
Abstract
Platinum is the model catalyst in fuel cells because of its high activity toward oxygen reduction and hydrogen oxidation. However, its applicability is limited due to the degradation of the catalyst under operating conditions. This degradation process has been extensively studied by repeatedly oxidizing and reducing the electrode, which leads to the roughening of the surface due to the nucleation and growth of platinum nano-islands. Although the general picture of this surface roughening is well known, the atomic details concerning the nucleation and early growth of the islands are still under debate. In this work, we use Density Functional Theory (DFT) to calculate formation energies and diffusion barriers of an adatom, in both the unoxidized and the oxidized state, with the aim to provide further insight into the nucleation phenomena. Moreover, we analyze from STM images obtained experimentally the shape of the nano-islands during the first stages of growth. Our results show not only that the islands form during the reduction of the surface, but also that they grow with a dendritic island shape, similarly to the platinum islands formed in vacuum by Molecular Beam Epitaxy (MBE).
Export citation and abstract BibTeX RIS
In heterogeneous catalysis, the surface structure of the catalyst plays a crucial role, as it determines not only the product selectivity and activity, but also influences its stability. 1–3 This is especially the case for electrochemical reactions that are highly surface sensitive, such as the Oxygen Reduction Reaction (ORR) on platinum, which activity in acid is higher on stepped surfaces than on Pt(111). 4,5
Platinum is the model catalyst to be used not only in fuel cells, due to its high activity toward ORR and hydrogen oxidation, 6–8 but also in electrolyzers to carry out the Hydrogen Evolution Reaction. 9 However, its economic feasibility is limited, as platinum is scarce, expensive and degrades upon fuel cell operation. 10 In order to obtain a better understanding of the reaction and the degradation mechanisms, it is crucial to observe the platinum surface structure under conditions as similar as possible to fuel cell operation. In the end, this would allow designing a better catalyst.
Cyclic oxidation-reduction of a platinum electrode is often used as a test for catalyst stability, as it simulates the switching On and Off of a fuel cell. 11–13 On Pt(111), this results in the formation of adatoms and vacancies, which leads to the nucleation and growth of platinum nano-islands (first only in 2D and later predominantly in height, thus 3D). 14–25 The island growth involves the creation of step sites at the expense of terrace sites, which leads to a higher electrochemical signal for hydrogen adsorption/desorption 25,26 and to a higher ORR activity, as specific sites near the step edges are more reactive toward the ORR. 1 The amount of step sites created depends not only on the number of islands present and their size, but also on their shape. The former two are correlated by the nucleation density: a larger number of nuclei requires the distribution of the adatoms among more islands that are, consequently, smaller. The nucleation density scales with F/M, where F is the flux of depositing atoms (i.e. in analogy to the deposition rate in Molecular Beam Epitxay (MBE)) and M is the adatom mobility. During growth, the 2D island shape differs from the standard equilibrium Wulff-shape: 27–29 while in thermodynamic equilibrium the 2D island shape is defined by the minimum of the total step free energy, 30 the (kinetic) growth Wulff-shape depends on the anisotropic step kinetics (i.e. different orientations along the island perimeter experience different growth speeds). 31,32 Therefore, the 2D growth shape significantly depends on the atom diffusion along the island perimeter, and thus the steps, as well as around the according kink and corner sites. 33–35 In addition, it also depends on the rate, with which the adatoms reach the island edge. 36,37 Enhancing the atom diffusion along the island perimeter, e. g. by raising the temperature, leads to more compact islands, while increasing the flux leads to islands with a larger number of branches, often called dendrites. 30,38,39
Previously, it was reported that the islands formed upon oxidation-reduction cycling of a Pt(111) electrode are hexagonal, and their steps and facets were identified and correlated with the different peaks in the hydrogen adsorption/desorption fingerprint (i.e. below 0.4 V in the Cyclic Voltammogram, see Fig. 1). 26 However, it is known that Pt islands formed by MBE at low temperatures (≤300 K) 30,37–39 grow dendritically. This dissimilarity is relevant, as the island shape effects the electrochemical activity: if the islands formed upon oxidation-reduction would also grow dendritically, the earlier assignation of the peaks in the hydrogen adsorption/desorption region to the different steps and facets surely would need to be reconsidered. Moreover, it is still a puzzle when these islands exactly nucleate, as they could, until now, only be observed with a Scanning Tunneling Microscope (STM) when the surface was reduced, but not during the oxidation, 24,40 in which additional atoms are pushed out onto the surface. 17,41

Figure 1. Oxidation-Reduction Cycles on Pt(111). The CV to only 0.85 V and the first ORC are shown in red and in orange, respectively, while the remaining ORCs are indicated according to the color scale. The OH adsorption, step oxidation, PErev, and PEirr regions are indicated, as well as the voltage at the critical coverage. The scan rate is 50 mV s−1.
Download figure:
Standard image High-resolution imageTo address these issues, here we study the thermodynamics and kinetics of island nucleation by using formation and diffusion barriers calculated with Density Functional Theory (DFT). Our results show that nucleation takes place during the cathodic sweep, rather than during oxidation. Moreover, we analyze the island shape during early growth by measuring their perimeter and area, from which we finally determine the island compactness and a factor describing the additional step lengths. Contrary to what was suggested in Ref. 26, our observations provide evidence that the growth of the islands results in dendritic island shapes.
Experimental
Electrochemistry
The cyclic voltammograms were recorded in a three-electrode glass cell using a reversible hydrogen electrode (RHE) as the reference, a Pt wire (MaTeck, Germany) as a counter, and a Pt(111) crystal (with a purity of 99.999% and polished <0.1deg, Surface Preparation Laboratory, The Netherlands) as the working electrode. Prior to the experiments, we left the glassware overnight in an acidified potassium permanganate solution and cleaned it afterwards with diluted piranha, before finally boiling it five times in ultrapure water (>18.2 MΩ cm, Millipore Milli-Q). Similarly to sputter cleaning and annealing in vacuum, we first etched the Pt(111) sample electrochemically (125 cycles at 50 Hz, ±2 V versus Pt) in an acidified 2.5 M CaCl2 solution, before flame annealing (3 min at ≈1250 K) and cooling down in a 1:4 H2/Ar mixture. We repeated this procedure three times before each experiment. To develop a denuded, (bulk) contamination free interface zone, we performed the last annealing step at slightly lower temperature (approximatelly 50 K less). The measurements were performed in an Ar-purged 0.1 M HClO4 solution (Merck Suprapur) using a potentiostat from Bio-Logic (VSP-300).
Electrochemical scanning tunneling microscopy
We recorded the STM images with a home-built Electrochemical Scanning Tunneling Microscope (EC-STM) described previously. 42–45 The reference electrode used was a RHE, the counter was a Pt coil, and the working electrode was the same Pt(111) crystal described above. The STM tips were electrochemically etched from a Pt90Ir10 wire and coated with electrophoretic paint (Clearclad HSR) as well as polyethylene to minimize the faradaic contributions in the tunneling current. The cleaning procedure of the glassware is described in Ref. 25. To reduce the oxygen concentration in the electrolyte, we de-aerated it with N2 before measuring for at least 3 hours. At the beginning of each experiment, we checked the surface quality and cleanliness first by cyclic voltammetry and then by scanning large surface areas with the STM.
DFT calculations
Using Density Functional Theory (DFT), we calculated the Gibbs free formation energy of a Pt-adatom and PtO2-adatom (i.e. strictly speaking, a PtO2-admolecule) on Pt(111) as well as the Gibbs free adsorption energy of oxygen onto Pt step and kink sites. The DFT simulations were performed using the Vienna Ab-Initio Simulations Package. 46–48 A plane wave basis set was used with an energy cutoff of 450 eV and the ion core potentials were modelled using the projector augmented wave (PAW) approach. The PW91 exchange-correlation functional was used. 49 We modeled the surface as a four-layer slab, with the bottom two layers frozen at the experimentally measured lattice constant of Pt (3.92 Å 50 ). The Pt(111) surface was modeled with 5 × 5 and 7 × 7 unit cells. We used the energy of an additional Pt atom attached directly to a kink site on Pt(632) as a reference for the formation energy determination of a PtO2-adatom. The Pt(632) surface was modeled in a 1 × 1 unit cell. Monkhorst-Pack mesh k-space sampling grids were used. 51 A 3 × 3 × 1 mesh grid was used for both the 5 × 5 and 7 × 7 Pt(111) unit cells, and a 7 × 7 × 1 grid was used for the 1 × 1 Pt(632) surface. We optimized the geometry until the forces on each atom were less than 0.02 eV Å−1, and included dipole correction in the surface normal direction. 52
To calculate the potential-dependent Gibbs free formation energy of a PtO2-adatom on Pt(111), we modeled the reactions shown in Eqs. 1–3 in the Results section. We calculated the Gibbs free energy change for each reaction from ΔG = ∑Gproducts − ∑Greactants , where G stands for the Gibbs free energy of each species calculated at 300 K and 1 bar. For the adsorbed species we accounted only for vibrational energy and entropy terms, whereas for the gas phase species (hydrogen and water) we included all contributions to the free energy consisting of vibrational, rotational, and translational energy. The Gibbs free energies of adsorption and formation were calculated relative to aqueous water and protons using the computational hydrogen electrode method 7 and procedures described previously. 53 The Gibbs free energy of the gas phase water was evaluated at its experimentally measured vapor pressure at 300 K; under conditions where it is in equilibrium with liquid (aqueous) water.
We calculated the oxygen adsorption at step as well as kink sites on Pt(632) as a function of oxygen coverage: the structures evaluated were based on those found to be most stable by Fantauzzi et al. 54
We calculated the diffusion barrier for a Pt-adatom on Pt(111) using the climbing-image nudged elastic band method. 55 The transition state was identified when, for the highest energy image, the forces on each of the atoms and the tangent force were below 0.02 eV −1 and the tangent forces on the preceding images were opposite in sign to the those on the images following the transition state image. The transition state vibrational modes were also examined, and showed one imaginary frequency along the reaction path. Diffusion was examined from the most stable, fcc, adsorption site to the second most stable, hcp, site.
Results and Discussion
Electrochemical measurements
Figure 1 shows in red the Cyclic Voltammogram (CV) of the pristine Pt(111) surface in 0.1 M HClO4 measured between 0.06 and 0.85 V, and in orange the first Oxidation-Reduction Cycle (ORC) measured up to 1.35 V. The region below 0.4 V, attributed to hydrogen adsorption/desorption, changes during the next 200 ORCs due to the creation of (111) and (100) steps (which more precisely should be formulated as 〈110〉/{111} and 〈110〉/{100}, that is, steps along a direction equivalent to [110], with either {111} or {100} microfacets). 26,56–60 The increase in the number of step sites with increasing cycle number also becomes evident from the potential window between 0.8 and 1.0 V, where step oxidation takes place, leading also there to an increase of the electrochemical current. 61,62
Focusing now on the terraces, the first step toward (but still prior to) terrace oxidation is the adsorption of OH that takes place between 0.5 and 0.8 V. 63,64 At higher potentials, the OHad is converted into Oad, which subsequently interchanges position with the platinum surface atoms in the so-called reversible Place Exchange (PErev): 65 concretely, at least 1, but most likely 2 oxygen atoms go subsurface underneath a Pt atom, which they lift vertically up to nearly one atomic distance in height. 41,66–72 This process results in the pronounced peak at around 1.06 V. 73,74 Its position shifts to higher potentials with faster scan rates, indicating that either the conversion of OHad to Oad, or the place-exchange, or both are kinetically limited. 41,75–77
The coverage of lifted PErev atoms rises with increasing electrode potential, leading to an increase of the surface stress. 65 If the upper (vertex) potential during the ORCs is held below 1.17 V 41 (or below 1.25 V, according to You et al. 78 ), no structural changes take place upon reduction: all hovering PErev atoms fall back into their vacancies, and the original, flat Pt(111) surface is fully restored. 41,72,77–79 This changes completely, if the upper potential is increased beyond the critical value, which implies also that a critical coverage of PErev atoms is surpassed. 65 At this moment the surface stress becomes so large that, instead of creating new PErev atoms, the next atoms created by the PE mechanism are no longer hovering above their vacancies; instead they are displaced on top of the terrace, thus becoming real adatoms, which we call PEirr atoms or PtO2-adatoms as they do get oxidized. 17,41,65 Upon reduction, the surface is no longer restored during the cathodic sweep, as the adatoms formed during the PEirr nucleate into islands before finding their way back to their original sites, i.e. before annihilating with their vacancies. 80 The reason for this is not only the easy nucleation into an island with other created PEirr atoms close by, but also due to the Ehrlich-Schwoebel barrier, 81,82 which makes it more difficult for the adatoms to hop-down the descending step and fall into the vacancy (although it is known that the Ehrlich-Schwoebel barrier breaks down for small terraces 83 and for downhill transport mediated by kinks 84,85 ). This irreversible regime is responsible for the roughening, the island formation, and thus the changes in the CV during the ORCs.
It was concluded that the PErev takes place randomly similar to a 2D adatom gas. However, the formation of a semi-ordered oxide structure, like local 1D chains, cannot be fully excluded. 41,71 The latter would match with the fact that rows of PtO2 units have been observed on Pt(111) with STM in "vacuum" under 1 bar of oxygen pressure and at 529 K. 86 In addition, the stability of rows at electrochemical potentials, similar to the ones for oxygen adsorption, was supported by DFT. 87 The formation of the rows during the PErev would also explain the existence of a critical coverage and potential, see above. It would result in an increase of the surface stress (i.e. as a consequence of the lattice mismatch between Pt(111) and PtO2), which becomes larger as the rows grow in length. Once the critical stress is surpassed, the PEirr starts, and one PEirr atom per row is pushed out onto the terrace, thereby decreasing the local surface stress. 65
Van Spronsen et al. 86 observed that these oxide rows, or "spokes", arrange in "spoke-wheel" structures with hexagonal symmetry: triangles of spokes delimiting triangular terraces. Figure 2a shows an ideal, fully ordered spoke-wheel structure, in which each of the spokes consists of 8 elevated PtO2 units (PErev atoms), at the expense of 9 Pt surface atoms. The length of a row consisting of 8 PtO2 coincides almost exactly with the length of a row of 9 Pt surface atoms, and thus the surface stress is minimum in this configuration. The "missing" Pt atom per spoke is pushed out onto the triangular terraces during the PEirr, creating a PtO2-adatom. On average, this results in 1.5 PtO2-adatoms formed in each triangle (i.e. two PtO2-adatoms in every second triangle, while only one adatom in the other triangles) and hence a total of 9 adatoms formed per spoke-wheel. However, in reality, the spokes might not arrange into perfect spoke-wheels: some degree of disorder is present that is either caused by entropy or by a kinetic limitation during their formation (see Fig. 2b). 86

Figure 2. The Irreversible Place-Exchange Regime: The Pt atoms of the original, flat terrace are colored in dark blue, the lifted PErev atoms in light blue, forming spokes or rows, the oxygen atoms in orange, and the PEirr atoms that are pushed onto the terrace, thus becoming PtO2-adatoms, in dark red. The PtO2-adatoms lead eventually to the growth of the nano-islands. (a) shows one fully ordered spoke-wheel structure without any disorder present. However, from Refs. 41, 71, 86 we know that, probably due to entropy, a certain degree of disorder is present. Therefore, (b) shows an impression of a disordered spoke-wheel phase: elements of the original triangles are still visible, in which the PtO2-adatoms, the PEirr atoms, are trapped.
Download figure:
Standard image High-resolution imageFormation of adatoms and adatom islands
As the PtO2-adatoms are formed during the PEirr, one naturally would assume that the formation of the adatom islands takes place also during this regime. However, two independent STM studies stated that the islands cannot be observed during the oxidation up to 1.3 V, but become visible only when the surface is subsequently reduced. 24,40 There are two possible explanations for this. The first one is that the PtO2 islands are actually present already at 1.3 V, but cannot be easily identified with an STM. This is because the Local Density of States (LDOS) of PtO2 is significantly lower than the LDOS of bare platinum, leading to a major decrease in the tunneling current, and hence a lower apparent height than the 1 ML height expected (i.e. if tunneling filled states). Moreover, it might be almost impossible to distinguish between PtO2-adatom islands and the PErev atoms, as the latter are also lifted nearly 1 ML from the surface, and also do have a different LDOS than the bare platinum. Wakisaka et al. reported that the Pt(111) surface becomes bumpy during the anodic sweep where tiny dots appear that are lifted around half a monolayer in height. It could be that these dots are the PErev atoms, but it might also very well be that they are irreversible, pushed out PtO2-adatoms, or even small PtO2-adatom islands that will become larger upon further growth. The second possible explanation, to why the islands could not be observed with an STM during the oxidation, is that indeed the islands are not formed until the surface is reduced. Even if the PtO2-adatoms are formed during the PEirr regime, whether these PtO2-adatoms nucleate into islands depends both on their concentration and their mobility, such that thermodynamic as well as kinetic considerations have to be taken carefully into account.
In order to provide further insight into the precise formation of the islands, we applied DFT to extract thermodynamic formation energies and kinetic diffusion barriers, all of which are important in the nucleation of Pt- as well as PtO2-adatoms into islands, on both an oxidized as well as reduced Pt(111) surface.
Thermodynamic requirements for nucleation
In order to study the nucleation of adatom islands, we need first to introduce the chemical potential of adatoms, which is described as follows: 88
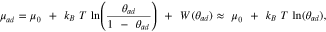
where μ0 is the ground state energy of an adatom (and thus the adatom formation energy), kB
is the Boltzmann constant, T is the temperature, θad
is the adatom concentration on the surface, and W(θad
) describes the interaction energy between adatoms. The entropy term  accounts for the number of free sites where an adatom can be placed (i.e. similarly to the entropy term in a Langmuir isotherm) and can usually be approximated to
accounts for the number of free sites where an adatom can be placed (i.e. similarly to the entropy term in a Langmuir isotherm) and can usually be approximated to  , as in most cases the adatom concentration is very low. Moreover, the adatom interaction term W(θad
) is small in comparison to the other terms such that it can typically be neglected (especially for low coverages).
, as in most cases the adatom concentration is very low. Moreover, the adatom interaction term W(θad
) is small in comparison to the other terms such that it can typically be neglected (especially for low coverages).
Thermodynamic equilibrium requires the equilibration of all chemical potentials such that the surface will maintain the equilibrium adatom concentration (θeq,ad ) by a balanced mass transport of adatoms from step- and, in particular, kink-sites to the terraces and back.
At constant temperature, changes in the actual adatom concentration (θad ) are only possible during surface growth (or etching), which originates from the increased (decreased) chemical potential created by the additional flux of adatoms (or vacancies) during, e.g. an MBE (or sputtering) experiment.
When surface growth (or etching) stops, the surface will pursue to reestablish the thermodynamic equilibrium. If θad is lower than θeq,ad , the equilibrium is restored by the formation of additional adatoms that detach from kinks and diffuse onto the terrace. If on the other hand, θad is higher than θeq,ad , adatoms on the terrace quickly diffuse toward the steps, where they finally get incorporated at kinks. At too high deposition flux, where the surface no longer will revert back to its lowest free energy configuration, adatoms find each other before encountering the steps during their random walk (which depends on the flux, the mobility, and their distance to the step), and nucleate into adatom islands to locally lower the enhanced chemical potential. Neglecting for the moment any kinetic arguments, we can, therefore, conclude that nucleation is only possible if θad ≫ θeq,ad .
Realizing the importance of the equilibrium Pt-adatom background pressure, θeq,Pt,ad , on the thermodynamics of nucleation, one can calculate θeq,Pt,ad by using the Boltzmann equation:
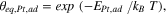
in which EPt,ad is the formation energy of a Pt-adatom, defined as the energy required to take an atom from a kink site and place it onto the terrace, and T is room temperature (T = 298 K).
As estimated from our DFT calculations, EPt,ad is 1.2 eV (see the orange line in Fig. 3a), independently of the electrode potential. This gives rise to a potential independent equilibrium adatom density of only θeq,Pt,ad = 5 · 10−21, shown with the orange line in Fig. 3b. Therefore, the use of the Boltzmann equation, which includes the approximation in the entropy term explained before and neglects the adatom-adatom interaction is well justified (see Supplementary Information for further details). As usual, the numbers obtained from DFT should be taken with some care, due to the inherent error margin of this methodology. However, the order of magnitude is typically captured correctly. In this work, the effect of near-surface solvation on the formation of a Pt- or PtO2-adatom has not been considered, and while we expect the effect of solvation on the difference in energy between a Pt- and a PtO2-adatom on Pt(111) to be small (as the magnitude of the surface-normal dipole moments are small), this should be evaluated in future work. In our case, this error does not change significantly the atomic picture, and hence the message is still the same, e. g. with a typical error of 0.2 eV, EPt,ad would be 1.0 eV instead of 1.2 eV, resulting in θeq,Pt,ad = 10−17, which is still very low.

Figure 3.
Formation Energies and Concentrations of Adatoms. (a) The formation energies of a Pt-adatom and a PtO2-adatom on Pt(111), and a PtO2-adatom on a Pt(111) with 4/25 ML Oads, are shown in orange, red, and blue, respectively. They are referenced with respect to the binding energy of an additional Pt atom attached directly to a kink site on Pt(632), which can be unoxidized (green dashed line) or oxidized (pink dashed line). The numbers in brackets indicate the reactions considered, explained in the main text. The dotted red and purple lines indicate the Pt-adatom oxidation potential and the potential at which  becomes zero, respectively. The first ORC is shown in dotted light blue and the different regions are indicated. (b) Equilibrium Pt- and PtO2-adatom concentrations calculated from (a). The dotted red and blue lines result from the Boltzmann equation with the approximation which match, respectively, the solid red and blue lines (considering the complete entropy term without approximation) at potentials <0.85 V. The dashed black line and the arrows indicate the path followed upon one ORC, while the vertical lines indicate the potentials that are provided also in Table I. (c) Zoomed-in graph of the region between 0.85 V and 1.00 V in (b). (d) Concentration ratios between PtO2- and Pt-adatoms.
becomes zero, respectively. The first ORC is shown in dotted light blue and the different regions are indicated. (b) Equilibrium Pt- and PtO2-adatom concentrations calculated from (a). The dotted red and blue lines result from the Boltzmann equation with the approximation which match, respectively, the solid red and blue lines (considering the complete entropy term without approximation) at potentials <0.85 V. The dashed black line and the arrows indicate the path followed upon one ORC, while the vertical lines indicate the potentials that are provided also in Table I. (c) Zoomed-in graph of the region between 0.85 V and 1.00 V in (b). (d) Concentration ratios between PtO2- and Pt-adatoms.
Download figure:
Standard image High-resolution imageFrom Fig. 1 we know that first the steps, and then the terraces oxidize. This is expected, as lower coordinated sites (i.e. the steps) bind an oxygen atom more strongly.
1
Similarly, considering the oxidation of Pt-adatoms, which, to our knowledge, has not been studied before, one would expect them to oxidize before 0.8 V, during the anodic sweep. Consequently, we have calculated, using DFT, the energy required to form a PtO2-adatom from a kink site. In general, we have to distinguish three different configurations: (1) a pure Pt adatom (see Fig. 4b) is created from an unoxidized step, thus kink-atom ( in Fig. 4a), that oxidizes immediately when diffusing onto the terrace leading to a PtO2-adatom (Fig. 4c); (2) a PtO2-adatom is created from a Pt atom at an oxidized step (Fig. 4d), thus kink (
in Fig. 4a), that oxidizes immediately when diffusing onto the terrace leading to a PtO2-adatom (Fig. 4c); (2) a PtO2-adatom is created from a Pt atom at an oxidized step (Fig. 4d), thus kink ( ), which takes with it two oxygens such that the remaining "empty" kink site has to be reoxidized also; (3) both the steps, thus kinks, and the terraces are oxidized, and the PtO2-adatom is created from a kink (
), which takes with it two oxygens such that the remaining "empty" kink site has to be reoxidized also; (3) both the steps, thus kinks, and the terraces are oxidized, and the PtO2-adatom is created from a kink ( ) by placing such a unit onto the terrace (see Fig. 4e). The reaction equations that describe these three processes are given as follows:
) by placing such a unit onto the terrace (see Fig. 4e). The reaction equations that describe these three processes are given as follows:
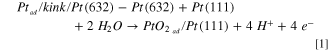
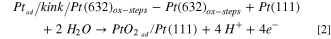
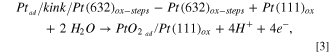
where the slash stands for "at" , e. g.  stands for a Pt-adatom at a kink site at a Pt(632) surface, and "ox" stands for "oxidized".
stands for a Pt-adatom at a kink site at a Pt(632) surface, and "ox" stands for "oxidized".

Figure 4. Ball Models of Step and Adatom Configurations: (a) A bare platinum step on a Pt(632) surface, which was used to calibrate the total energy scale via the Pt-adatom at the kink site at the step. (b) Pt-adatom on an fcc site on a Pt(111) terrace. (c) PtO2-adatom on a bridge site on a Pt(111) terrace. (d) A fully oxidized step forming PtO2 oxide chains similar to the spokes on the terraces. 89,90 (e) PtO2-adatom on a bridge site on a Pt(111) terrace that is covered with a perfectly ordered 4/25 ML Oads superstructure.
Download figure:
Standard image High-resolution imageFigure 3a shows, in red, the potential dependent formation energy of a PtO2-adatom on Pt(111). As this energy is the same for both reaction pathways, creation from an unoxidized step (Eq. 1) and from a fully oxidized step (Eq. 2), we consider both cases together and name the corresponding formation energy  . We used the energy of an additional Pt atom attached directly to a kink site on Pt(632) as a reference for the formation energy determination of a PtO2-adatom. Please note that we find that the formation energy of an adatom is nearly the same whether we reference an oxidized or a non-oxidized kink site on Pt(632). Therefore, we have decided to independently consider adatom formation and Pt(632) step/kink oxidation. This leads to the oxidized kink and the non-oxidized kink reference states having the same energy in Fig. 3a, because we find the oxidation of the step does not significantly affect the adatom formation energy and because we are calculating the adatom formation energy with a constant oxygen coverage in our reference state. We have separately plotted the thermodynamics of the oxidation of the step/kink sites on Pt(632) as a function of oxygen coverage along the step in Fig. S2 in the Supplementary Information. A 3-dimensional plot would be required to simultaneously consider varying the adatom coverage and the oxygen coverage; we leave this for future work, but do not expect this to affect our conclusions, again given that the adatom formation energy from an oxidized kink at constant oxygen coverage is not significantly different to that from a bare kink.
. We used the energy of an additional Pt atom attached directly to a kink site on Pt(632) as a reference for the formation energy determination of a PtO2-adatom. Please note that we find that the formation energy of an adatom is nearly the same whether we reference an oxidized or a non-oxidized kink site on Pt(632). Therefore, we have decided to independently consider adatom formation and Pt(632) step/kink oxidation. This leads to the oxidized kink and the non-oxidized kink reference states having the same energy in Fig. 3a, because we find the oxidation of the step does not significantly affect the adatom formation energy and because we are calculating the adatom formation energy with a constant oxygen coverage in our reference state. We have separately plotted the thermodynamics of the oxidation of the step/kink sites on Pt(632) as a function of oxygen coverage along the step in Fig. S2 in the Supplementary Information. A 3-dimensional plot would be required to simultaneously consider varying the adatom coverage and the oxygen coverage; we leave this for future work, but do not expect this to affect our conclusions, again given that the adatom formation energy from an oxidized kink at constant oxygen coverage is not significantly different to that from a bare kink.
As the formation energies  cross EPt,ad
at 0.59 V, this defines the equilibrium potential, at which the Pt-adatoms oxidize to PtO2-adatoms, according to:
cross EPt,ad
at 0.59 V, this defines the equilibrium potential, at which the Pt-adatoms oxidize to PtO2-adatoms, according to:
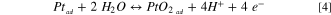
Equipped with the potential dependent formation energy,  , we calculate the equilibrium concentration of PtO2-adatoms,
, we calculate the equilibrium concentration of PtO2-adatoms,  , again by using the Boltzmann equation. This approach is justified because, similarly to the Pt-adatoms formed from kink-sites, PtO2-adatom formation happens in the same way with the simultaneous oxidation of either the Pt-adatom or the kink site, see Eqs. 1 and 2. Note that even if the number of free adsorption sites on the terrace would change like, e.g., during the creation of the spokes and the PErev atoms, the Boltzmann distribution still describes correctly the percentage of PtO2-adatoms on the remaining sites. As
, again by using the Boltzmann equation. This approach is justified because, similarly to the Pt-adatoms formed from kink-sites, PtO2-adatom formation happens in the same way with the simultaneous oxidation of either the Pt-adatom or the kink site, see Eqs. 1 and 2. Note that even if the number of free adsorption sites on the terrace would change like, e.g., during the creation of the spokes and the PErev atoms, the Boltzmann distribution still describes correctly the percentage of PtO2-adatoms on the remaining sites. As  decreases with potential,
decreases with potential,  increases, see the red line in Fig. 3b. Consequently, above 0.59 V, the ratio
increases, see the red line in Fig. 3b. Consequently, above 0.59 V, the ratio  , which gives an indication for the driving force to oxidize a Pt-adatom, becomes larger the higher the potential is: while it is equal to 1 at 0.59 V (the concentration of Pt-adatoms and PtO2-adatoms is equal), it is already a factor 8000 higher at 0.65 V, see the red line in Fig. 3d. As the potential increases further,
, which gives an indication for the driving force to oxidize a Pt-adatom, becomes larger the higher the potential is: while it is equal to 1 at 0.59 V (the concentration of Pt-adatoms and PtO2-adatoms is equal), it is already a factor 8000 higher at 0.65 V, see the red line in Fig. 3d. As the potential increases further,  becomes larger, up to the point that the Boltzmann equation results in
becomes larger, up to the point that the Boltzmann equation results in  at 0.90 V, and even adatom concentrations larger than 1 ML at potentials above (see the dotted red line in Fig. 3b). Obviously, the Boltzmann equation does not hold at these high adatom coverages, which can be, in first instance, solved by using the full entropy term in the adatom chemical potential equation,
at 0.90 V, and even adatom concentrations larger than 1 ML at potentials above (see the dotted red line in Fig. 3b). Obviously, the Boltzmann equation does not hold at these high adatom coverages, which can be, in first instance, solved by using the full entropy term in the adatom chemical potential equation,  ,instead of the approximation to
,instead of the approximation to  (see the Supplementary Information). The equilibrium adatom concentration is then given by:
(see the Supplementary Information). The equilibrium adatom concentration is then given by:
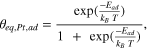
In the Supplementary Information we evaluate the error one makes when using the Boltzmann approximation and show that it is as low as 6 · 10−4 for  up to 0.85 V.
up to 0.85 V.
By using the equation with the full entropy term, we now see in Fig. 3b that the equilibrium adatom concentration asymptotically approaches 1 ML with increasing potential. However, one has to realize that at these high coverages the definition of the adatom gas is no longer valid, even if the Fermi statistics without approximation still holds. Single, individual adatoms do only exist up to a coverage of 0.25 ML: at higher coverage one connects at least 3 into a cluster. As the relative error between the Boltzmann approximation and the full solution is only 25% at 0.25 ML, which is 0.25 ± 0.06 ML, the equilibrium adatom density is (for most applications) sufficiently well described already by the Boltzmann approximation.
Nevertheless, it is conceptually still interesting to consider the meaning of reaching a coverage of 1 ML. This situation is probably most comparable to the roughness transition on surfaces, where at high enough temperatures the step free energy crosses zero and the surface can create as many steps as desired. 91–95 The roughening transition is a phase transition of infinite order, and although it is surely different, something similar must happen here during the oxidation. Derived from the chemical potential with a kink site as reference, 1 ML of coverage at 0.94 V implies the formation of a complete oxide layer. It is striking that the PErev starts around this potential, which describes the first part of the terrace oxidation. 78
We know from Fig. 1 and Refs. 61, 62 that the steps begin to oxidize at 0.8 V, and that full oxidation happens as late as 1.05 V, probably leading to oxide chains as observed in vacuum.
89,90
Therefore, we also consider the situation in which a PtO2-adatom is formed from a fully oxidized step (kink). As the formation energy, as explained before, is the same, independently of whether the step is unoxidized or fully oxidized, it is still described by the red line in Fig. 3a. However, even if the thermodynamics of a PtO2-adatom formation do not change, the kinetics surely do. As a fully oxidized step forms a closed 1D-PtO2 row, we expect that detaching a PtO2-kink atom from such a step involves overcoming a significant barrier, and is thus kinetically limited (i.e. the PtO2-kink has to "diffuse" out of the step). Consequently,  will not be reached (although, strictly speaking, it always would when given enough time) and, as then
will not be reached (although, strictly speaking, it always would when given enough time) and, as then  <
<  , nucleation will not occurr. Furthermore, even if this kinetic barrier is small, in order to reach 1 ML PtO2-adatoms the steps would need to retract with the same width. This would involve a large mass transport and thus require a very long time, i.e. 1.5 · 105 PtO2 kink atoms need to be detached from the step to create 1 ML PtO2-adatoms on a 100 × 100 nm2 terrace.
, nucleation will not occurr. Furthermore, even if this kinetic barrier is small, in order to reach 1 ML PtO2-adatoms the steps would need to retract with the same width. This would involve a large mass transport and thus require a very long time, i.e. 1.5 · 105 PtO2 kink atoms need to be detached from the step to create 1 ML PtO2-adatoms on a 100 × 100 nm2 terrace.
At 1.17 V, the PEirr starts, leading to the formation of PEirr atoms. As the terrace is then oxidized, we calculated also the formation energy of a PtO2-adatom on a terrace covered with 4/25 ML Oads, which we named  , see the blue line in Fig. 3a. By comparing
, see the blue line in Fig. 3a. By comparing  with
with  , it is evident that, once the terraces oxidize, it becomes energetically more expensive to form PtO2-adatoms. Consequently, the equilibrium PtO2-adatom concentration
, it is evident that, once the terraces oxidize, it becomes energetically more expensive to form PtO2-adatoms. Consequently, the equilibrium PtO2-adatom concentration  is lower than
is lower than  at every given potential. Based on this result, we expect that the potential dependent formation energy of the PtO2-adatoms shifts to higher potentials the more the terrace is oxidized.
at every given potential. Based on this result, we expect that the potential dependent formation energy of the PtO2-adatoms shifts to higher potentials the more the terrace is oxidized.
As there is a huge driving force toward adatom oxidation ( =2·1020 at 1.17 V), see Table I, which provides also an overview of our results), the PEirr atoms are surely oxidized, thus PtO2-adatoms. There are two possible pathways for the formation of PtO2-adatoms from the PEirr: either the newly lifted PErev atoms are pushed out onto the terrace, where they immediately oxidize forming a PEirr adatom, or the PErev atom first tries extending a spoke/row, for which it needs 2 extra oxygen atoms, which it takes with it, when pushed onto the surface forming a PEirr adatom, due to the too high stress in the row. At 1.17 V as well as at 1.35 V, the upper (vertex) potential during our ORCs,
=2·1020 at 1.17 V), see Table I, which provides also an overview of our results), the PEirr atoms are surely oxidized, thus PtO2-adatoms. There are two possible pathways for the formation of PtO2-adatoms from the PEirr: either the newly lifted PErev atoms are pushed out onto the terrace, where they immediately oxidize forming a PEirr adatom, or the PErev atom first tries extending a spoke/row, for which it needs 2 extra oxygen atoms, which it takes with it, when pushed onto the surface forming a PEirr adatom, due to the too high stress in the row. At 1.17 V as well as at 1.35 V, the upper (vertex) potential during our ORCs,  is practically 1 ML. However, the flux (concentration) of PtO2-adatoms formed during the PEirr is only 0.0245 ML, obtained from a fit of the roughening evolution during 170 ORCs up to 1.35 V.
80
As 0.0245 ML is much lower than the 1 ML of critical coverage, the PtO2-adatoms created will not nucleate into islands; not even at 1.35 V.
is practically 1 ML. However, the flux (concentration) of PtO2-adatoms formed during the PEirr is only 0.0245 ML, obtained from a fit of the roughening evolution during 170 ORCs up to 1.35 V.
80
As 0.0245 ML is much lower than the 1 ML of critical coverage, the PtO2-adatoms created will not nucleate into islands; not even at 1.35 V.
The situation changes completely during the backward (cathodic) sweep. Following the dashed black line in Fig. 3b, from right to left,  decreases with decreasing potential. At 0.94 V,
decreases with decreasing potential. At 0.94 V,  becomes equal to
becomes equal to  , thus 0.0245 ML. Therefore, upon decreasing the potential,
, thus 0.0245 ML. Therefore, upon decreasing the potential,  has to decrease in order to adapt to the decreasing equilibrium concentration
has to decrease in order to adapt to the decreasing equilibrium concentration  , and hence the nucleation of PtO2-adatoms into PtO2-islands is now thermodynamically favorable. However, this still does not ensure that nucleation takes place, as one should also consider the kinetics, thus the mobility, of the PtO2-adatoms: if the PtO2-adatom diffusion is very slow, it will take a significant amount of time for the nucleation to occur. Moreover, as the potential decreases further, the equilibrium PtO2-adatom concentration (
, and hence the nucleation of PtO2-adatoms into PtO2-islands is now thermodynamically favorable. However, this still does not ensure that nucleation takes place, as one should also consider the kinetics, thus the mobility, of the PtO2-adatoms: if the PtO2-adatom diffusion is very slow, it will take a significant amount of time for the nucleation to occur. Moreover, as the potential decreases further, the equilibrium PtO2-adatom concentration ( , or
, or  once the terraces are reduced) drops also further, thus rising the driving force for nucleation. This situation holds until 0.59 V, when the PtO2-adatoms reduce back to Pt-adatoms and the equilibrium adatom concentration becomes constant. Anyhow, although θeq,Pt,ad
is much lower (5·10−21) than the coverage of PtO2-adatom created during the PEirr, 0.0245 ML, the nucleation will still depend on kinetics, now on the mobility of the Pt-adatoms. Therefore, in order to solve the complete riddle, we also need to study the diffusion rates of both Pt- and PtO2-adatom species. This is discussed in more depth in the next section.
once the terraces are reduced) drops also further, thus rising the driving force for nucleation. This situation holds until 0.59 V, when the PtO2-adatoms reduce back to Pt-adatoms and the equilibrium adatom concentration becomes constant. Anyhow, although θeq,Pt,ad
is much lower (5·10−21) than the coverage of PtO2-adatom created during the PEirr, 0.0245 ML, the nucleation will still depend on kinetics, now on the mobility of the Pt-adatoms. Therefore, in order to solve the complete riddle, we also need to study the diffusion rates of both Pt- and PtO2-adatom species. This is discussed in more depth in the next section.
Table I.
Pt- and PtO2-adatom Equilibrium Concentrations and their Ratios. We only provide relevant values at each of the potentials: if the terraces are not oxidized, we consider  , while we consider
, while we consider  if the terraces are oxidized. PZC stands for potential of zero charge of Pt(111).
96,97
if the terraces are oxidized. PZC stands for potential of zero charge of Pt(111).
96,97
| 0.36 V (PZC) | 0.59 V | 0.90 V | 1.17 V | 1.35 V | |
|---|---|---|---|---|---|
| θeq,Pt,ad | 5·10−21 | 5·10−21 | − | − | − |

| 4·10−36 | 5·10−21 | 0.5 | − | − |

| 9·10−17 | 1 | 1·1020 | − | − |

| − | − | − | 1 | 1 |

| − | − | − | 2·1020 | 2·1020 |
Kinetic requirements for nucleation
As the probability for nucleation does not only depend on the supersaturation, thus how much the adatom concentration exceeds the equilibrium adatom concentration, we will in the following consider also the effects of (limited) adatom mobility. For nucleation to occur during the oxidation sweep, the PtO2-adatoms must diffuse over the terrace, find each other, and overcome the critical nucleus size, as only clusters larger than the critical nucleus size do not decay. 37 If the diffusion of the PtO2-adatoms is slow, then it will take longer times for the adatoms to find each other and therefore the nucleation will be delayed. In the extreme limit, when the diffusion would be completely kinetically hindered, nucleation would never take place.
Using DFT, we calculated the diffusion barrier of a Pt-adatom on a Pt(111) surface, EPt,ad,Diff , to be 0.28 eV. This value is close to the 0.26 eV obtained in two different studies, one which combines STM with MC simulations 98 and the other one based on Field Ion Microscopy measurements, 99 and not significantly far from the 0.16 eV calculated by an early Effective Medium Theory study. 100 Using this value, we can calculate an approximation of the Pt-adatom jump rate by using an Arrhenius-type equation:
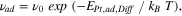
in which νad is the jump rate and ν0 is the attempt frequency, given by the vibrational frequency (1013 Hz) for single metal atoms. As a result, we obtain a Pt-adatom jump rate of 108 jumps/s.
We have also calculated the relative energy of a PtO2-adatom on a Pt(111) surface at different adsorption sites: bridge1, bridge2, atop, fcc and hcp (see Fig. 5). The most stable configuration is bridge1, which was used as reference to compare with the energetics of the others. Within the configurations calculated, a PtO2-adatom in bridge1 has an energy that is at least 0.78 eV lower than all other configuration considered. While these are just thermodynamic arguments, and further DFT calculations on the kinetics of PtO2-adatom diffusion would be beneficial (but are left for future work), the change in energy when moving from one site to another gives an estimate on the diffusion barrier, within the error margins that are characteristic from DFT. We expect the diffusion barrier of a PtO2-adatom to be at least as high as 0.78 eV, and obtain a PtO2-adatom jump rate of only 0.28 jumps/s, which is approximately 109 times less than the jump rate of a Pt-adatom. To demonstrate how slow this PtO2-adatom diffusion is, we estimate how many jumps one PtO2-adatom can perform during the cathodic sweep. Decreasing the potential from 1.35 V to 0.59 V (where PtO2-adatoms reduce to Pt-adatoms) with 50 mV s−1 leads to a reduction time of 15.2 s. In this time, a PtO2-adatom can only do 4 jumps. This is not enough to encounter other PtO2-adatoms, and therefore nucleation will not occur.

Figure 5. Potential Energy Landscape of a PtO2 -adatom (PEirr -adatom) on Pt(111): (a) bridge1, (b) atop, (c) fcc, (d) hcp, (e) bridge2. The lowest energy state, the energy of a PtO2-adatom at bridge1, is used as reference. The diffusion barrier of a PtO2-adatom must be at least 0.78 eV.
Download figure:
Standard image High-resolution imageIn addition to this extremely low jump rate, as the PErev involves the formation of spoke-wheels, the PtO2-adatoms formed during the PEirr will be "trapped" within the triangular areas. The reason for this is that the spokes, delimiting the triangles, are lifted nearly one monolayer high above the terrace, preventing the PtO2-adatoms from diffusing across. 65 Even if the spoke-wheels are distorted, like in Fig. 2b, a PtO2-adatom can only "escape" from a triangle if it finds the open pathway between the spokes. This requires a significant amount of time, as the PtO2-adatom needs several attempts during its random walk to find this open path. Even if nucleation is thermodynamically favourable, it will take quite a long time to occur. This limited diffusion also extremely hinders mass transport between the terrace sites and the kinks, thus preventing a quick equilibration of the chemical potentials and the required adatom concentration.
If we now consider that the critical nucleation cluster would be a single adatom, meaning that the smallest stable cluster would be a dimer, and that diffusion between triangles is fully prohibited, nucleation would only occur in those triangles that have received two PtO2-adatoms: thus in 50% of the triangles, as there is, on average, the formation of 1.5 PtO2-adatoms per triangle. In the case that a dimer would be the critical nucleus size, one additional PtO2-adatom would be needed for all the dimers to form a stable nuclei. As this only can come from neighboring triangular areas, a single PtO2-adatom has to "escape" its own triangular area, finding its way through the open paths between the spokes. Statistically, this would lead to every second triangle with a stable trimer, while the others would not have any adatom.
Once the spokes reduce during the broad cathodic peak at around 0.8 V (see Fig. 1), the PtO2-adatoms can freely diffuse over the Pt(111) terrace, although, as commented before, only with 0.28 jumps/s (compare to the Pt-adatoms with 108 jumps/s). Therefore, even though, below 0.94 V,  has decreased such that now
has decreased such that now  is larger (i.e.
is larger (i.e.  = 0.0245, as this is the flux of PtO2-adatoms created during the PEirr
80
), nucleation will not occurr within the time needed to reach 0.59 V during the cathodic scan. At 0.59 V, the PtO2-adatoms reduce to Pt-adatoms, which probably explains the cathodic peak starting at around this potential. As the Pt-adatoms diffuse with 108 hops per second, instant nucleation occurs. Interestingly, Wakisaka et al. reported that they could not observe the islands until the potential was reduced to 0.49 V,
24
which would point out that Pt-adatoms, and not PtO2-adatoms, are the species involved during nucleation.
= 0.0245, as this is the flux of PtO2-adatoms created during the PEirr
80
), nucleation will not occurr within the time needed to reach 0.59 V during the cathodic scan. At 0.59 V, the PtO2-adatoms reduce to Pt-adatoms, which probably explains the cathodic peak starting at around this potential. As the Pt-adatoms diffuse with 108 hops per second, instant nucleation occurs. Interestingly, Wakisaka et al. reported that they could not observe the islands until the potential was reduced to 0.49 V,
24
which would point out that Pt-adatoms, and not PtO2-adatoms, are the species involved during nucleation.
Due to the non-random character of both the PErev and the PEirr processes, the islands resulting from the nucleation are distributed within a certain distance to each other, as observed in Ref. 65. More concretely, there are two possible reasons for this. The first one is that the PtO2-adatoms are formed within a certain distance between each other leading therefore, statistically, also to nuclei with a homogeneous distance distribution. The second one is that the PtO2-adatoms are retained so long within their triangular areas of origin that, once the spokes are reduced, the statistical distance between the PtO2-adatoms is equal, leading to nuclei that show (statistically) the same distances to each other: the average number of local random walks on the surface is the same before a nucleation event occurs. Once the islands are formed, processes such as Ostwald ripening (i.e. atom diffusion from the smaller to the larger islands) and/or Smoluchowski ripening (i.e. smaller islands diffusing as a whole unit and getting incorporated into the larger islands nearby, which are less mobile) 101–103 become important, and result in a larger averaged island size at the expense of a lower island density.
Dendritic islands and their step length
Figure 6a shows an STM image (reproduced with permission from Ref. 40) of islands created by stepping the potential from 1.0 V to 1.3 V and holding it there for 10 s, before stepping back to 1.0 V, and finally sweeping with 50 mV s−1 to 0.05 V to completely reduce the oxide. Subsequently, the Pt(111) sample was taken out of the electrolyte, dried, and mounted inside a vacuum-STM chamber to perform the imaging. Interestingly, the islands created in this way are dendritic, and not hexagonal as reported in the electrochemical STM study in Ref. 26.

Figure 6. Dendritic Islands on Pt(111): (a) Vacuum-STM image of dendritic islands formed after a potential step from 1.0 to 1.3 V, holding 1.3 V for 10 s, and stepping back to 1.0 V before finally sweeping to 0.05 V at 50 mV s−1. The image is reproduced with permission from T. Maagaard. 40 (b), (c) and (d) are the upper, the best, and the lower estimate, respectively, of the area of the island marked with a red square in (a). (e) Dendritic islands observed with our EC-STM after holding the potential at 1.35 V for 200 s, and then reducing to 0.3 V with 25 mV s−1. This image was recorded with US = 300 mV, Ut = 310 mV, and It ≈300 pA. (f), (g) and (h) are the respective upper, best, and lower estimate of the area of the island marked in red in (e).
Download figure:
Standard image High-resolution imageFigure 6e shows an electrochemical STM image (i.e. taken in the electrolyte) of the islands formed by sweeping the potential from 0.3 to 1.35 V at 25 mV s−1 and holding it there during 200 s before sweeping back to 0.3 V at 25 mV s−1 to reduce the oxide. Upon closer inspection of the islands, one can see that also these islands are dendritic in shape, although more compact than the ones measured afterwards in vacuum, shown in Fig. 6a.
The origin of the dendritic islands requires a growth instability that is caused by the Kink Ehrlich-Schwoebel Effect (KESE). The reason for this energetic barrier is manifested in the lower coordination of an atom diffusing around a kink when it is at the transition state. Its similarity with the one dimensional higher (3D instead of 2D) Ehrlich-Schwoebel barrier 81,82 was realized for the first time by Pierre-Louis. 104,105 Figure 7 sketches the general mechanism for the formation of dendritic islands. The arrival of adatoms to an island edge, given by a step, leads to the nucleation of a 1D island step-row (in full similarity to island nucleation on a terrace). In analogy to mound formation in 3D, 80,106 these 1D islands grow in the perpendicular direction to the step, because of the limited diffusion around the kinks introduced by the KESE. The faster the arrival of the adatoms (i.e. the higher the flux) toward the island edge, the higher the growth speed becomes in the perpendicular direction to the step and, as the diffusion barriers are flux independent, there is simply less time for the arriving atoms to try hopping around the corner and thus overcome the KESE. In other words, the remaining growing triangles do become shorter at their base, and it might be even possible to nucleate two 1D rows, thus two triangles, at one step edge. Therefore, a sudden large flux of adatoms that are released during the reduction of the PtO2-adatoms significantly favors the formation of even smaller branches that are characteristic for the dendritic shape. Once these branches are formed, they ramify by the incorporation of new adatoms to the edges of the branches, thus once again forming 1D islands that grow.

Figure 7. Scheme of the Growth Instability (KESE) resulting in Dendritic Islands: (a) Step-adatom (1D) diffusion resulting in the nucleation of 1D-island rows. (b) The KESE makes it more difficult for the atom to diffuse around the kink, leading to the nucleation and growth of a second row (c). (d) The arrival of more adatoms leads eventually to the growth of triangles (branches) perpendicular to the step. (e) Nucleation of a new row on the side of a triangle, which finally results in the bifurcation of the branch (f).
Download figure:
Standard image High-resolution imageThe island shape, especially the ratio of the perimeter to area, thus the step sites to terrace sites, crucially effects the ORR activity as well as the catalyst degradation. 107,108 Calle-Vallejo et al. 1 reported that the ORR activity measured at 0.9 V increased more than three times after roughening a Pt(111) electrode by performing 10 ORCs up to 1.72 V, which is likely below the 2D to 3D transition growth state of the islands. 25
In order to measure the perimeter and the area of the dendritic islands, we first need to delimit the island boundaries. This is far from trivial, as STM images suffer inherently from tip convolution and this effect is more severe the smaller the feature sizes and the larger the tip radius. Realizing the dendritic island shape with its tiny branches, we obviously need a careful approach for the proper determination of the perimeter. The approach chosen for the analysis and the effect of tip convolution could be the reason why in Ref. 26 it was suggested that the islands were hexagonal.
Starting with the more branched islands measured in vacuum, we decided to look for the upper, the best, and the lowest estimate of the island as shown for the particular example displayed in Fig. 6(b–d). In order to determine the different estimates, we first changed the color scale to gray scale, which delivers higher contrast. Then, using a threshold routine that displays everything above the threshold in white and below in black, we generated the images. The upper estimate was generated by decreasing the threshold until the shallow trenches within the island were almost filled (note the trench indicated with the orange arrow in Fig. 6b as an example). For the lower estimate we increased the threshold until one of the parts clearly forming the island almost disappeared (see the part marked with the orange arrow in Fig. 6d). The lower estimate ensures that we account also for tip deconvolution. Even if a very tiny feature on the surface, like a single atom, images in reality the very broad and large tip, this feature cannot disappear completely, thus, in our case, it must still be part of the island. In the best estimate, which is the one used in the further analysis, we adjusted the island area as accurate as possible to the upper part of the island (step) height. The upper, the best, and the lowest estimates of the area of the islands measured in the electrolyte were obtained in the same way (see Figs. 6f–h as an example).
To quantify the degree of the dendritic shape, we defined a roughness factor of the island edge ( r
) as follows:
r
) as follows:
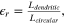
where Ldendritic is the perimeter (step length) of the dendritic island in the best estimate and Lcircular is the perimeter of a circular island of exactly the same area, calculated as:
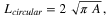
where A is the area of the dendritic island in the best estimate.
Figure 8 shows the statistical distributions of r
obtained from analyzing 200 islands imaged in vacuum (a) and 250 islands imaged in the electrolyte (b). The maximum in the fit of the r
distribution for the vacuum measurement has a clearly higher value, r
= 1.96, than the maximum in the fit of the r
distribution for the measurement in electrolyte, r
= 1.63. This is in agreement with the optical impression one gets, when simply looking to the images in Fig. 6. One possible explanation for the lower value would be the STM resolution at the island edges, which is lower in electrolyte than in vacuum, as the work function is approximately three times larger in the latter case.
44
One could try to compensate this effect by increasing the tunneling current, hence decreasing the distance between tip and sample, but this would also lead to a worse signal-to-noise and more instabilities in the feedback loop of the STM, as the same mechanical noise would be also exponentially increased. Next to this purely technical reason, the precise experimental details for the island creation also determines the degree of the dendritic shape and thus the measured r
, as we explain in the following.

Figure 8.
Statistical Distribution of
r
for the Best Estimate of the Island Shapes: (a) measured in vacuum (analyzed from Fig. 6a and Fig. S3 in the Supplementary Information) and (b) in electrolyte (analyzed from Fig. 6e and Fig. S5 in the Supplementary Information). The peak maximum of the statistical distributions, obtained by fitting with a Pearson IV function, are indicated with red dashed lines. The lower and upper bound are indicated with the orange arrow, while the statistical error, derived via the possible shift within the statistical error bars, is indicated with the green arrow.
Download figure:
Standard image High-resolution imageThe size of the islands is dictated by the ratio F/M that was described in the Introduction. A higher flux leads to smaller islands, while a higher mobility leads to larger islands.
17,25,80
While both the higher upper potential and the longer oxidation time, for the EC-STM measurements, result in a larger effective flux, the slower scan rate during the cathodic sweep results in a higher mobility (i.e. the adatoms have more time to diffuse before nucleating into islands). As becomes clear from the above, these two effects point toward opposite directions: a decrease and an increase, respectively, of the island area. However, by comparing the islands in Fig. 6a with the ones in Fig. 6e, one can see that the latter are significantly smaller (note the different image sizes), and therefore, the increase of the upper potential and the oxidation time must outweigh the higher mobility introduced by slower sweeping. Smaller islands are more difficult to analyze accurately than larger ones; recall the above explained tip convolution. This leads to a lower measured r
, as the small scale roughness at the island edges cannot easily be captured. As explained before, the faster the arrival of adatoms toward the island edge, the more pronounced the dendritic growth becomes. Therefore, a faster scan rate during the cathodic sweep, and thus faster reduction of the PtO2-adatoms to Pt-adatoms (which have a higher diffusion rate) results in a larger r
.
As delimiting the island edges is subject to error, we used the three estimates measured for each island to calculate the lower and upper errors of r
. This was done by dividing, respectively, the smallest perimeter with the largest area, and the largest perimeter with the smallest area, before averaging over all the islands. This resulted in a lower and an upper error of 23% and 25%, respectively, for the islands measured in vacuum. For the islands measured in electrolyte, we found an upper and lower error of 21% and 30%, respectively. Realizing both upper and lower errors, one could argue that there is no difference in the r
between both measurements, as the difference in the two peak maximum values falls within this error margin. This is clearly observed in Fig. 8, where both upper and lower boundaries are shown. However, the upper and lower bounds are larger than the natural statistical error that is given by the shifts of the distribution of r
to the left and to the right, taking into account the error of  , where N is the number of counts for each bin. This is not surprising as the upper and lower bounds describe the maximum total errors. The statistical errors, however, intrinsically project the real randomness of the island shape. Considering this error for both measurements, we conclude that, although there is a difference between the two peak maximum values of r
, the difference is very small: while the peak of r
from the measurement in vacuum (a) could be shifted down to 1.81, it could be shifted up to 1.7 in the measurement in the electrolyte (b), thus only 0.11 difference between them.
, where N is the number of counts for each bin. This is not surprising as the upper and lower bounds describe the maximum total errors. The statistical errors, however, intrinsically project the real randomness of the island shape. Considering this error for both measurements, we conclude that, although there is a difference between the two peak maximum values of r
, the difference is very small: while the peak of r
from the measurement in vacuum (a) could be shifted down to 1.81, it could be shifted up to 1.7 in the measurement in the electrolyte (b), thus only 0.11 difference between them.
In order to study further not only the effect of the experimental oxidation-reduction conditions chosen, but also the evolution of the island shape during growth, we created islands by performing ORCs between 0.06 V and 1.35 V and at 50 mV s−1. We then obtained the distributions of r
of the islands after 1, 6 and 7 ORCs, which we compared with the distribution of r
for the EC-STM measurement presented above in Fig. 6e, in which the potential was held at 1.35 V for 200 s. Figure 9 shows the four EC-STM images that we used for the comparison, and Fig. 10 shows the according statistical distributions of r
obtained for each case.

Figure 9. Degree of Dendritic Island Shape as Function of ORC-number. (a) As a reference, after holding the potential at 1.35 V for 200 s before reducing to 0.3 V with 25 mV s−1. (b) After the first ORC between 0.06 V and 1.35 V. (c) After 6 ORCs. (d) After 7 ORCs. All ORCs are performed with a scan rate of 50 mV s−1. All images are 80 × 80 nm2. Larger scale images are provided in the Supplementary Information.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10.
Evolution of the Statistical Distributions of
r
upon Oxidation-Reduction Cycling (a) Comparison of the statistical distributions obtained from the measurement described before in which the potential was held at 1.35 V for 200 s (blue dotted line), with the distributions obtained after 1, 6, and 7 ORCs, in green, orange and purple, respectively. The latter three distributions are also shown in (b), (c), and (d), respectively, where we also provide: a curve fit to highlight the peak maximum, error bars based on the statistical error at each bin, and a green arrow representing the shift of the peak maximum due to the statistical error.
Download figure:
Standard image High-resolution image{kind=link}
The first we note is that all islands in Figs. 9a–9d are dendritic, independently on the electrochemical treatment. The statistical distribution resulting from the potential hold experiment shows a larger r
value of 1.63 in comparison to the the r
= 1.57 of ORC #1. The reason for this is that at longer waiting times, more PEirr atoms are created, due to less disorder in the spoke-wheel like structures.
65,71
However, even if we handle the same "waiting" time, like it is within the ORCs, the distributions show that the islands become more compact with increasing ORC number: note the shift between ORC #1 and ORC #6, but also the slight shift to smaller values from ORC #6 to ORC #7. Although these shifts fall within the statistical errors of the distributions and hence should be treated with caution, the trend matches very well with the basic atomic details underlying the island growth, as provided in the following.
We have concluded earlier that above 0.8 V the steps oxidize, and that there is a large driving force to form PtO2-adatoms from the kinks. We have evidence that, in analogy to the PEirr on the terrace, the oxide chains at the steps also push PtO2 units up onto the upper terrace, thus becoming PtO2-adatoms. It was observed in Ref. 25 that the transition from 2D to 3D island growth (ocurring between the 20th and the 30th ORC) results in a significant decrease in the growth exponent. This can only be explained by a pronounced decrease of the flux, which can only be due to a change in the roughening (growth) mechanism. 109 The flux has two contributions, the PEirr atoms created on the terraces, and the PtO2-adatoms formed by oxidizing the steps. The mechanism, in which the latter takes place, could be similar to the PErev and the PEirr on a terrace: it starts with lifting PtO2 units (PErev atoms) along the step and, once a critical stress along the step-row is reached, one unit per certain row length is pushed onto the upper terrace. While the terrace width decreases considerably as the growth transitions from 2D to 3D (i.e. the terrace width of the 3D mounts becomes as small as 2 platinum distances 26 ), the step density simultaneously increases to its maximum. Therefore, the decrease in the flux must be due to the change of these two contributions. However, the flux does not go to zero, as the roughening (growth) continues to at least up to 170 cycles. As then the terraces in the 3D mounts cannot accommodate the oxide spokes, the dominating flux of PtO2-adatoms must come from the steps. Furthermore, as the growth of the mounts occurrs predominantly in height, the PtO2-adatom flux from the steps must go uphill. We have more evidence for this uphill flux from recent measurements at stepped surfaces.
Coming back to the explanation for the decrease of r
upon cycling, the uphill flux from the steps results in PtO2-adatoms on the island. These PtO2-adatoms (or Pt-adatoms, upon reduction), instead of nucleating, hop down the step and get incorporated in the island edge. This is because the Ehrlich-Schwoebel vanishes for small terrace widths, like the branches. For example, the critical terrace width observed on Ag(111) and Cu(111) is six atomic units.
83–85
Consequently, the "fjords" between the branches get filled. This effect is more dominant at the center of the islands, as the adatoms are bounced back several times, before they finally overcome the Ehrlich-Schwoebel barrier, leading to more compact shapes. This "annealing" is repeated during each ORC and competes with the dendritic growth in the perpendicular direction of the step. If the filling of the fjords is faster than the dendritic growth, r
will decrease after each cycle, meaning that the islands become more compact.
Finally, another cause for the decrease of r
upon cycling originates from the increase of the island perimeter upon growth. As this leads to longer steps with less kinks, the KESE becomes less relevant for the step-adatom diffusion along the island edge: the step-adatoms have more distance to diffuse along the step before finding a kink. This leads to an increase of the width of the branches at their base (see Fig. 7), and hence less dense branching, which results in a lower r
.
Conclusions
In this work, we have studied with DFT both the stability and the mobility of two different adatom species, Pt-adatom and PtO2-adatom, on Pt(111) by calculating their formation energies and diffusion rates at different electrode potentials, with the aim to get a better understanding on the nucleation of adatom islands upon oxidation-reduction cycling. Our results show that, although during the forward (anodic) scan the irreversible place-exchange that starts at 1.17 V, leads to the creation of PtO2-adatoms, nucleation into PtO2-islands does not occurr. The reason for this is that the equilibrium concentration of PtO2-adatoms reaches practically 1 ML at 0.94 V, and thus at these high potentials the complete surface wants to be oxidized. Nucleation into PtO2-adatom islands does not occur with ORCs up to 1.35 V and 50 mV s−1 scan rate, because there are only 0.0245 ML PtO2-adatoms created during the irreversible regime of the place-exchange, which is significantly lower than 1 ML. During the subsequent cathodic scan, the equilibrium concentration of PtO2-adatoms decreases exponentially with the potential and, below 0.94 V, it becomes lower than the actual PtO2-adatom concentration of 0.0245 ML. Therefore, nucleation becomes thermodynamically favorable. However, the PtO2-adatoms are "trapped" in between the oxide rows that are created during the reversible part of the oxidation. As these rows are lifted almost 1 ML high from the surface, nucleation is hindered by too low mobility. In addition, PtO2-adatoms diffuse very slow, 0.28 jumps/s, and hence they do not have enough time to find each other before they reduce to Pt-adatoms. Pt-adatoms diffuse much faster (108 jumps/s) and therefore, once they are formed at 0.59 V, they will rapidly find each other and nucleate into adatom islands, as also their actual concentration is much higher than their equilibrium concentration. These results match the observations from Wakisaka et al. 24 who reported that the adatom islands were not formed until the potential was decreased to 0.49 V.
As not only the electrocatalytic activity of platinum, but also its degradation is highly dependent on the concentration of the different surface sites, we also analyzed the shape of the islands formed from vacuum STM and EC-STM images. We quantified the island perimeter and the area to finally determine the island compactness and a factor that describes the additional step length respect to a circular island. Contrary to what was suggested in the literature, the islands formed upon oxidation reduction are dendritic, and not hexagonal, which matches the observations from MBE experiments in vacuum. Finally, we studied the shape evolution during the island growth upon oxidation-reduction cycling. Although within the statistical error bars of the distributions, we have indications that the dendritic islands change toward more compact shapes with increasing cycle number. This trend nicely matches our insight into the atomic picture during growth and can be explained by: (1) the uphill flux originating from the island edges, and the downhill interlayer diffusion of adatoms that end up filling the "fjords" between the island branches; and (2) longer step length for larger islands on which a step-adatom can diffuse without encountering a kink leading to less branching and thus less dendritic growth.
Acknowledgments
This work is part of the research programme TOP with project number 716.017.001, which is financed by The Netherlands Organisation for Scientific Research (NWO).
The authors acknowledge LVH Coatings for supplying their Clearclad electrophoretic paint.
The authors gratefully thank T. Maagaard and I. Chorkendorff for providing the STM images measured in vacuum as well as the permission for republication.
The authors gratefully acknowledge L. Jacobse, who skillfully performed high-resolution measurements at our EC-STM during his time as a Ph.D. student in Leiden. We used some of his images for our analysis. He is now a staff member at the Deutsches Elektronen-Synchrotron (DESY, Hamburg, Germany).
I. T. McCrum gratefully acknowledges start-up funding provided by Clarkson University. Some of the computational modeling was performed on the ACRES cluster at Clarkson University. We would like to thank Clarkson University and the Office of Information Technology for providing computational resources and support that contributed to these research results. These resources are supported in part by the National Science Foundation under Grant no. 1 925 596. This work also used the Extreme Science and Engineering Discovery Environment (XSEDE) which is supported by National Science Foundation grant number ACI-1548562.
Supplementary data (0.1 MB ZIP)