
Abstract
The electrolytes of the today omnipresent lithium-ion batteries (LIBs) have for more than 25 years been based upon 1 M LiPF6 in a 50:50 EC:DMC mixture—commonly known as LP30. The success of the basic design of the LP30 electrolyte, with many variations and additions made over the years, is unchallenged. Yet, some molecular level fundamentals of LP30 are surprisingly elusive: the structure of the first solvation shell of the Li+ cation is still a topic of current debate; the details of the dynamics are not fully understood; the interpretation of structural and dynamic properties is highly dependent on the analysis methods used; the contributions by different species to the ion transport and the energetics involved are not established. We here apply dynamic structure discovery analysis as implemented in CHAMPION to molecular dynamics simulation trajectories to bring new light on the structure and dynamics within LP30 and especially the (Li+) ion transport to rationalize further development of LIB electrolytes.
Export citation and abstract BibTeX RIS
There is today much reason to celebrate the success of the lithium-ion battery (LIB), both scientifically and technologically as well as for its societal impact. This was not the least acknowledged by the Nobel Prize in Chemistry in 2019. 1 It is, however, to be noted that the successfully commercialized cells of Sony in 1991 were based on an anode of coke. While graphite was already in 1978 2 identified as a suitable intercalation anode for LIBs, it came into play first in the mid-1990's. This extremely important change, paving the way for vastly improved cell energy density, was made possible by: (i) the seminal study by Dahn et al. showing that lithium intercalation into graphite was possible by the use of a cyclic carbonate electrolyte solvent; ethylene carbonate (EC), 3,4 and (ii) the electrolyte formulation launched by Tarascon and Guyomard based on LiPF6 dissolved in EC combined with a linear carbonate as co-solvent; dimethyl carbonate (DMC). 5,6 Finding a co-solvent to EC, to extend the liquid range and lower the viscosity, and thereby a much wider operating temperature window and higher ionic conductivities, had been pursued for quite some time, 7–9 until the EC:DMC mixture finally enabled the electrochemical stability window needed. The 1 M LiPF6 dissolved in a 50:50 EC:DMC mixture electrolyte, commonly known as LP30, today remains the baseline LIB electrolyte formulation, even if functional electrolytes 10–12 use a cocktail of various additives to solve/remedy/mitigate different practical shortcomings. 13 In all, the role of LP30 in the LIB success saga can hardly be overestimated.
The LP30 electrolyte is also truly the Drosophila of battery research; it is the benchmark for electrolyte development and electrode testing as well as for new analysis methods, studies of battery degradation and life-time, etc. Hundreds of other non-aqueous/aprotic solvents and solvent mixtures, and more than a handful of Li-salts, 14,15 have been the subject of extensive R&D, but LP30 and its extended family remain unchallenged.
It is notable that in different roadmaps the key performance indicator (KPI) of LIB electrolyte room temperature ionic conductivity most often has a target value of no more than 1 mS cm−1 for 2030 16 —one magnitude lower than that attainable today, ca. 10 mS cm−1. The hesitation in pushing this KPI further and its popularity originates in its simplicity as a proxy metric, while to properly and unambiguously assess the useful ion transport/mobility/migration of the charge carrier of interest (Li+) is much more difficult.
Hence, it might be constructive to consider what is actually known about the (Li+) ion transport in LP30. What is known about the structure and dynamics of the Li+ cation at the molecular level and how do different species and transitions between them contribute to the (bulk electrolyte) ion transport ? The bulk electrolyte energetics and the composition of the Li+ solvation shell also affect the solid electrolyte interphase (SEI) 17 and the Li+ cation de-solvation prior to intercalation constitutes ca. 3/4 of the energy barrier to overcome. 18
Time-resolved experimental techniques able to directly capture the LP30 electrolyte ion transport mechanism are missing, which is why much of the research performed is rather optimization efforts; numerous modes of correlations and statistics are employed to decipher how different macroscopic properties (permittivity, viscosity, glass transition temperature, etc.) affect the ionic conductivity. 19 Many important features can be explained at the phenomenological level: mass transport and diffusion limitations, temperature dependencies, power rate limits, etc., and be integrated in e.g. Newman style modelling 20,21 and the Advanced Electrolyte Model. 22 However, no detailed understanding of the ion transport mechanism itself emerges, beyond, at least in hindsight, obvious notions: the Li+ cations are solvated by the solvents, while the anions are much less strongly solvated, and therefore the cation transport number (t+) is inferior to the anion transport number (t-) due to the larger size of the solvated cation vs anion; the extent of cation-anion ion-pairing/aggregation vary by salt, solvent, and concentration, why weakly coordinating anions, such as PF6 −, and high permittivity solvents are employed. This all connects back to theoretical frameworks such as Fuoss and Onsager on the conductance of dilute electrolytes, 23 Bjerrum's criteria of ion-pairing, 24 and the Stokes-Einstein and Nernst-Einstein equations for ion mobility and conductivity. 25
To provide a deeper insight into the Li+ solvation and dynamics within LP30 we here apply our unique dynamic structure discovery analysis 26 as implemented in CHAMPION (Chalmers Hierarchical Atomic, Molecular, Polymeric & Ionic analysis toolkit) 27,28 and commercialized by Compular. 29 From classical molecular dynamics (MD) simulation trajectories we highlight the complexity in local structure and dynamics and reveal how/whether the global electrolyte structure and the diffusive (Li+) ion transport is coupled, including transitions involved and individual contributions to both vehicular and non-vehicular (Li+) ion transport.
Computational
MD simulation methodology and set-up
A classical MD simulation of 1 M LiPF6 in EC:DMC (50:50 by weight) was performed employing a many-body polarizable APPLE&P force field 30–32 that was modified by refitting charges of EC and DMC to the electrostatic potential obtained at MP2/aug-cc-pVTZ level, and using repulsion parameters between Li+ and O/C/H of EC and DMC sampled on a grid around EC and DMC using the Perdew–Burke–Ernzerhof (PBE) functional with the 6–31 + G(d,p) basis set. The intramolecular interactions between charges and induced dipoles within EC, PF6 − and DMC were excluded with the exception of CH3−....CH3 groups of DMC. The MD simulation cell contained 312 EC, 306 DMC and 48 liPF6. After 2 ns equilibration at 333 K and 22 ns equilibration in NPT ensemble at 298 K, 100.8 ns NVT runs were performed at 298 K and used for calculating viscosity using stress tensor and diffusion coefficients via the mean squared displacement (MSD). A time reversible (RESPA) integrator was used with the following time resolutions: (i) the contribution from bonds and angles to the forces were calculated at any 0.5 fs, (ii) the contribution of dihedrals and non-bonded forces within 8 Å cut-off were updated at any 1.5 fs, and (iii) the remainder of the forces, reciprocal space Ewald and non-bonded forces within 16 Å cut-off, were updated at any 3 fs. A Nose-Hoover thermostat was used for temperature control with an associated frequency of 0.01 fs−1 and two chains. Induced dipoles (μind) were found self-consistently every 3 fs using tolerance of Δμ2 < 10–8 (e*Å)2, where e is the electron charge. Coordinates were saved every 2 ps for analysis. A stress tensor was saved every 9 fs and used for calculating viscosity using the Einstein relation, including both diagonal and non-diagonal elements to enhance statistics. 30,33
CHAMPION analysis
We analyzed three widely separated 2 ns intervals of the MD trajectory, that all were consistent, and the detailed dynamic structure discovery analysis is based on one of these. First, all covalent and coordination bonds were identified, using the dynamic structure discovery algorithm 26–28 (Fig. 1). Bound pairs of atoms are detected based on the dynamics of their pairwise distances:
- a)Time windows are identified wherein the distance between a pair of atoms is less than
 0.8 times the sum of their van der Waals radii. The pair forms a candidate bond during this time window.
0.8 times the sum of their van der Waals radii. The pair forms a candidate bond during this time window. - b)For each candidate bond, the average distance is compared with the first peak of the partial radial distribution function (pRDF) for the pair of species, and the candidate is eliminated if it falls outside a tolerance of 10% of the full-width at half-maximum (FWHM) around the peak.
- c)To eliminate next-nearest neighbors, an exclusion cone is formed along each shorter bond involving one of the atoms in the candidate bond, expanding out at a 50° angle from the common atom.

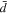
If all criteria pass, the bond is considered to form the first time the distance goes below the pRDF peak and break the last time this distance is exceeded.

Figure 1. Criteria for whether a candidate bond is accepted: (a) during a time window, two atoms are closer than  0.8 times the sum of their van der Waals radii, (b) during the same window, their average distance is within 10% of FWHM of
0.8 times the sum of their van der Waals radii, (b) during the same window, their average distance is within 10% of FWHM of  the first peak in their pRDF, (c) the candidate bond does not lie within a 50° exclusion cone around a shorter bond involving one of the atoms.
the first peak in their pRDF, (c) the candidate bond does not lie within a 50° exclusion cone around a shorter bond involving one of the atoms.
Download figure:
Standard image High-resolution imageSubsequently, time-dependent bond graphs were constructed based on the time-dependent bonds, where the global structure is characterized by connected components of the global bond graph (Fig. 2a) and the 1st solvation shells of the Li+ ions are identified with the graph neighborhoods of Li+ (Fig. 2b) up to 4 bonds out. We collectively refer to these topologically defined structures as structural species. Different structural species are distinguished by their graph topology. The structural species are assigned probabilities based on the time-averaged fraction of Li+ ions in each structure.

Figure 2. Schematic illustration of two ways of partitioning the global bond graph. (a) Connected components, (b) neighborhoods of the green species, one bond out from the central species.
Download figure:
Standard image High-resolution imageThe components and solvation shells were then treated statistically to enable analysis of: the global and local structures and their lifetimes, the transitions between them, and their contributions to diffusive ion transport. With respect to global and local structure, this enabled a detailed analysis of the global structure as well as the cation 1st solvation shell composition and dynamics including the Li+ (partial) solvation numbers ((p)SNs) and coordination numbers ((p)CNs), where the former is the number of ligands, and the latter the number of coordination bonds, of a Li+ ion, and partial denotes that only one type of ligand is considered. Note that (p)SN and (p)CN are identical when only monodentate bonding takes place but differ when bidentate bonding is present.
Transport mechanisms were distinguished based on an additive decomposition of the instantaneous velocity vector of an ionic or molecular species (e.g. Li+) into a vehicular part given by the center-of-mass velocity of its connected component, and a non-vehicular part, making up the remainder: 
For the vehicular transport mechanism, defined as transport without changes in the graph neighborhood (and thus also including independent motion of uncoordinated ions and molecules), the additive contribution,  from each Li+ structural species
from each Li+ structural species  to the total Li+ diffusivity,
to the total Li+ diffusivity,  was computed by
was computed by

where  is a time interval,
is a time interval,  is a point in time,
is a point in time,  is the position of atom
is the position of atom 
 is an indicator function denoting whether
is an indicator function denoting whether  is part of structural species
is part of structural species  at time
at time  and
and  is the vehicular velocity of atom
is the vehicular velocity of atom  The expression in angle brackets is averaged over all atoms
The expression in angle brackets is averaged over all atoms  of the molecular species, starting points
of the molecular species, starting points  and time intervals
and time intervals  beyond the onset of the diffusive regime,
beyond the onset of the diffusive regime,  Summing up
Summing up  over all
over all  gives the total vehicular diffusivity.
gives the total vehicular diffusivity.
The effective diffusivity  of a structural species was obtained by dividing
of a structural species was obtained by dividing  by the probability
by the probability  of Li+ being in structure
of Li+ being in structure  —it is thus a measure of its mobility in terms of its contribution to Li+ diffusivity in particular.
—it is thus a measure of its mobility in terms of its contribution to Li+ diffusivity in particular.
We similarly computed the additive contributions to the Li+ diffusivity from transitions from structural species  to
to 
 by
by

where  is an indicator function active between the midpoints of when
is an indicator function active between the midpoints of when  is in structure
is in structure  and
and  and
and  is the non-vehicular part of the velocity. Summing over all
is the non-vehicular part of the velocity. Summing over all  gives the total non-vehicular contribution, and summing over both
gives the total non-vehicular contribution, and summing over both  and
and  gives the total diffusivities.
gives the total diffusivities.
 is in turn a product of several factors,
is in turn a product of several factors,  where
where  is the probability of structure
is the probability of structure 
 is the transition rate from
is the transition rate from  to
to  and
and  is the contribution to the Li+ MSD associated with each transition event. Taking the square root of the latter gives
is the contribution to the Li+ MSD associated with each transition event. Taking the square root of the latter gives  which is a measure of the average contribution from the transition to the total net distance traveled by Li+ in the long-time limit. The product of the probability and the transition rate together make up the frequency of the transition,
which is a measure of the average contribution from the transition to the total net distance traveled by Li+ in the long-time limit. The product of the probability and the transition rate together make up the frequency of the transition, 
The effective diffusivities  of structural species containing Li+ and PF6
− were further utilized to compute the contributions to ionic conductivity from each species,
of structural species containing Li+ and PF6
− were further utilized to compute the contributions to ionic conductivity from each species,

where  is the contribution to the conductivity of ionic species
is the contribution to the conductivity of ionic species  from structural species
from structural species 
 Faraday's constant,
Faraday's constant,  the gas constant,
the gas constant,  the temperature,
the temperature,  the concentration of species
the concentration of species 
 its net charge, and
its net charge, and  the charge contribution from ionic species
the charge contribution from ionic species  Consequently, summing over
Consequently, summing over  for a given
for a given  gives the total cationic or anionic conductivity and summing over
gives the total cationic or anionic conductivity and summing over  for a fixed
for a fixed  gives the total contribution to ionic conductivity from structural species
gives the total contribution to ionic conductivity from structural species 
Based on the calculations of diffusivities and the contribution of each ionic species to the total ionic conductivity, we can define two different measures of the transport number,  one based on the relative diffusivity of Li+:
one based on the relative diffusivity of Li+:
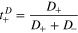
and one based on its relative conductivity:

When computing means, variances and correlations over the population of structural species and transitions, there is a need to weight the statistical moments with the amount of data on which each sample is based, since a large fraction of the individual structures and transitions are based on very few exemplars, and thus their estimated properties very uncertain. For moments related to the properties of individual structures we therefore used their probabilities  as weights, and for properties related to transitions, we weighted by their frequency
as weights, and for properties related to transitions, we weighted by their frequency  A more thorough description of the algorithms employed is provided in the CHAMPION method paper.
27,28
A more thorough description of the algorithms employed is provided in the CHAMPION method paper.
27,28
Results and Discussion
We first consider both the global and local structure of LP30, especially the composition and structure of the 1st solvation shell of Li+. We then turn to the dynamics of the solvation shell by the residence times of all species: PF6 –, EC and DMC, which is integrated with the overall issue of transport properties, by evaluating the diffusivity as well as partitioning into vehicular and non-vehicular contributions, and then connect back to frameworks such as the Stokes-Einstein equation. From this we create qualitative pictures of the Li+ ion structure, dynamics and transport mechanism, and can (semi-)quantitatively rank the most important contributors to the (Li+) ion transport w.r.t. local structure, dynamics and transitions.
Structure
Considering first the overall structure of LP30 visually (Fig. 3), we find that most of the Li+ ions have tetrahedral 1st solvation shells dominated by, and in turn also surrounded by, solvent molecules. There are some anions involved, but most are free from Li+ coordination. The overall picture is that the numerical preponderance of solvent molecules in LP30, being a 1 M electrolyte, effectively screens the ions from interacting directly with each other, precluding the aggregation which is observable for higher salt concentrations from the same kind of analysis. 34

Figure 3. A snapshot of the MD simulation cell. Connected components containing Li+ ions are emphasized by thicker bonds. Element colors: purple: Li; orange: P; cyan: F; red: O; grey: C; white: H.
Download figure:
Standard image High-resolution imageMore quantitatively the distributions of species for Li+ containing connected components show that there are hardly any aggregates present,  with
with  denoting the number of Li+ ions in a connected component, with only a very negligible contribution from
denoting the number of Li+ ions in a connected component, with only a very negligible contribution from  (Fig. 4a). Thus, the structure in terms of Li+ containing connected components is in essence equivalent to the Li+ 1st solvation shells. Indeed, also the solvation shell composition (Figs. 4b–4c) and especially the pCN distributions (Fig. 4b) are almost identical to the connected component species distributions (Fig. 4a). In more detail, two-thirds of the PF6
– ions are free (pCNPF6−= 0), with pCNPF6− = 1 making up most of the remainder. The solvents, EC and DMC, both form bell-shaped pCN distributions, centered around 2 and 1, respectively. The average composition of the Li+ 1st solvation shells (Fig. 4c) has EC > DMC > PF6
–, and a total CN of 4.0. This is at large in agreement with the literature, even if qualitative and quantitative differences exist, often due to the different methods applied. Many studies do support a much more preferential Li+ solvation by EC in bulk LP30. Experimentally ca. 90% being EC was obtained by ex situ ESI-MS,
35
leaving most of the DMC non-coordinated. Computationally, already in 1994/1995, Ue
36
and Blint
37
modelled [Li(EC)x
]+ complexes by ab initio methods, and experimentally, again by ex situ ESI-MS analysis, a predominance of x = 2–3 was obtained.
38,39
The more expected CN, closer to 4–5, was arrived at later by e.g. a combined Raman spectroscopy and density functional theory (DFT) study showing a preference for a [Li(EC)4]+ complex of S4 symmetry i.e. CN = 4.
40
Some differences may arise due to the very criteria used to define the connected components; Chen et al.
32
arrived at 2% of aggregates and 38% of ion-pairs. They used a Li-P cutoff of 4.4 Å, while the CHAMPION analysis is based on detecting Li-F coordination bonds and on how their lengths change over time,
27,28
and the former does thus not cover anion rotation the same way.
(Fig. 4a). Thus, the structure in terms of Li+ containing connected components is in essence equivalent to the Li+ 1st solvation shells. Indeed, also the solvation shell composition (Figs. 4b–4c) and especially the pCN distributions (Fig. 4b) are almost identical to the connected component species distributions (Fig. 4a). In more detail, two-thirds of the PF6
– ions are free (pCNPF6−= 0), with pCNPF6− = 1 making up most of the remainder. The solvents, EC and DMC, both form bell-shaped pCN distributions, centered around 2 and 1, respectively. The average composition of the Li+ 1st solvation shells (Fig. 4c) has EC > DMC > PF6
–, and a total CN of 4.0. This is at large in agreement with the literature, even if qualitative and quantitative differences exist, often due to the different methods applied. Many studies do support a much more preferential Li+ solvation by EC in bulk LP30. Experimentally ca. 90% being EC was obtained by ex situ ESI-MS,
35
leaving most of the DMC non-coordinated. Computationally, already in 1994/1995, Ue
36
and Blint
37
modelled [Li(EC)x
]+ complexes by ab initio methods, and experimentally, again by ex situ ESI-MS analysis, a predominance of x = 2–3 was obtained.
38,39
The more expected CN, closer to 4–5, was arrived at later by e.g. a combined Raman spectroscopy and density functional theory (DFT) study showing a preference for a [Li(EC)4]+ complex of S4 symmetry i.e. CN = 4.
40
Some differences may arise due to the very criteria used to define the connected components; Chen et al.
32
arrived at 2% of aggregates and 38% of ion-pairs. They used a Li-P cutoff of 4.4 Å, while the CHAMPION analysis is based on detecting Li-F coordination bonds and on how their lengths change over time,
27,28
and the former does thus not cover anion rotation the same way.

Figure 4. (a) Probability distribution for Li+ to be in a connected component with n exemplars of each species, (b) pCN distributions for Li+, (c) average composition of Li+ 1st solvation shells.
Download figure:
Standard image High-resolution imageAll of the above strategies and methods are very dependent on the time- and length-scales probed, and more or less provide static, averaged or superimposed, pictures of the very local structure. In contrast, we here show that beyond the Li+ 1st solvation shells, 66% of the PF6 –, 68% of the EC, and 76% of the DMC are free from direct cation interaction. Furthermore, the detailed structure and composition of the most common Li+ 1st solvation shells reveal that only three types make up more than half of them: [Li(EC)2(DMC)2]+, [Li(EC)3(DMC)]+, and [Li(PF6)(EC)2(DMC)]0 (Fig. 5), in accordance with cluster-continuum DFT calculations. 41 Notably the above quoted dominant [Li(EC)4]+ contributes by less than 5% and thus the picture arrived at by Johansson et al. 40 for LiClO4 in EC/PC is an oversimplification that does not necessarily hold for LP30. Overall, there is quite a diversity in the solvent shell compositions; the top 10 make up no more than 84% and twice as many are needed to reach 97%. As expected, both EC and DMC coordinate to Li+ exclusively by their carbonyl groups and furthermore no bidentate coordination is observed, neither for solvents nor anions (why SN ≡ CN).

Figure 5. The 20 most common Li+ 1st solvation shell structures, compositions, and Li+ probabilities (in percentages). Element colors: purple: Li; orange: P; cyan: F; red: O; grey: C; white: H.
Download figure:
Standard image High-resolution imageOverall, most of the top 20 1st solvation shells (11) have a CN = 4, 5 of them a CN = 3, and 4 of them a CN = 5. Scrutinizing the data more, the CN = 4 is, however, in reality even more dominant; the top 6 1st solvation shells all have a CN = 4. There is also a clear trend for PF6 –; the top 10 has only pCNPF6- = 0 or 1, thus there are rather few anion-containing 1st solvation shells in LP30, consistent with the literature and the notion of very few contact ion-pairs (CIPs) observed spectroscopically.
Dynamics and transport
To connect with the structural analysis above, we start by analyzing the local dynamics of the 1st solvation shells (Fig. 4a) by the survival probabilities of the coordination bonds and computed mean residence times of the Li+ ligands (Fig. 6). When Li...F contacts are considered, F(PF6 –), PF6 – has by far the shortest residence time, 12 ps. The residence time of P(PF6 –) is double that, 25 ps, implying that the PF6 – ions rotate between on average two F atoms before (momentarily) losing their coordination with Li+ (Fig. 6a). Allowing for breakage of coordination bonds followed by re-entry at most 20 ps later (when 93% of eventual re-entries have occurred), increases the calculated average lifetimes of ion-pairs by an order of magnitude to 246 ps (Fig. 6b). In fact, defining the residence time as the time spent within a cut-off distance of 4.4 Å, gives a residence time as long as 678 ps for P(PF6 –). This basically means that even when the cation-anion coordination bonds are broken, the ions in the pair stick together at relatively close proximity and very often reunite after a short hiatus. Indeed, over a 2 ns trajectory, the average number of re-entries of broken bonds was found to be 19.8 for P(PF6 –) and 3.9 for F(PF6 –), further strengthening this picture.

Figure 6. Survival probabilities for coordination bonds between Li+ and EC/DMC/PF6 –(F)/PF6 –(P).
Download figure:
Standard image High-resolution imageThe residence times of EC and DMC are easier to interpret. Without considering re-entries, EC and DMC have average residence times of 122 and 161 ps, respectively. With re-entries, they both roughly double, to 246 and 327 ps, with that of EC indistinguishable from that of P(PF6 –) determined by the same method. It is notable that EC has the shortest residence time considering that its binding energy is larger than that of DMC to a Li+ ion 41 and that EC is only ca. 2% more numerous to DMC overall in the LP30 electrolyte, but is ca. 40% more common in the 1st solvation shell (Fig. 4c). The overall picture emerging is thus fast ligand exchange for EC, resulting in a new (or old) EC entering rather than DMC (or PF6 −). Considering the number of re-entries per broken coordination bond, they are significantly lower than for Li..P(PF6 – ), and only slightly higher, 1.6, for EC than for DMC, 1.2.
Data points are samples, solid lines stretched exponential fits, and dotted lines mean residence times.(a) Including only contiguous bonds as registered by the bond detection algorithm, (b) Including also bonds that are broken and reformed within 20 ps.
For the analysis of the ion transport, we start by the traditional measure of the MSDs of the different species, where the high degree of linearity points to the MD simulation having reached a diffusive regime for all species (Fig. 7a), and from these we also compute their diffusivities (Fig. 7b). Based on DLi+ =  m2s−1 and DPF6–
m2s−1 and DPF6–
 m2s−1 we obtain a cation transference number
m2s−1 we obtain a cation transference number  = 0.47, which is much higher than expected based on experimental experience, but a rather common feature in computational studies. Analysis of the full trajectory of 100.8 ns yielded self-diffusion coefficients DLi+, DPF6, DEC and DDMC in good agreement with pfg-NMR spectroscopy derived: 2.3 vs 2.45 × 10–10, 3.0 vs 3.5 × 10–10, 4.95 vs 4.59 × 10–10 and 5.5 vs 5.97 × 10–10 m2 s−1, respectively.
42
In addition, and as expected, EC and DMC have similar diffusivities, DDMC > DEC, and are both roughly twice as diffusive as the ions.
= 0.47, which is much higher than expected based on experimental experience, but a rather common feature in computational studies. Analysis of the full trajectory of 100.8 ns yielded self-diffusion coefficients DLi+, DPF6, DEC and DDMC in good agreement with pfg-NMR spectroscopy derived: 2.3 vs 2.45 × 10–10, 3.0 vs 3.5 × 10–10, 4.95 vs 4.59 × 10–10 and 5.5 vs 5.97 × 10–10 m2 s−1, respectively.
42
In addition, and as expected, EC and DMC have similar diffusivities, DDMC > DEC, and are both roughly twice as diffusive as the ions.

Figure 7. (a) MSDs with the 95% confidence intervals as lighter bands around the means, (b) diffusivities, subdivided into vehicular (dark) and non-vehicular (light) contributions, (c) diffusivities of PF6 –, EC and DMC subdivided between "free" (left) and Li+-coordinated (right), and further subdivided into vehicular (dark) and non-vehicular (light) contributions. The percentages in b and c are the vehicular fractions.
Download figure:
Standard image High-resolution imageUsing the effective diffusivities of the species together with concentrations and charges we also computed and decomposed the ionic conductivity. We predicted a total ionic conductivity  mS cm−1, in excellent agreement with experimental values.
43
We further decomposed this into the conductivities of Li+, 4.0 mS cm−1, and PF6
−, 6.9 mS cm−1, giving a transference number
mS cm−1, in excellent agreement with experimental values.
43
We further decomposed this into the conductivities of Li+, 4.0 mS cm−1, and PF6
−, 6.9 mS cm−1, giving a transference number  which still is somewhat larger than usually expected, but notably lower than that based only on the MSD data (0.47).
which still is somewhat larger than usually expected, but notably lower than that based only on the MSD data (0.47).
By the CHAMPION analysis we decompose these total diffusivities into vehicular and non-vehicular contributions (Fig. 7b). The former is dominant for all species, but significantly less so for Li+; one-third of the transport is non-vehicular, similar to conclusions made for an LiTFSI-EC electrolyte. 44 While this is not often being a transport mechanism at all considered for the LP30 electrolyte this stands to reason; for Li+ a CN = 4 is most common, whereas the anions and solvents both mostly are "free"/non-coordinated and tend to coordinate to at most 1 li+, and hence there are many more opportunities for Li+ to be involved in structural rearrangements, some of which may lead to net ion transport. It is also logical and consistent that EC has a higher fraction of non-vehicular transport than DMC, due to its shorter residence time (Fig. 6).
Considering separately the diffusivities of free and Li+-coordinated anions and solvent molecules (Fig. 7c), the latter in general levies a penalty on the diffusivities. This effect is weakest for PF6 –, which can be understood from its already rather low diffusivity. For EC the decrease is somewhat larger (46%) than for DMC (34%), which correlates with its larger binding energy to Li+, 41 but a complicating feature is the ligand exchange mechanism outlined above, i.e. that some "free" EC molecules keep close to Li+ even if not contained in its 1st solvation shell per se.
Finally, we decompose also these above separately handled diffusivities into vehicular and non-vehicular contributions, showing that for all, the non-vehicular contribution increases upon Li+ coordination. While again expected, since coordinated species have more opportunities for structural rearrangements, it also shows that PF6 – and DMC are almost exclusively having a vehicular transport mechanism when "free," whereas EC has 12% non-vehicular diffusion even when "free." This may seem counterintuitive, but gives further support to the observation that "free" EC stays in the vicinity of the Li+ 1st solvation shell and its transport is facilitated by frequent solvation and de-solvation events, in a way not being the case for neither PF6 – nor DMC.
To further elucidate the ion transport mechanism(s), the individual contributions to the Li+ diffusivity are ranked (Figs. 8–9), starting with the vehicular transport contributors (Fig. 8). Overall, this ranking of vehicular diffusivity contributions amongst the 1st solvation shell structures is very similar to the ranking of their probabilities (Fig. 5); the top 20 are the very same structures, albeit in slightly different orders.

Figure 8. The 20 largest contributors to the vehicular Li+ diffusion with: rank ("〈number〉.), probability rank ("#〈number〉", cf Fig. 5), chemical formula ('[...]'), relative contribution to total Li+ diffusivity ("〈number〉%"), and Li+ probability ("(〈number〉%)"). Element colors: purple: Li; orange: P; cyan: F; red: O; grey: C; white: H.
Download figure:
Standard image High-resolution image
Figure 9. The 9 transitions giving the largest contributions to Li+ diffusivity with: rank, probability ranks of the structures (cf Fig. 5), and relative contributions to non-vehicular and total Li+ diffusivities.
Download figure:
Standard image High-resolution imageThe dominance of the vehicular ion transport mechanism is clear from considering also the 9 transitions with the largest contributions to Li+ diffusivity (Fig. 9). The most important non-vehicular contribution only ranks as the 11th most important transport contribution overall and accounts for merely 1.5% of the total transport. In addition, it also accounts for only 4.4% of the total non-vehicular diffusivity, indicating a much more diverse non-vehicular mechanism, which is expected since the number of possible transitions should be proportional to the square of the number of structures. 34 Again, EC solvent exchange is seen to contribute much more to the ion transport than does DMC solvent exchange, while PF6 – exchange does not contribute to any extent at all. Furthermore, there seems to be no strong preference for neither associative nor dissociative mechanisms.
All of the above can be discussed in a context of and contrasted with the Stokes-Einstein relation, often suggested to be (at least partly) fulfilled by "simple" electrolytes such as LP30. To this end, we performed a linear regression of the effective diffusivity as a function of the inverse radius of the structures, weighted by their probability (Fig. 10a). Using the slope of the fitted line as input to the Stokes-Einstein equation renders a dynamic viscosity of 0.37 mPa*s, which is almost an order of magnitude lower than we obtained by a more conventional calculation, 2.8 mPa*s, in fair agreement with the experimentally determined 3.2 cP (cP ≡ mPa*s) at 30°C. 45 It should be noted that we do not expect a very good estimate for the viscosity from the Stokes Einstein method, for two main reasons: (i) It is not clear what measure of radius that would be relevant for the solvation shells—we here used a very naive measure; the mass-weighted root-mean-squared distance to the center-of-mass, and (ii) The whole method of estimation is predicated on the assumption that the Stokes-Einstein equation holds, which the rather modest correlation coefficient found (r = 0.34) puts into question. Nevertheless, taken together, the clear but modest correlation in Fig. 10a and the more or less numerically reasonable prediction for the viscosity, allow us to conclude that the Stokes-Einstein relation indeed is a useful qualitative framework for understanding the dynamics and Li+ ion transport in LP30. Figure 10a also clarifies why most structures contribute to the total Li+ diffusion more or less in proportion to their probability; the radii range is rather narrow for the Li+ 1st solvation shells. In stark contrast, the inverse radii and diffusivities of PF6 –, EC, and DMC are all scattered around the regression line (Fig. 10b) and though within 50% of the viscosity corresponding to the regression, the Stokes-Einstein relation is clearly not by itself enough to fully explain their transport. We therefore argue that the different local viscosities "felt" by the different species, i.e. frictions, matters. PF6 –, being an anion, experiences greater friction than do the neutral EC and DMC solvent molecules, and EC, with its higher permittivity, greater than DMC. That the Li+ containing structures are intermediate can be explained by their exteriors, which are mixtures of EC, DMC and PF6 –. Thus, the Maxwell-Stefan model, including pair-wise friction coefficients between the species, may give a more accurate description.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Effective diffusivity as a function of the inverse radius, with the probability weighted average (+) and the regression line: (a) only Li+ containing connected components, and (b) including "free" PF6 – (olive), EC (black), and DMC (red). The opacities of the Li+ containing connected components data points increase as a function of their probability.
Download figure:
Standard image High-resolution image{kind=link}
We finally investigate the effect of the Li+ ligand exchange on the non-vehicular transport mechanism (Table I). Amongst the different species the solvents clearly dominate quantitatively; EC exchange is involved in 58%, DMC in 37% and PF6
− in 16%. Overall, the contributions are very equally distributed between addition and removal of ligands. Expanding  into its constituents, the most striking feature is that the probability (
into its constituents, the most striking feature is that the probability ( ) and the transition rate (
) and the transition rate ( ) are strongly negatively correlated (
) are strongly negatively correlated ( ). This is, however, quite logical; the more stable structures with lower transition rates last longer and hence accumulate in the population. In even more detail,
). This is, however, quite logical; the more stable structures with lower transition rates last longer and hence accumulate in the population. In even more detail,  correlates positively with
correlates positively with  (
( ) and negatively with
) and negatively with  (
( ), thus a high probability for the precursor is more important than rapid dynamics. Furthermore, the transition frequency accounts for the greater contribution from EC exchange as compared to DMC, having very similar
), thus a high probability for the precursor is more important than rapid dynamics. Furthermore, the transition frequency accounts for the greater contribution from EC exchange as compared to DMC, having very similar  In contrast, the lesser contribution from PF6
− exchange is mainly explained by its shorter
In contrast, the lesser contribution from PF6
− exchange is mainly explained by its shorter  being roughly only half of the distance associated with the solvent exchange, which is in line with its diffusivity being roughly half the solvent diffusivity. The rather large difference in frequency between addition and removal of PF6
− we can only ascribe to statistical fluctuations, as there is no overall trend towards aggregation throughout the trajectory.
being roughly only half of the distance associated with the solvent exchange, which is in line with its diffusivity being roughly half the solvent diffusivity. The rather large difference in frequency between addition and removal of PF6
− we can only ascribe to statistical fluctuations, as there is no overall trend towards aggregation throughout the trajectory.
Table I.
Relative diffusivity contributions (
 ), fractions of total frequency (
), fractions of total frequency ( ), and root mean squared displacement (
), and root mean squared displacement ( ) contributions of transitions involving all exchanges, as well as decomposed into additions and removals, of EC, DMC and PF6
−.
) contributions of transitions involving all exchanges, as well as decomposed into additions and removals, of EC, DMC and PF6
−.
| Exchange | D*/D [%] | νrel [%] |
 [Å] [Å] | |
|---|---|---|---|---|
| EC | Total | 58 | 45 | 0.54 |
| Addition | 29 | 25 | 0.51 | |
| Removal | 29 | 21 | 0.56 | |
| DMC | Total | 37 | 23 | 0.60 |
| Addition | 18 | 12 | 0.58 | |
| Removal | 19 | 11 | 0.62 | |
| PF6 − | Total | 16 | 35 | 0.32 |
| Addition | 8 | 22 | 0.29 | |
| Removal | 7 | 13 | 0.35 |
Concluding Remarks
There is somewhat of a false paradigm within the battery research community that the development of the "inactive" electrolyte follows from and must be made to adhere to the requirements set by the "active" electrodes. Therefore, it is a sobering thought that the success of the LIB largely was made possible by the LP30 electrolyte, but at the same time it is discouraging that it was made despite how little we knew, and still know, about (the origins of) its ion transport properties.
The analyses made here demonstrate how looking at structures in a time-resolved manner can reveal how the Li+ ion transport originates from a wide variety of distinctly different local structures and dynamic transitions between them contributing differently and gives pictures and details beyond averages. It definitively serves to verify that a vehicular transport mechanism for a small/fixed set of local structures is simply a too simple description.
An increased understanding of the ion transport mechanism in the bulk, by the rather modest means here used, is also needed to be able to tackle the electrolyte/electrode interfaces/interphases (and hence assist also in the development of electrodes). The latter, being instrumental for progressing LIBs further, is the target for, for example, the very large efforts currently being made within the BIG-MAP consortium 46 where standard liquid electrolytes are the first in line to study. Therein, and elsewhere, the CHAMPION style of analyses presented herein can assist, complement, and be part of both multi-scale modelling (MSM), 47 and different artificial intelligence/machine learning (AI/ML) 48,49 based developments of new LIB electrolytes.
Acknowledgments
R. A. and P. J. gratefully acknowledge the funding received through the European Union's Horizon 2020 research and innovation programme under Grant Agreements No. 666221 (HELIS) and No. 957189 (BIG-MAP), the latter being part of Battery 2030+, and from the Swedish Energy Agency through "Batterifonden"; grants P43525–1 and P39909–1. R.A. and P.J. have conflict of interest w. r. t. Compular 29 as co-founders and co-owners. P.J. acknowledges the immense support from the Swedish Research Council's Distinguished Professor Grant and from Sweden's Innovation Agency (VINNOVA) through Battery Alliance Sweden (BASE). We also acknowledge the support from the Theory and Modelling scheme of Advanced User Support from Chalmers Areas of Advance: Materials Science to R.A. The MD simulations performed by O.B. at ARL were supported by the Assistant Secretary for Energy Efficiency and Renewable Energy, Vehicle Technology Office of the U.S. DOE through Applied Battery Research for Transportation (ABRT) program via interagency agreement 89243322SEE000018 supporting contract No. DE-SC0012704.