
59
The binary electrolyte with LiFSI (ILE3) has no  upon cooling, but a
upon cooling, but a  appears at −89.9 °C and cold-crystallization and melting peaks appear at −52 °C and −25 °C, respectively. Very similar Li0.2EMI0.8FSI electrolytes were investigated by Kerner et al.
40
and Matsumoto et al.
59
who observed
appears at −89.9 °C and cold-crystallization and melting peaks appear at −52 °C and −25 °C, respectively. Very similar Li0.2EMI0.8FSI electrolytes were investigated by Kerner et al.
40
and Matsumoto et al.
59
who observed  's of −97 °C and −91 °C, respectively. They also observed cold-crystallization at −50 °C and melting peaks at −30/−25 °C.
's of −97 °C and −91 °C, respectively. They also observed cold-crystallization at −50 °C and melting peaks at −30/−25 °C.
Neat P111i4FSI crystallizes at −37.0 °C and melts at 15.3 °C, with corresponding exo- and endothermic solid-solid transitions at −100.1/−97.8 °C. Salem et al.
60
and Girard et al.
61
reported slightly lower melting points of ∼12.5 °C, and Girard et al. also reported an endothermic peak at −101 °C. When 0.79 m LiFSI is added (ILE7), no crystallization is seen but a  appears at −88.1 °C. Small solid-solid phase transition and/or cold crystallization and melting peaks are observed at −24.0 °C (exo), −18.2 °C (exo) and −4.6 °C (endo). Girard et al.
61
investigated the heat flow of similar electrolytes with 0.5 m and 1.0 m LiFSI and found
appears at −88.1 °C. Small solid-solid phase transition and/or cold crystallization and melting peaks are observed at −24.0 °C (exo), −18.2 °C (exo) and −4.6 °C (endo). Girard et al.
61
investigated the heat flow of similar electrolytes with 0.5 m and 1.0 m LiFSI and found  at −96 °C and −92 °C and
at −96 °C and −92 °C and  at 5 and −18 °C, respectively. In addition, an exothermic peak was observed at −51.8 °C for the 0.5 m LiFSI electrolyte.
at 5 and −18 °C, respectively. In addition, an exothermic peak was observed at −51.8 °C for the 0.5 m LiFSI electrolyte.
The standard carbonate electrolyte (STD2) had a  of −33.2 °C, a
of −33.2 °C, a  of −88.5 °C and
of −88.5 °C and  of −23.4 °C, although endothermic peaks that can be ascribed to gradual melting of carbonates are observed also at −51.8 °C and −18.3 °C. Large endothermic heat flows are observed from ∼70 °C, which is attributed to evaporation of the carbonates. This demonstrates one weakness of carbonate electrolytes, their high vapor pressure, which can be problematic in volume-constrained applications such as LIBs. The neat ILs and their binary electrolyte mixtures with LiFSI can, on the other hand, be seen to not exhibit any change in heat flow upon heating to 200 °C, the limit of these experiments.
of −23.4 °C, although endothermic peaks that can be ascribed to gradual melting of carbonates are observed also at −51.8 °C and −18.3 °C. Large endothermic heat flows are observed from ∼70 °C, which is attributed to evaporation of the carbonates. This demonstrates one weakness of carbonate electrolytes, their high vapor pressure, which can be problematic in volume-constrained applications such as LIBs. The neat ILs and their binary electrolyte mixtures with LiFSI can, on the other hand, be seen to not exhibit any change in heat flow upon heating to 200 °C, the limit of these experiments.
Observed heat signatures for the neat ILs and electrolytes investigated in this work are generally in good agreement with those available in the literature, although the observed heat flows tend to occur at higher temperatures than what had previously been reported, both upon cooling and heating. Such deviations are likely caused by differences in the purity of both the neat ILs and the LiFSI salt (purer substances generally have higher melting points), the LiFSI concentration of the ILEs, or differences in the measurement conditions and procedure (e.g. open/closed pan, volume of sample, rate of temperature change, thermal history).
From lowest to highest  the electrolytes rank ILE2 < ILE1 < ILE3 < STD2 < ILE7. A lower
the electrolytes rank ILE2 < ILE1 < ILE3 < STD2 < ILE7. A lower  generally implies lower viscosities at room temperature, and the viscosity is correlated to the conductivity of ILs, even if there are no simple relations.
62
This implication does not hold true for the electrolytes studied herein, as will be elucidated in the next section on ionic conductivity. The low
generally implies lower viscosities at room temperature, and the viscosity is correlated to the conductivity of ILs, even if there are no simple relations.
62
This implication does not hold true for the electrolytes studied herein, as will be elucidated in the next section on ionic conductivity. The low  of ILE2 is especially surprising, considering it is by far the least conductive electrolyte at all temperatures. All ILEs, except ILE2, have wide liquid ranges which is important for the applicability in batteries. The superior stability of the ILEs at high temperatures compared to the carbonate electrolyte (STD2) is clearly seen in Fig. 3.
of ILE2 is especially surprising, considering it is by far the least conductive electrolyte at all temperatures. All ILEs, except ILE2, have wide liquid ranges which is important for the applicability in batteries. The superior stability of the ILEs at high temperatures compared to the carbonate electrolyte (STD2) is clearly seen in Fig. 3.
The higher crystallinity of ILE2 compared to ILE1 illustrates the effect of the different anion in these otherwise similar electrolytes. The larger TFSI− anion is known to have stronger interactions with cations (Li+ and IL cation) than FSI−, which can probably explain this difference. 63,64
Electrolyte transport properties
Figure 4 shows an Arrhenius plot of the total conductivity of electrolytes ILE1, ILE2, ILE3, ILE7 and STD2 in the temperature range 80 °C to −20 °C. Measurements were conducted in slightly narrower temperature range for ILE2 (0–80 °C) and STD2 (−20–60 °C) to ensure measurements on a homogeneous mixture, based on the phase changes observed by DSC. All values are provided in Table SIII.

Figure 4. Arrhenius plots of the total ion conductivity of electrolytes ILE1 (LiFSI:PYR13FSI), ILE2 (LiFSI:PYR13TFSI), ILE3 (LiFSI:EMIFSI), ILE7 (LiFSI:P111i4FSI) and STD2 (LiFSI:EC:DMC:FEC:VC). Coloured solid lines represent the best fit of the logarithmic VFT equation and dotted lines a linear fit. Parameters for both fittings can be found in Table SIV.
Download figure:
Standard image High-resolution imageThe highest room temperature conductivity was found for the carbonate electrolyte (STD2) at 12.2 mS cm−1, with the imidazolium based ionic liquid (ILE3) achieving only a slightly lower conductivity of 10.5 mS cm−1 and the other ILEs had conductivities between 1 and 5 mS cm−1. At 60 °C, the conductivity of the electrolytes ILE1, ILE2, ILE3, ILE7 and STD2 were 142, 225, 130, 164 and 67% higher than at 20 °C, respectively. In general, the conductivity of the electrolytes follows the trend STD2 > ILE3 > ILE1 ≥ ILE7 > ILE2 in the measured temperature range, with the exception being that above 40 °C, ILE3 outperforms STD2. The trend in conductivity between the electrolytes does not follow the trend in glass transition temperatures from the DSC measurements.
The temperature dependency of the conductivity in 4 exhibit a convex curved profile, and were therefore fitted to a Vogel-Fulcher-Tammann (VFT) type equation: 65–67
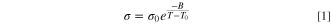
Or rather, its logarithmic version
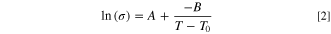
and the calculated values of parameters 
 and
and  are given in Table SIV.
are given in Table SIV.
The preexponential factor  (or
(or  ) is related to the number of charge carriers, B is related to the activation energy for conduction (pseudo-activation energy) and
) is related to the number of charge carriers, B is related to the activation energy for conduction (pseudo-activation energy) and  is the VFT temperature of structural arrest/onset of motion, often called the ideal glass transition temperature (a non-crystalline state of lowest energy).
68,69
The VFT equation describes the deviation of a temperature dependent transport property (like ionic conductivity) from the well-known Arrhenius equation (Eq. 3), which is often the case at "low temperatures,"
is the VFT temperature of structural arrest/onset of motion, often called the ideal glass transition temperature (a non-crystalline state of lowest energy).
68,69
The VFT equation describes the deviation of a temperature dependent transport property (like ionic conductivity) from the well-known Arrhenius equation (Eq. 3), which is often the case at "low temperatures," 
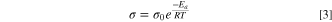
Angell proposed a classification of liquids along a "strong" to "fragile" scale, where a strong liquid is close to Arrhenius behavior, whereas a fragile liquid will deviate from the Arrhenius equation showing a curved line. The physical meaning of fragility is the ability of a substance to structurally reorganize upon a change in temperature; more fragile means more rapid restructuring.
70
A parameter  called the Angell Strength parameter, is often used as a relative measure of fragility, where a
called the Angell Strength parameter, is often used as a relative measure of fragility, where a  is taken to represent a fragile liquid.
40
is taken to represent a fragile liquid.
40
 describes the sharpness of the deviation from Arrhenius behavior, where a value of D equal to infinity shows Arrhenius behaviour.
71
The D value is given in Table SIV for the studied electrolytes.
describes the sharpness of the deviation from Arrhenius behavior, where a value of D equal to infinity shows Arrhenius behaviour.
71
The D value is given in Table SIV for the studied electrolytes.
Empirically,  is expected to be approximated fairly well by
is expected to be approximated fairly well by  43
meaning that for systems like the ones studied in this work with
43
meaning that for systems like the ones studied in this work with 
 is expected to be around 140 K. This is not the case for most of the ILEs studied (Table SIV), although ILE2 and ILE3 are closer to the expected value than ILE1 and ILE7. This implies that the temperature dependency of conductivity is different from the one expected for such liquids, following a trend with a lower degree of "curvature" in the Arrhenius plot than what would be expected, or that the approximation
is expected to be around 140 K. This is not the case for most of the ILEs studied (Table SIV), although ILE2 and ILE3 are closer to the expected value than ILE1 and ILE7. This implies that the temperature dependency of conductivity is different from the one expected for such liquids, following a trend with a lower degree of "curvature" in the Arrhenius plot than what would be expected, or that the approximation  is a poor one for these ILEs.
is a poor one for these ILEs.
The slightly higher degree of linearity in the conductivity data reported here is also evident when comparing the results to other literature data, such as data on pyrrolidinium based electrolytes similar to ILE1 by Rüther et al. 72 and imidazolium based electrolytes similar to ILE3 presented by Matsumoto et al. 59 and Kerner et al. 40 (Fig. S2). The deviation seen in the data and corresponding VFT fits in this work from that in the literature is probably explained by minor differences in composition or purity of the electrolytes and differences in the experimental setup, as well as the investigated temperature region.
The standard carbonate electrolyte (STD2) is clearly not well described by the parameters of the VFT equation (unphysical negative value of  ), and the Arrhenius equation is better suited. The logarithmic Arrhenius equation was fitted to the conductivity data of all electrolytes as an alternative to the VFT equation, providing approximate values for the activation energy (
), and the Arrhenius equation is better suited. The logarithmic Arrhenius equation was fitted to the conductivity data of all electrolytes as an alternative to the VFT equation, providing approximate values for the activation energy ( ) for conduction in the investigated temperature region. The electrolytes are ranked, from highest to lowest
) for conduction in the investigated temperature region. The electrolytes are ranked, from highest to lowest  ILE2 > ILE7 > ILE1 > ILE3 > STD2.
ILE2 > ILE7 > ILE1 > ILE3 > STD2.
These results show that the ionic liquid electrolytes generally have a much lower ionic conductivity than the standard carbonate electrolyte, except for imidazolium based ILE. However, ionic conductivities significantly above 1 mS cm−1, could be achieved in a quite broad temperature range for all the FSI− based ILEs. This is relevant as  is often deemed a requirement for LIB applications,
73
although in these electrolytes
is often deemed a requirement for LIB applications,
73
although in these electrolytes  because the lithium ion transport number can be expected to be lower than unity. At moderately high temperatures (∼60 °C), the pyrrolidinium-FSI (ILE1) and phosphonium (ILE7) electrolytes achieve ionic conductivities comparable to the room temperature conductivity of STD2, while the imidazolium (ILE3) electrolyte exceeds it. The beneficial effect of temperature on electrolyte conductivity combined with the higher temperature stability (3) means that operation at higher temperatures might enable the use of these ILEs in LIBs without sacrificing rate capability.
because the lithium ion transport number can be expected to be lower than unity. At moderately high temperatures (∼60 °C), the pyrrolidinium-FSI (ILE1) and phosphonium (ILE7) electrolytes achieve ionic conductivities comparable to the room temperature conductivity of STD2, while the imidazolium (ILE3) electrolyte exceeds it. The beneficial effect of temperature on electrolyte conductivity combined with the higher temperature stability (3) means that operation at higher temperatures might enable the use of these ILEs in LIBs without sacrificing rate capability.
As indicated above, it is not the total ionic conductivity that determines the electrolyte's effect on the overall rate capability of a battery, but the conductivity/mobility of the Li+ ions. As a means of quantifying the lithium mobility of the electrolytes, programmed-current chronopotentiometry was performed on Li∣∣Li cells at 20 °C and 60 °C. The limiting current density is taken as the value at the onset of the large polarization of the cell, as seen in Figs. S3 and S4, with values summarized in Tables SV and SVI.
Assuming effects like lithium plating/dissolution are not limiting, the limiting current density will correspond to the maximum lithium ion flux that can be obtained under the potential applied, and can thus be taken as a relative measure of lithium ion mobility in the different electrolytes.
74
This is a reasonable approximation if the thickness of the Nernst diffusion layers are the same between electrolyte systems. The measured values of the limiting current densities do not differ significantly and not according to a  relationship
75,76
for separators of different thickness (i.e. l = 260 μm and 1.55 mm give very similar values for ilim). This indicates that the Nernst diffusion layers are thinner than 260 μm. ilim values are therefore compared directly between cells with differing separator thickness.
relationship
75,76
for separators of different thickness (i.e. l = 260 μm and 1.55 mm give very similar values for ilim). This indicates that the Nernst diffusion layers are thinner than 260 μm. ilim values are therefore compared directly between cells with differing separator thickness.
Figure 5 presents the relative mobility values of all electrolytes, where the values are normalized by the limiting current density of a regular "LP40" carbonate electrolyte at 20 °C. The ILEs have significantly lower Li+ mobilities than both the LP40 and STD2 carbonate electrolytes at 20 °C. For STD2 (LiFSI in carbonate), although uncertain (cell prone to shorts, see Fig. S3e), the limiting current is considerably higher than for LP40 (LiPF6 in carbonate), in line with the better conductivity and rate performance of LiFSI as reported by Han et al. 39

Figure 5. Relative mobility values for electrolytes LP40, ILE1 (LiFSI:PYR13FSI), ILE2 (LiFSI:PYR13TFSI), ILE3 (LiFSI:EMIFSI), ILE7 (LiFSI:P111i4FSI) and STD2 (LiFSI:EC:DMC:FEC:VC). Values are calculated by normalizing the mean limiting current found in Li∣Li cells with programmed current derivative chronopotentiometry by the mean limiting current of the commercial carbonate electrolyte LP40. Details and E-I plots are given in the supplementary information.
Download figure:
Standard image High-resolution imageIt is worth noting that although the order of relative Li+ mobility between the ILEs is the same as for the total ionic conductivity (ILE3 > ILE1 > ILE7 > ILE2), the differences between the relative Li+ mobilities of the electrolytes are smaller than the differences in total ionic conductivities at 20 °C. This applies in particular to ILE1 which has a relative Li+ mobility very close to ILE3. The results indicate that the lithium transference number might be higher in the ILEs with pyrrolidinium or phosphonium cations compared to imidazolium at this temperature. At 60 °C, the ILEs all show an increase in relative Li+ mobility compared to 20 °C which is largest for ILE2 with a >120% increase, significant for ILE3 and ILE7 with >50% increase and a statistically insignificant 11% increase for ILE1. The relative Li+ mobility of ILE3 at 60 °C is on par with the LP40 electrolyte at room temperature.
Looking solely at the contribution of the transport properties of the electrolytes to the overall rate performance of a LIB cell, cells with electrolytes in the order STD2, ILE3, ILE1, ILE7 and ILE2 at room temperature are expected to perform the best (highest retained capacity at higher rates). The improved transport properties for the ILEs at higher temperatures indicates that an improvement in the rate performance could be possible, but this is of course dependent on other effects of increased temperature such as e.g. increased decomposition of electrolyte and electrodes. The next section presents results from rate tests in LIB pseudo-full cells where this is illuminated.
Rate performance
Figure 6 presents the discharge capacity and coulombic efficiency (CE) vs cycle data from rate tests of μSi∣∣LFP pseudo-full cells with all electrolytes at both 20 °C (a) and 60 °C (b). ILE2 was not rate tested at 20 °C as it could barely achieve any reversible capacity even at a C/20 rate. As stated in Ref. 42, the ILE2 electrolyte has a high viscosity compared to the other electrolytes, which is typical for TFSI-based ionic liquids. The poor capacity of the anodes achieved at 20 °C in Ref. 42 (only 217 mAh g−1 could be achieved during the first cycle at C/20) was attributed to the poor conductivity, as is also shown in Fig. 4, in combination with poor wetting of the electrode. The latter was evident based on the low capacitance of electrodes in ILE2, measured by impedance spectroscopy. 42

Figure 6. Rate test cycling of pseudo-full cells of μMG-Si (≈2.68 mAh cm−2, ∅ 12 mm) vs LFP (≈3.5 mAh cm−2, ∅ 14 mm) with electrolytes ILE1 (■), ILE2 (●), ILE3 (▲), ILE7 (◆) and STD2 (★) at a) 20 °C and b) 60 °C. Galvanostatic cycling program between 2.4 and 3.4 V (≈1.05–0.05 V on silicon) with rates as depicted. First cycle with a constant voltage step at the end of both charge and discharge until the current is halved. *First cycle in b) performed at 20 °C. Capacities shown are the average discharge capacities of at least three cells shown with error bars representing one standard deviation. ILE2 was not rate tested at 20 °C because of poor performance even at lower rates.
Download figure:
Standard image High-resolution imageAt 20 °C
All cells achieve initial capacities in the vicinity of 3000 mAh g−1 at C/20, but the carbonate and imidazolium electrolytes (STD2 and ILE3, respectively) have a markedly more rapid decay in capacity in the following cycles at this rate, compared to ILE1 and ILE7. A lower CE is observed, indicating that more severe electrolyte degradation and active material isolation/insulation might be responsible for the rapid capacity loss.
At a higher rate of C/5 the capacity falls by a significant amount, ordering the cell performance as predicted from the conductivity and relative mobility experiments on the electrolytes; STD2 > ILE3 > ILE1 > ILE7(>ILE2). The cells still deliver from 1500 to 2000 mAh gSi −1. At the next rate step of C/2 a real difference between the carbonate and ionic liquid electrolytes is observed. While the cell with STD2 maintains >1500 mAh/gSi, ILE3 falls to ∼700 mAh/gSi, and ILE1 and ILE7 loses almost all reversible capacity only maintaining ∼200 mAh/gSi. Stable CEs of ∼99% are seen for STD2, while it is over 100% for the ILEs, indicating a release of trapped lithium from the previous cycles upon delithiation.
At the high discharge rates of 1 C and 2 C the cells with STD2 only loses a few hundred mAh/gSi, still achieving >1250 mAh/gSi, while ILE3 falls to 500 mAh/gSi and ILE1 and ILE7 stabilize slightly below 200 mAh/gSi. Upon returning to a slow rate (C/20), the cells with ILEs regain capacities of over 2500 mAh/gSi, while STD2 achieves between 2200–2000 mAh/gSi. The lower capacity retention of cells with STD2 is likely explained by higher degradation connected to the larger utilization of the active material during the high rate cycles.
The cumulative capacity loss (CCL) during the rate testing program at 20 °C is presented in Fig. 7a). The graph shows the higher degradation seen for STD2 and ILE3 during the initial slow cycles and a stabilization of the losses for the cells with ILEs as the reversible capacity falls at higher rates. The overall losses are below 2000 mAh/gSi. Active lithium loss is always lower than the CCL as shown by Holtstiege et al. 77 due to parasitic reactions not involving lithium. In addition lithium can be trapped both irreversibly (within the SEI, isolated active material or plated) and kinetically (within active material, but unavailable under a certain rate), both contributing to the CCL. 78,79 The active lithium loss in these cells is therefore considered well within the capacity of the oversized LFP, ruling out any negative effects from loss of lithium inventory. Further details regarding the cycling at 20 °C is provided in Rogstad et al. 42

Figure 7. Cumulative capacity loss (CCL) for rate cycling of pseudo-full cells of μMG-Si (≈2.68 mAh cm−2, ∅ 12 mm) vs LFP (≈3.5 mAh cm−2, ∅ 14 mm) with electrolytes ILE1 (■), ILE2 (●), ILE3 (▲), ILE7 (◆) and STD2 (★) at a) 20 °C and b) 60 °C. CCCV program between 2.4 and 3.4 V (≈1.05–0.05 V on silicon) with a rate of C/5 except for the first and every tenth/twentieth cycle, at C/20, with a constant voltage step at both charge and discharge until the current is halved. Average AIC of at least three cells shown with error bars representing one standard deviation.
Download figure:
Standard image High-resolution imageAt 60 °C
Figure 6b) presents data from the rate tests at 60 °C. The initial (formation) cycle was performed at 20 °C to allow for the formation of an SEI equal to the cells rate tested at 20 °C (Fig. 6a). The lower initial capacity for cells with ILE1 and ILE7 in b) compared to a) is likely due to a slight difference in wetting caused by pressure differences between the pouch-cells used for the 60 °C experiments and the PAT-cells used for the 20 °C experiments. The large standard deviation is also an indication of inhomogeneous wetting. Capacities and CEs are however mostly consistent with a), not differing in a statistically significant manner.
From the first C/20-cycle at 60 °C, the temperature has a major effect on the reversible capacity of the cells. Compared to cycling at 20 °C, only the cell with ILE7 can maintain similarly high capacities around 3000 mAh/gSi, although it is reduced to 2500 mAh/gSi within the first four cycles. ILE2 follows with impressive capacities around 2500 mAh gSi −1, up from only 500 mAh/gSi at room temperature. This is likely explained by the improved wetting of the electrodes at this temperature and the improved Li+ transport properties of the electrolyte. 44 The cells with STD2 comes in with the third highest capacities but shows by far the largest capacity loss during the four cycles. Cells with ILE1 and ILE3 only achieve modest, but "stable" capacities around 1750 mAh/gSi, down by over 1000 mAh gSi −1 compared to the slow cycling at 20 °C. Looking at the CE (full CE range in Fig. S5), it is evident that ILE3 has the overall lowest CE during the cycles at C/20 (<80%), with STD2 also performing worse than the rest of the electrolytes. All electrolytes have lower CEs than the corresponding cycling at 20 °C.
At a C/5 rate, the performance difference between cells with ILE7 and the other electrolytes becomes clearer. Cells with ILE7 maintain capacities only slightly below 2000 mAh/gSi, a significant improvement over the performance at 20 °C and higher than cells with the other electrolytes, achieving between 1500 and 750 mAh/gSi with large and overlapping standard deviations. The CE is improving at this rate and is more similar between electrolyte systems.
At C/2 the capacity of all cells drops further, with ILE7 still performing the best (∼1000 mAh/gSi). Cells with ILE1 and ILE2 have CEs above 100%, indicating release of trapped lithium, which is not the case for the other electrolytes. At the higher rates of 1 C and 2 C the capacity is further lowered, and CEs are quite stable around 97%–99%, except for cells with ILE2 where the CE drops significantly at 2 C. Upon comparison of the rate performance at 20 °C and 60 °C, it may be noted that the capacity with ILE7 is still high at 2 C (close to 1000 mAh/gSi), implying a significant increase from around 250 mAh/gSi at 20 °C. The improvement of the rate performance for ILE3 is less (increase from around 500 to 650 mAh/gSi), while for ILE1, there is only a moderate increase (from ca 250 to 400 mAh gSi −1). For NMC811 half cells, 44 a dramatic increase was observed for the rate performance in combination with an electrolyte of 1.2 M LiFSI and Pyr13FSI (ILE1) (from ∼60 mAh g−1 to ∼220 mAh g−1), suggesting that this system experience a quite different temperature effect on the electrode resistance compared to the system studied here. In a recent work of Araño et al., 32 good rate performance was demonstrated for highly concentrated LiFSI salts (3.2 m) in triethyl (methyl) phosphonium (P1222FSI) and Pyr13FSI at 50 °C and C/2 for Li/Si cells, as has also been observed for highly concentrated LiFSI (3.2 m) in P111i4FSI. In full cells, with NCA cathodes, the silicon electrodes delivered around 600 mAh gSi −1 at 2 C rate and 50 °C. In this work the good performance was attributed to a combination of high Li+ transport number in the electrolyte as well as the formation of a relatively stable SEI in the highly concentrated LiFSI ionic liquid.
On further cycling of these cells at the slow rate of C/20, the cells all have CEs over 100% for the first cycle, again indicating release of trapped lithium. Cells with ILE7 and ILE2 regain capacities around 2000 mAh gSi −1, cells with ILE1 and ILE3 around 1500 mAh gSi −1 and the carbonate cells around 1000 mAh gSi −1. The CEs are low (∼90%–95%), and a larger decay in capacity can be seen with each cycle compared to the higher rates, even at similar capacities. It is evident that the higher rate of decay is more dependent on cycling rate than degree of lithiation of the active material (volume change). Slower rates allow time for all reactions to happen, including parasitic side reactions that do and do not consume lithium (SEI formation, electropolymerization). The combination of slow rates and a higher temperature seem to be detrimental to the cells, leading to a poorer cycling stability for these cells compared to those cycled at 20 °C.
Similar findings were reported by Smith et al. 80,81 for different graphite∣∣LiM(P)Ox (M = Co, Fe, Mn) cells as well, where it was concluded that time of exposure, not cycle count, becomes the dominant contributor to degradation at higher temperatures. Thus, the apparent higher degradation and lower CEs for cycles performed at lower C-rates compared to at the higher rates is to be expected. The exact nature of the degradation reactions is not known and could in principle involve all components in the cell, including electrolyte, active material, conductive additives, SEI layers and binder. Rodrigues et al. cycled graphite electrodes at even higher temperatures (90 and 120 °C) with LiTFSI in Pyr13TFSI, and reported coloumbic efficiencies of 99.4% at 90 °C at a rate of C/5. 47 This is higher than CEs obtained with the FSI-based system studied here, indicating possibly even better stability for LiTFSI-based salts.
The effect of the accelerated side reactions at 60 °C can be seen in the overall CCL in Fig. 7b), with losses up to three times higher than for cycling at 20 °C. Cells with ILE3 have the highest losses (4000 mAh gSi −1) after the rate test and cells with ILE1 have the lowest (2000 mAh gSi −1). Still, the losses do not amount to more than the available lithium in the LFP cathode, thus excluding negative effects of lost lithium inventory also at this temperature.
It can be expected that the side reactions and corresponding large losses lead to higher internal resistance in the cells. Fig. S8 shows Nyquist plots of electrochemical impedance spectroscopy (EIS) performed on cells after the rate test program at both 20 °C and 60 °C. Based on the impedance data, the total electrode resistance was evaluated by fitting to the equivalent circuit shown in Fig. 2, and results are shown in Table III for the ILE1, ILE3 and ILE7 ionic liquid electrolytes. After cycling at 20 °C, the total electrode resistance is very similar for the ionic liquid electrolytes, and also higher than for the carbonate electrolyte, which is around 12 Ohm. In spite of some scatter in the experimental data, the total electrode resistance after cycling at 60 °C is clearly significantly lower for the phosphonium electrolyte (ILE7) compared to ILE1 and ILE3, which are experiencing a significant increase.
Table III. Total electrode resistance evaluated based on fitting to an equivalent circuit as depicted in Fig. 2.
| Electrolyte | Total electrode resistance after cycling at 20 °C [Ω] | Total electrode resistance after cycling at 60 °C [Ω] |
|---|---|---|
| ILE1 | 58.5 ± 17.9 a) | 224.5 ± 66.3 a) |
| ILE3 | 73.9 ± 13.9 b) | 265.1 ± 14.1 b) |
| ILE7 | 60.5 ± 20.6 b) | 123.9 ± 11.5 c) |
a)Average of 4 cells. b)Average of 2 cells. c)Average of 3 cells.
The most surprising result from the high temperature rate test is the apparent superiority of ILE7, especially compared to ILE1. The two electrolytes performed very similarly at 20 °C, but at 60 °C the phosphonium electrolyte (ILE7) achieved a much greater rate performance. This is not explained by the increase in ionic conductivity with temperature.
Figure 4, but ILE7 does have a larger gain in relative Li+ mobility (Fig. 5) which might be part of the explanation. A more thorough investigation of the Li+ transport properties as a function of temperature for these electrolytes should be undertaken. The EIS data (Table III, Fig. S8) from cycled cells indicates that cells cycled with ILE7 have lower charge transfer and SEI resistances than cells cycled with ILE1. Seemingly higher CEs for cells with ILE7 (although overlapping standard deviations prevent a firm conclusion on this) indicates that the passivating property of the SEI in these cells is not compromised to the same degree as for cells with the other electrolytes. A less resistive (to Li+ transport) SEI combined with improved Li+ mobility in the electrolyte is therefore the probable explanation for the superior rate performance of cells with ILE7.
Figure 8 shows differential capacity plots of the third cycle of each C-rate for both the cells tested at 20 °C and 60 °C. The differential capacity is plotted against the calculated silicon electrode potential, assuming  This assumption was tested by cycling three-electrode cells at 20 °C (see Figs. S6 and S7) which showed deviations to mostly be within ∼0.05 V in the relevant voltage range (around the peaks). Peaks assignments corresponding to the different lithiation (charge) and delithiation (discharge) reactions between lithium and silicon are adapted from the work by Ogata et al.
82
to the pseudo-full cells in this work, and are summarized in Table IV.
This assumption was tested by cycling three-electrode cells at 20 °C (see Figs. S6 and S7) which showed deviations to mostly be within ∼0.05 V in the relevant voltage range (around the peaks). Peaks assignments corresponding to the different lithiation (charge) and delithiation (discharge) reactions between lithium and silicon are adapted from the work by Ogata et al.
82
to the pseudo-full cells in this work, and are summarized in Table IV.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Differential capacity plots of pseudo-full cell rate test cycling of μMG-Si electrode with an LFP cathode and electrolytes (a) ILE1 (LiFSI:PYR13FSI), (b) ILE2 (LiFSI: PYR13TFSI), (c) ILE3 (LiFSI:EMIFSI), (d) ILE7 (LiFSI:P111i4FSI) and (e) STD2 (LiFSI:EC:DMC:FEC:VC) at −1) 20 °C and −2) 60 °C. ILE2 was not rate tested at 20 °C because of poor performance even at C/20. C∣D = Charge∣Discharge. Data chosen from the individual cell for each electrolyte that was most representative of the average cell data shown in Fig. 6. The silicon electrode potential (EWE = ESi) was calculated from the cell voltage assuming a constant LFP electrode potential of 3.45 V (vs Li/Li+). The accuracy of this approximation will vary with electrolyte and rate, and it will be more inaccurate at low potentials on the LFP electrode (high on Si) due to polarization of the LFP (see Figs. S5 and S6). Arrows of similar color within each subfigure are guides to the eye to follow the development of corresponding lithiation and delithiation peaks from low to high C-rate (e.g. orange following c3/d2 and green following c2/d4 in Fig. a1), whereas black arrows crossed by an "x" indicate where there is no further (de)lithiation of silicon (peaks "disappear" at higher rate). Peak assignments c2, c3, c4, d2, d3 and d4 are adapted to the pseudo-full cells used in this work from the excellent work by Ogata et al. 82 and correspond to the reaction processes summarized in Table IV.
Download figure:
Standard image High-resolution image{kind=link}
Table IV. Differential capacity plot peak assignments corresponding to the different alloying and dealloying reactions between lithium and silicon. Adapted from the work by Ogata et al. 82
| Peak name | Reaction | Peak name | Reaction | ||
|---|---|---|---|---|---|
| c1 |

| d4 |
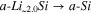
| ||
| Lithiation ↓ | c2 |

| Delithiation ↑ | d3 |

|
| c3 |

| d2 |

| ||
| c4 |

| d1' |
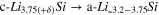
| ||
| d1 |
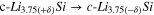
|
c-Si: Crystalline silicon, a-Si: Amorphous silicon.
When comparing the differential capacity curves from cells cycled at 20 °C and 60 °C, several differences are evident. In general, cells cycled at 60 °C have:
- i.Lower overpotentials (i.e. ∼20–30 mV at C/20).
- ii.A more pronounced d3 peak.
- iii.Poorer capacity at slow rates and poorer capacity retention over the course of the program (as also seen in Fig. 6).
The lower overpotentials seen at C/20 (i) is explained by the improved Li+ transport properties in the electrolytes at higher temperatures (figure and figure ) and better wetting of the electrodes.
44
This means that lithiation to the higher lithiated silicide phases (characterized by the c3/d2 and c4/d3 peaks) is now to a larger extent available within the voltage operating window. The presence of the highly lithiated crystalline silicide phase  (c4) is especially noteworthy. The delithiation of this phase (
(c4) is especially noteworthy. The delithiation of this phase ( ) has a high overpotential, leading to the characteristic d3 peak around 0.45 V (ii). This asymmetric delithiation reaction is associated with large inhomogeneous volume changes of the silicide as the two-phase reaction proceeds,
82,83
which is more detrimental to particle integrity and the SEI than the symmetric c2/d4 and c3/d2 reactions. The presence of this reaction can thus be part of the explanation for the poorer cycle stability observed in cells with ILE1, ILE3, ILE7 and STD2 compared to ILE2, and compared to cycling at 20 °C, as elucidated below.
) has a high overpotential, leading to the characteristic d3 peak around 0.45 V (ii). This asymmetric delithiation reaction is associated with large inhomogeneous volume changes of the silicide as the two-phase reaction proceeds,
82,83
which is more detrimental to particle integrity and the SEI than the symmetric c2/d4 and c3/d2 reactions. The presence of this reaction can thus be part of the explanation for the poorer cycle stability observed in cells with ILE1, ILE3, ILE7 and STD2 compared to ILE2, and compared to cycling at 20 °C, as elucidated below.
When comparing the C/20 cycle from early cycling (3rd cycle) to the C/20 cycle towards the end of the program (23rd cycle) for the cells at 60 °C, the peak positions change very little, but the height is generally reduced. This indicates that for most cells, the internal resistance is not substantially changed from the 3rd to the 23rd cycle, but there is loss of active material (iii). There are some notable exceptions to this:
- 1.Cells with ILE2 see lower overpotentials for all peaks, both on lithiation and delithiation, and while the area under the c2/d4 peaks is reduced, it is increased for the c3/d2 peaks. The lower overpotentials indicate lower internal cell resistance at the end of the rate test than in the beginning and is likely explained by better wetting of the electrode as well as a more amorphous and available silicon structure. The reduced c2/d4 and increased c3/d2 peak intensities means there is a reduction in lithium poor silicides and an increase in lithium rich silicides.
- 2.Cells with STD2 have the c2 and c3 peaks shifted to slightly lower potentials (from the 3rd to the 23rd cycle), meaning there is an increase in the internal resistance large enough to shift the potential of the lithiation reactions even at this slow rate (C/20). The presence of a highly resistive SEI is supported by the impedance response of the cell after cycling (Fig. S8e–2).
Regarding the previously discussed difference in rate performance between cells cycled with electrolytes ILE1 and ILE7, it can now clearly be seen that cells with ILE7 have a substantially lower overpotential at higher rates (8 a-2 vs d-2).
The cells were previously characterized by XPS after one cycle in the same electrolytes. 42 One of the most evident differences between the spectra was the higher fraction of Si found for the ILE7 electrolyte, taken as an indication of a thinner SEI for this electrolyte. Furthermore, higher fractions of N, S and F were observed in the spectra of both ILE and ILE3, as compared to ILE 7, with the highest fractions found for the ILE3 electrolyte. The ratio of N/S was also the highest for this electrolyte. This is a strong indication of reductive decomposition of the EMI+ cation, which is supported by the presence of a significant peak in the differential capacity plot around 0.6 V. 42 Reductive decomposition of EMI has also been reported in other works with 0.5 M LiFSI in EMIFSI in combination with graphite at 0.6 V vs Li/Li+, 84 and at a slightly higher voltage (0.9 V vs Li/Li+) in combination with metallic Li. 37
There is no conclusive evidence of decomposition of the cations of the ILE1 or ILE7 electrolytes. Excellent stability has also previously been demonstrated for the P111i4 + cation, with an electrochemical reduction potential of around −0.1 V vs Li/Li+. 61 This work showed also, through a combination of FTIR and XPS measurements, that undecomposed P111i4 + cations were incorporated into the SEI of the Li metal anodes. In Ref. 42, the same P2p feature, corresponding to undecomposed P111i4, was apparent in the XPS spectra of the Si electrode, thus indicating the incorporation of the cation in the SEI layer.
As seen in Ref. 42, the C1s spectra for the pristine Si electrode has a very distinct peak at 286.7 eV, attributed to the binding energy of an O–C–O feature, which must originate from the binder. For all electrodes cycled in the ionic liquids, this particular feature is significantly reduced compared to the other C1s features, and even more so for the ILE7 electrolyte. The exact reason for this is not known, but might be an indication of surface reactions involving the binder.
To summarize the findings for the μMG-Si vs LFP pseudo full cells: At room temperature, the ionic liquid electrolytes are only viable at low rates between C/20 and C/5, where discharge capacities of between 3000 and 1500 mAh/gSi could be achieved. At higher rates up to a 2 C discharge, only the standard carbonate electrolyte (STD2) could retain useable capacities >1200 mAh/gSi. Among the ILEs, the imidazolium-FSI (ILE3) had the best rate performance. The performance of the cells could be explained mainly by the Li+ transport properties of the electrolytes.
At 60 °C, cells cycled with the phosphonium-FSI electrolyte (ILE7) performed the best at all rates, delivering discharge capacities of around 1000 mAh/gSi even at a 2 C rate. This could be explained mainly by lower polarization of the cell and lower loss of active material thought to originate from a more stable and ion conductive SEI. In addition, the fact that both the coulombic efficiency and the discharge capacity are higher for this electrolyte clearly indicates a better passivating SEI. In addition, the Li+ transport properties are also the highest for this electrolyte at the elevated temperature.
ILE2 becomes more viable as an electrolyte at this temperature, but only at a C/20 rate, where discharge capacities around 2500 mAh/gSi can be obtained. The apparent stability of the cells with ILE2 can mainly be explained by the low utilization of the active material during high C-rate cycling, but it is probable that the presence of the thermally and electrochemically more stable TFSI− anion positively impacts the cycling stability as well. 85
Conclusions
Four selected ILs were investigated for their thermal stability, and binary mixtures with LiFSI were investigated for their temperature-dependent ionic conductivity and Li+ mobility. The ionic liquids were based on pyrrolidinium, imidazolium and phosphonium cations and bis(fluorosulfonyl)imide (FSI) and bis(trifluoromethanesulfonyl)imide (TFSI) anions.
The electrochemical performance of these ionic liquid electrolytes was tested in cells with a micrometer-sized metallurgical grade silicon (μMG-Si) anode and a capacitively oversized LFP cathode at 20 °C and 60 °C, with a state-of-the-art carbonate electrolyte as reference.
The ionic liquid electrolytes were stable up to at least 200 °C in dynamic differential scanning calorimetry, whereas the carbonate electrolyte started evaporating at ∼70 °C. FSI− based ionic liquid electrolytes behaved more glass like than the TFSI− based ionic liquid electrolytes, and the carbonate electrolyte with no crystallization upon cooling, thus displaying very wide liquid ranges which can be important for applications.
The total ionic conductivities of the ILEs follow a VFT temperature relation while the carbonate electrolyte follows an Arrhenius relation. Significant differences in the apparent activation energies were observed among the ionic liquid electrolytes, with the highest observed for LiFSI in Pyr13TFSI followed by LiFSI in P111i4FSI, PYR13FSI, EMIFSI and the mixture of EC:DMC with additions of FEC and VC. At 60 °C, the conductivity of the ILEs more than doubled compared to at 20 °C, and the pyrrolidinium-FSI (PYR13FSI) and phosphonium (P111i4FSI ) electrolytes achieved ionic conductivities comparable to the room temperature conductivity of the carbonate electrolyte (11.4 mS cm−1, 10.1 mS cm−1).
The lithium mobility in the pyrrolidinium-TFSI (Pyr13TFSI), imidazolium-FSI (EMIFSI) and phosphonium-FSI (P111i4FSI) electrolytes increases substantially at 60 °C, while only a moderate increase is observed for the pyrrolidinium-FSI (PYR13FSI).
μMG-Si∣∣LFP cells with the phosphonium-FSI electrolyte (P111i4FSI) achieved a significant improvement in the rate performance at a higher temperature (60 °C). Discharge capacities around 1000 mAh/gSi were retained even at a 2 C rate, outperforming the carbonate electrolyte. The improved performance is explained by faster Li+ transport in the electrolyte at the higher temperature and a reasonably passivating SEI layer with a higher Li-ion conductivity. The imidazolium electrolyte (EMIFSI) was on par or slightly better than the carbonate electrolyte at 60 °C.
Further studies should include long term cycling, as well as the evaluation of the high temperature degradation process in the phosphonium FSI ionic liquid, in order to verify the potential for operation of cells at elevated temperatures with this electrolyte.
Acknowledgments
This work was performed within MoZEES, a Norwegian Centre for Environment-friendly Energy Research (FME), co-sponsored by the Research Council of Norway (project number 257653) and 40 partners from research, industry and public sector. Institute for Energy Technology (IFE) is acknowledged for providing the Si electrodes. The Research Council of Norway is acknowledged for the support to the Norwegian Micro- and Nano-Fabrication Facility, NorFab, project number 245963/F50.
Supplementary data (4.2 MB PDF)