
Abstract
The recent development of single-molecule sensors (SMS), which detect individual targets one at a time, allows determination of ultra-low concentrations of structurally similar compounds from a complex matrix. Protein nanopores are one of the earliest methods able to resolve the signal from a single molecule, and have already been successfully employed in commercial DNA sequencers. The protein nanopore based SMS, however, remains challenging, largely because the quantitative single-molecule analysis requires recording a sufficient number of signals for statistical significance within a reasonable time frame, thus restricting the lower limit of detection. This review aims to critically evaluate the strategies developed in this field over the last two decades. The measurement principle of nanopore SMS is first elucidated, followed by a systematic examination of the eight common protein pores, and a comprehensive assessment of the major types of sensing applications. A particular emphasis is placed on the intrinsic relationship between the size and charge of protein nanopores and their sensing capabilities for different kinds of analytes. Innovative approaches to lift the performance of nanopore SMS are also analyzed in detail, with a prediction at the end of the most promising future applications.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Single-molecule sensors (SMS) are an emerging type of sensors with the ultimate sensitivity and detection limit for chemical or biological assays. 1,2 By measuring the individual target molecules and counting their exact number, SMS may enable the accurate identification of rare but crucial species in complex samples, 3 as well as the potentially calibration-free determination of analyte concentration. 4 Nanopore is one of the earliest methods that can detect single molecules under ambient conditions. 5 Thanks to its ultra-high spatiotemporal resolution, the proof-of-principle demonstrations of nanopore SMS were first realized in 1999–2000 by Hagan Bayley. 6–8 However, despite the early success, the progress of developing practical nanopore SMS was unfortunately slow, in stark contrast to the single-molecule nanopore DNA sequencers that have now become commercially available. 9 Compared to the discrimination of nucleobases along a DNA strand, nanopore sensing concerns not only the recognition of tiny structural variation, but the quantification of individual molecules as well. It is worth noting that nanopore technology is essentially a near-field measurement (see the "Sensing Principle" section below for more details), and thus its lower limit of detection is restricted by the capture rate of analytes, 10 which is one of the biggest challenges to meet when designing high-performance nanopore SMS.
Over the past decades, many different types of nanopores have been applied for single-molecule studies, including:
- a)
- b)
- c)
- d)
According to the fabrication methods, biological and biomimetics nanopores (also called membrane nanopores 51 ) apply in most cases the bottom-up approach, 52 i.e., pores are assembled from different forms of building blocks, and then inserted into, e.g., the lipid bilayers. On the contrary, synthetic nanopores take the top-down approach to drill a hole on solid-state membranes or to shrink a capillary, followed with post-synthesis modifications. Therefore, it is clear that the inherent advantage of membrane nanopores lies in the facile engineering of pore structures with atomic precision, while in comparison, solid-state nanopores feature greater flexibility in pore dimensions, easier integration of other sensing techniques, and potentially greater long-term stability.
This review aims to systematically examine the strategies that have been proposed for protein nanopore (a subclass of the biological nanopores) based SMS. Protein pores are the first type of nanopores adopted for single molecule analysis 53 and are still widely used to date. The key benefits of using protein nanopores lie in their precise and stable open-pore structures. In fact, almost all the single-molecule studies on a new protein pore 11,13–18 were initiated only after the respective structures were solved. 54–63 Such a well-defined structural basis of protein pores enables a broad range of engineering approaches, namely the site-directed mutagenesis, 6 covalent 8 and non-covalent 7 modifications, unnatural amino acid mutagenesis, 64 native chemical ligation, 65 directed evolution, 66 truncation, 67 bio-orthogonal functionalization. 68 The highly controllable pore lumens facilitate the determination of analyte positions in protein nanopores, and furthermore, the analyte transport behaviors, which is crucial to the rational engineering of ultrasensitive protein nanopore based SMS.
In the remaining part of this review, the unique sensing principles of nanopore SMS are first elucidated, followed by a critical examination of the strengths and weakness of the eight common protein pores. After that, the major types of analytes and SMS applications based on protein nanopores are comprehensively analyzed, covering metal ions, small molecules, nucleic acids, peptides and proteins. Finally, the remaining challenges for protein nanopore based SMS and the promising future research directions are summarized. Note that this review focuses specifically on the progress and prospects of single-molecule sensing by protein pores, many other exciting studies are therefore not included, such as the nanopore-enabled sequencing of DNA 69 or protein, 70 measurement of dynamic interaction, 71,72 and analysis of reaction mechanism. 73,74 In the meantime, for readers who are interested in the solid-state or other types of membrane nanopores, please check the recent reviews by Tim Albrecht 10 and Stefan Howorka. 51
Sensing Principles
As shown in Fig. 1, nanopore SMS measure essentially the through-pore ionic current under a constant potential, since the velocity of ions (typically K+ and Cl−) passing through a nanopore is much slower than the redox reaction kinetics on electrodes (e.g., a pair of Ag/AgCl). In this regard, when no analyte is present, the baseline or steady-state open-pore current depends on the structure and charge of the nanopore employed; while after adding a certain amount of analyte, the transient current blockade appeared can be used to identify and quantify the target molecules. Assuming that each blockade is caused by the translocation of an individual molecule, the magnitude and dwell time of current signal are therefore determined by the structure of analyte molecules, and their frequency is correlated with the concentration. In other words, for nanopore SMS, the nanopore itself provides a tiny signal transduction region to temporally accommodate the analyte molecules one after another, while the flowing K+ and Cl− ions serve as a sub-molecular probe to gradually reveal the structure of those confined analytes. It is thus clear that the size comparison between ions, analytes and nanopores, as well as their charge density and distribution, is crucial to improve the spatial resolution of such a "ternary" system. Besides, the maximum capability of structural determination, i.e., the chirality 75 and stereoisomers 76 discriminability, can be achieved if the analytes are kept sufficiently long in the pore lumen, either via the covalent bond formation 77 or by non-covalent interactions. 78
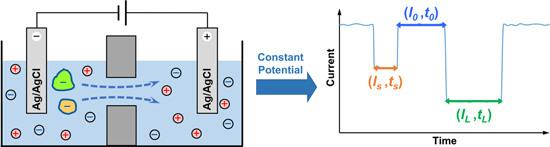
Figure 1. A simplified scheme of single molecule nanopore sensing (left) and the respective current signal (right). In this particular system, Ag/AgCl electrodes are used for both cathode and anode. Driven by a constant applied voltage, two negatively charged species can enter and pass through the nanopore on after another, causing two distinct current blockades (highlighted in orange and green). Herein, I0 refers to the open-pore current when there is no analyte is in the nanopore. Is and IL are the characteristic residual current of two different analytes, while ts and tL are the respective dwell time, both of which can be used for identification. For example, the greater blockade and longer residence of the green molecule than the orange one indicates a larger size, stronger interaction with the nanopore, and/or less charge. In addition, the interval time between two signals, t0, is affected by the concentration of target analyte, and thus can be used for quantification.
Download figure:
Standard image High-resolution imageIt is of no doubt that data analysis is at the heart of nanopore SMS. To extract meaningful information from the current trace, the raw signals are often turned into a pair of current-time (I, t) data set to describe various states of the system, e.g., the open pore, the trapping of an A molecule, or the translocation of another B molecule. Accurate classification of each state or the analyte behavior is therefore crucial. Most of the protein pore groups use a commercial software, Clampfit, 79 for current level identification, which defines the half distance between baseline and signals (or two different signal levels) as the threshold. In order to evaluate the short current pulse correctly, some groups have developed their own threshold-based algorithms 80–82 or software 83 that set the threshold at, typically four- or five-times of the standard deviation of the baseline current. Several other algorithms have been applied for the fitting of multi-level events, such as HMM (hidden Markov model), 84,85 CUSUM (cumulative sum), 86–88 or GMM (Gaussian mixture model). 89
Apart from the signal processing, the establishment of a quantitative physical model for nanopore SMS is beneficial to improving the signal-to-noise ratio (SNR) and response time as well. In particular, it is imperative to understand:
- 1)how ions are transported in a nanopore, which corresponds to the open-pore conductance;
- 2)how analyte molecules are captured into a nanopore, which affects the throughput of current signal;
- 3)how analyte molecules interact with the nanopore interface, which influences the patterns of signal.
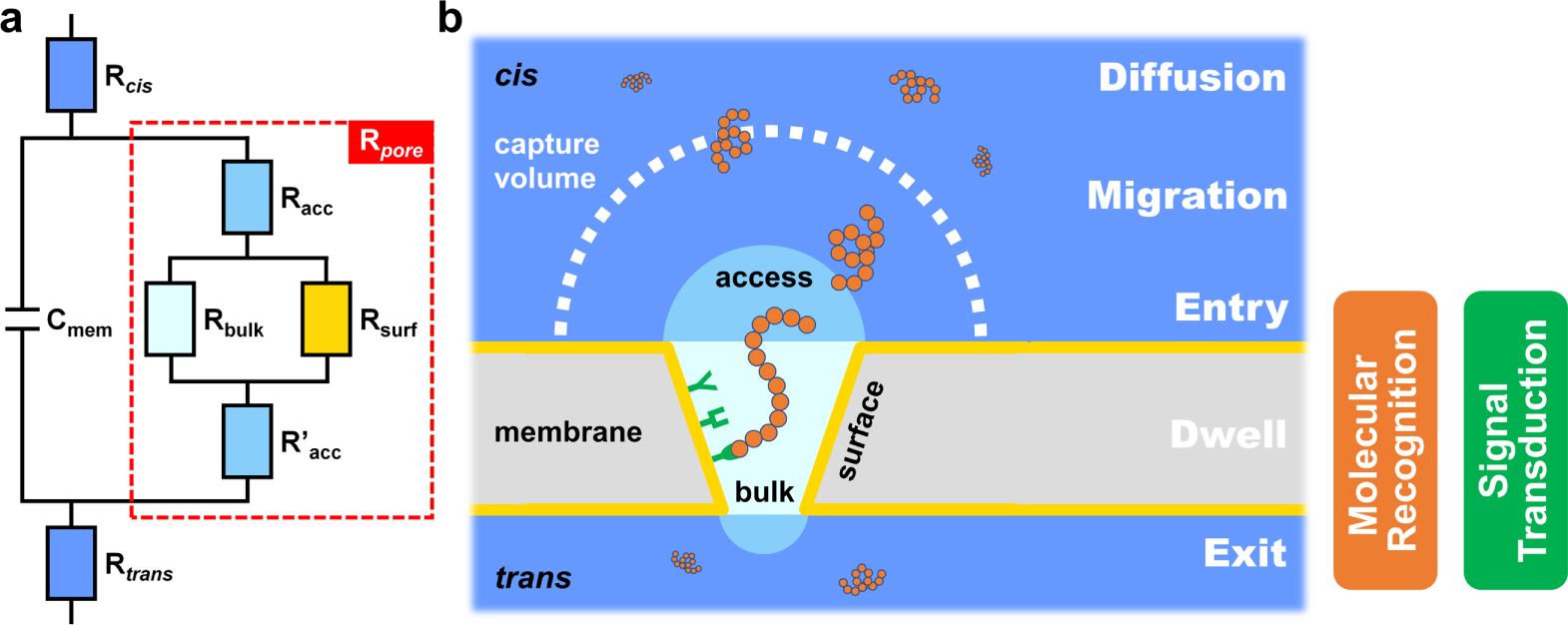
To start with, a higher baseline current (I0 ) or open-pore conductance suggests that more ions are available to probe the structure of analyte molecules, which has been proven useful to enhance nucleotide discrimination in commercial nanopore sequencing. 53 A simplified equivalent circuit of nanopore SMS is given in Fig. 2a to elucidate the origin of open-pore conductance, or alternatively, resistance. Herein, Rcis and Rtrans represent the solution resistance for the cis and trans compartment of a nanopore system, respectively, which is much smaller than the nanopore resistance (Rpore , highlighted by a red dashed box) but not negligible, mostly in the range of 0.1–1 kΩ. 90 Cmem is the capacitance of a free membrane, typically within 10–100 pF for a 50–200 μm functional lipid bilayer. The resulting RC time constant suggests that the maximum bandwidth of nanopore SMS can be over MHz, 90 although most of the reports set the cutoff frequency at 5–10 kHz so as to reach an acceptable noise level.
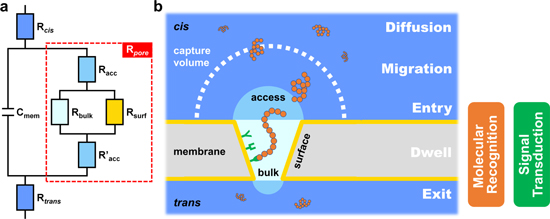
Figure 2. The design and working principle of a nanopore SMS: (a) a simplified equivalent circuit, and (b) the five steps for a typical translocation event of a single molecule, including diffusion, migration, entry, dwell, and exit. Here, Rcis and Rtrans are the solution resistance, Cmem is the capacitance of membrane (i.e., the lipid bilayer for the biological nanopore system), and Rpore is the resistance of nanopore, which can be further broken into bulk (Rbulk), surface (Rsurf) and access (Racc and R'acc) resistance. Rbulk and Rsurf reflect the influence of size and charge of the nanopore, respectively, while Racc and R'acc arise from the entrance of ions from bulk electrolyte into the nanopore. The boundary between the diffusion- and the migration-controlled region defines the capture volume of a certain combination of analyte and nanopore. For most of the protein pores, both molecular recognition and signal transduction elements are embedded in the pore lumen, which makes nanopore technique a near-field measurement.
Download figure:
Standard image High-resolution imageThe nanopore resistance can be further broken down into three components, namely the bulk (Rbulk ), surface (Rsurf ) and access (Racc and R'acc ) resistance. Rbulk is determined by the nanopore geometry, Rsurf accounts for the interaction of ion flux with the inner charged pore walls, and Racc and R'acc arise from the entrance of ions from bulk electrolyte into a nanopore. 91 Hence, Rbulk and Rsurf are connected in parallel, then in series to Racc and R'acc . For an idealized cylindrical nanopore, Rpore can be calculated as:
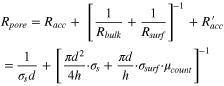
where σs is the solution conductivity, d and h are the diameter and length of a nanopore, σsurf is the surface charge density along the inner pore wall, and μcount is the electrophoretic mobility of the neutralized counterions. 10 Based on the above expression, it is noted that the contribution of access resistance to the total resistance increases as the length of nanopore is reduced, and may even become dominant for 2D nanopores such as graphene. 41 In addition, the charge distribution on inner pore walls induces the ion selectivity, which is particularly pronounced for a narrow nanopore, and may facilitate the specific transport of target analytes. 92
After the baseline current of nanopore SMS is discussed, the next thing to consider is the interval between two signals (t0 ). Distinct from other ensemble sensing techniques, a critical feature of single-molecule detection is to ensure the statistical significance of signal. Consequently, a sufficient amount of translocation events has to be recorded within the practical response time, e.g., 1,000 signals in 10 min. Since nanopore SMS is a typical near-field method, i.e., both of its molecular recognition and signal transduction happen in pore lumen, the requirement on signal frequency equals to the minimum capture rate of analytes. In order to understand the underlying factors, a five-step model for analyte translocation is shown in Fig. 2b, including diffusion, migration, entry, dwell, and exit. A simplified free energy profile of the above process has been proposed by Murugappan Muthukumar. 93 Briefly speaking, the diffusion of analyte is mainly governed by the concentration gradient and Brownian motion, while migration is more affected by the external applied bias, e.g. the electrophoretic force (EPF) and electroosmotic flow (EOF). 94 A critical radius, Rc , at which the diffusion and migration are of the same magnitude, is used to describe the effective "capture" volume of nanopore SMS. For a cylindrical nanopore, Rc is expressed as:
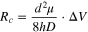
where μ and D are the electrophoretic mobility and diffusion coefficient of the analyte molecules, and ΔV is the applied voltage. When the capture process is diffusion limited, the frequency of analyte molecules (with a concentration of C0 ) to enter capture volume (kdiff ) can be quantified by the Smoluchowski theory: 95
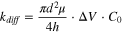
Taking into account of the required minimum capture rate, the above equation defines the lower limit of detection for nanopore SMS. 1 More importantly, it is worth noting that reaching the opening of a nanopore does not guarantee the entry of those molecules—in some cases, an additional energy barrier (e.g., entropic and/or electrostatic) needs to be overcome before they can thread into the nanopore. Such a barrier-limited capture rate (kbar ), according to the Kramers theory, 96 is written as:
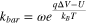
where ω is the rate constant, q is the effective charge of the analyte molecules, U is the energy barrier under zero external voltage for analyte threading, kB is the Boltzmann constant, and T is the temperature. Furthermore, even for those molecules that can enter the nanopore, they do not always cause a signal, because the translocation may be too rapidly to be detected. 97
The last element related to signal components is the current response induced by analyte translocation (I, t), which depends on the size and charge distribution of both analytes and nanopores. For a high concentration electrolyte (e.g., 1 M KCl), of which the Debye length is small, current blockade (1 − I/I0 ) is mainly determined by the steric exclusion effect; 98 while at a low salt condition (e.g., ≤0.1 M KCl), the number of mobile counterion introduced may even exceed the amount excluded by the analyte, leading to an overall increase in ionic current. 99 Besides, current blockade is also affected by the position of analytes within a nanopore. 100
With regard to the translocation dwell time (t), an average value can be estimated, to a first approximation, by the following expression:
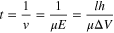
assuming that the length of analyte (l) is much greater than the pore (h), and the applied voltage (ΔV) is sufficiently high. Herein, ν and μ are the effective velocity and electrophoretic mobility of analyte inside the nanopore, rather than in a free solution, and E is the strength of electric field in a nanopore. Alternatively, if l is significantly shorter, as is the case for small molecules, the respective mean residence time is:
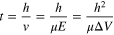
Note that the translocation speed of analytes is strongly associated with the resolution of the ionic current signal, i.e., whether the mobile ions move fast enough to capture sufficient information of the "stationary" target molecules within a nanopore, 101 two important implications are made based on the above estimation:
- 1)
- 2)The reciprocal relationship between t and ΔV is commonly adopted as evidence to verify the successful translocation of analyte molecules. However, the reduction of ΔV may not always be feasible to nanopore SMS as it lowers down the capture rate and SNR as well. A more practical approach is to decouple the dwell time and the throughput of signal by designing a nanopore system in which the exit barrier is (slightly) higher than the entry one. 104 As a result, when the ΔV is just high enough to overcome the exit barrier, both rapid capture and long translocation can be achieved.
Since a large number of translocation events are required to be recorded, the whole picture of nanopore SMS signal must include the distribution of current blockade and dwell time, shown as a 2D scatter plot with marginal histograms. In most cases, the FWHM (full width at half maximum) of current blockade, together with peak separation, decide the resolution of a nanopore system, but studies on the origin 105 or the reduction 104 of signal variance are very limited at the moment. Apart from the signal component, the bandwidth dependent current noise is also crucial to the performance of nanopore SMS. For those who wish to know more, there is a critical review 106 provided recently by Cees Dekker to assess the influence of noise in biological and solid-state nanopores, as well as the possible solutions to improve SNR.
Types of Protein Nanopores
The protein nanopores that have already been applied for single-molecule research are generally separated into two categories: pore-forming toxins (PFTs) and β-barrel porins; and the formers are further classified as β-PFT and α-PFTs, based on the secondary structures of their membrane spanning domains. 107 Here, the major families of β-PFTs include α-hemolysin (α-HL), 6–8,11 aerolysin (AeL), 12,108,109 and membrane attack complex/perforin, 110 while α-PFTs are divided into cytolysin A (ClyA) 13,111,112 and actinoporin. 14,113 Meanwhile, quite a few β-porin families have also been studied, such as mycobacterial porin, 15,114,115 general bacterial porin, 16 sugar porin, 116 outer membrane protein G (OmpG), 17,117 outer membrane receptor, 18 outer membrane auxiliary, 118 and curli transporter. 53 The difference between PFTs and β-porins lie in that, at a high salt or sometimes physiological condition, PFTs are able to remain open, but β-porins may exhibit spontaneous gating due to the movement of loop or cork. 119 Hence, loop deletion/pinning or cork removal are needed to form a quiet β-porin. 17,18,120,121 In the following paragraphs, the structure and functions of the eight major protein pores (Fig. 3) are critically evaluated to elaborate the design principles of nanopore SMS.

Figure 3. Side and top view of common types of protein nanopores, including beta-barrel pore-forming toxins (β-PFTs): α-hemolysin (α-HL, PDB ID: 7AHL 54 ) and aerolysin (AeL, PDB ID: 5JZT 122 ); alpha-helical pore-forming toxins (α-PFTs): cytolysin A (ClyA, PDB ID: 2WCD 55 ) and Fragaceatoxin C (FraC, PDB ID: 4TSY 56 ); and beta-barrel porins (β-Porins): Mycobacterium smegmatis porin A (MspA, PDB ID: 1UUN 57 ), outer membrane porin F (OmpF, PDB ID: 2OMF 58 ), outer membrane protein G (OmpG, PDB ID: 2IWW 60 ), and ferric hydroxamate uptake component A (FhuA, PDB ID: 1BY3 62 ). The secondary structures of the protein nanopores are highlighted in different colors: "yellow", β-sheet; "red", α-helix; and "green", loop.
Download figure:
Standard image High-resolution imageα-HL
The atomic-resolution structure of α-HL nanopore was the first to be resolved among all PFTs. 54 It is a mushroom-shaped heptamer of 100 Å long and is divided into a vestibule and a β-barrel stem of 48 and 52 Å, respectively. The cis vestibule entry and trans stem exit are 26 and 24 Å in diameter, with a central constriction of 14 Å. The geometry of α-HL nanopore indicates that a single-stranded DNA (ssDNA) can thread into the β-barrel stem, but not the double-stranded DNA (dsDNA), while a folded protein may not even be able to enter the vestibule. In this regard, the dsDNA/ssDNA 123 and protein/ssDNA 124 hybrid structures have been frequently used to probe the recognition sites and sensing capability of α-HL, 125 to facilitate the sensitive and selective capture of target analytes, 126 and to examine the translocational unfolding of adjacent protein 127 or unzipping of dsDNA. 128 Besides, the wild-type (WT) α-HL is weakly anion selective at pH 7.5, 129 which enables the electro-osmotic capture of neutral β-cyclodextrin (βCD) 7 from the trans side of α-HL nanopore at negative potentials.
AeL
The X-ray structure of AeL monomer was the first β-PFT to be identified in its solution form, 130 but the structure of AeL pore was only recently realized by cryo-EM (electron microscopy) at a near-atomic resolution. 122 The total length of AeL heptamer is comparable to α-HL (ca. 100 Å), but the diameter of its β-barrel is narrower, with two constriction regions < 10 Å. The long, narrow pore lumen of AeL makes it extremely sensitive to detect and discriminate both short oligonucleotides 109 and the positively charged homo-oligopeptides 131 as well. The WT AeL is slightly more anion-selective than α-HL, 132 possibly due to the narrower pore lumen as well as a greater number of charged residues along the β-barrel. 133 The resulting cis-to-trans EOF under a positive applied bias can be enhanced by remote pH modulation to increase the diffusion-limited capture of longer oligonucleotides (e.g., >10 bases). 134
ClyA
The X-ray structure of dodecameric ClyA nanopore was the first α-PFT to be determined. 55 The total length of the cylindrical pore lumen is 130 Å, with a cis entry of 70 Å and a trans constriction of 35 Å. Notably, tridecameric and tetradecameric ClyA can also be experimentally isolated via directed evolution, 66 of which the structures were recently solved by cryo-EM. 135 The combination of a huge cis opening and a trans steric hinderance not only facilitates the capture of folded proteins that remain functional, and also prolongs their residence in ClyA nanopore. 136 The formed "internal adapter" further enables the detection of other small ligands that can be specifically bound to the immobilized protein. 111 It is noted that the internal surface of the commonly used dodecameric ClyA-AS (C87A/C285S) is highly negatively charged, leading to a strong cis-to-trans EOF under negative potentials. 66 As a result, an EOF dominant capture has been used to trap proteins with a negative charge, as the backward EPF can extend their dwell time in ClyA. 100
FraC
The X-ray structure of octameric Fragaceatoxin C (FraC) was the second α-PFT nanopore to be identified. 56 It forms a funnel-shaped pore geometry with a total length of 70 Å, a cis entry of 60 Å and a trans constriction of 16 Å. The oligomerization of FraC is triggered by the presence of lipid sphingomyelin and thus nanopore needs to be pre-formed before inserted into the bilayers. 14,56 The population of FraC heptamer and hexamer can be promoted by weakening the monomer-lipid interactions during oligomerization. The resulting narrower heptameric or hexameric FraC nanopore exhibits a higher ion selectivity and EOF than the octameric counterpart. 137 The inverted conical shape of FraC, as well as the moderate cation selectivity, allows the EOF dominant capture if a wide variety of proteins with different size and charge. 113 Furthermore, despite the fact that the trans exit of WT FraC octamer is wide enough to accommodate ssDNA, the strong negative charge at the constriction (D10) prevents the direct translocation. 14 A double mutant ReFraC (D10R/K159E) has thus been proposed, which allows not only ssDNA but dsDNA as well to pass through the nanopore, indicating that the α-helical trans constriction of FraC may be elastically deformed. 14
MspA
The first described nanopore structure of the mycobacterial porin family, MspA (Mycobacterium smegmatis porin A), has a unique octameric goblet-like conformation with a total length of 96 Å and cis entry of 48 Å. 57 The most distinctive feature of MspA is its thin trans constriction of 6 Å long and 12 Å in diameter, which was predicted correctly by Jens H. Gundlach to show better spatial resolution for nucleotide identification than the ∼50 Å long sensing region of α-HL. 15 In order to enable the translocation of ssDNA, three aspartate residues at the constriction of WT MspA were replaced with asparagine (M1MspA, D90N/D91N/D93N). 138 Meanwhile, three additional negative amino acids at the cis entry and mid-section of M1MspA were further changed into the positive ones (M2MspA, D134R/E139K/ D118R) to facilitate the capture of DNA and to prolong its residence. 15
OmpF
The X-ray structure of OmpF (matrix porin) trimer was one of the first two β-barrel porins to be solved. 58 Each subunit is composed of a 16-stranded β-barrel of ca. 50 Å long, with a cross section of 15 × 22 Å2 and a central constriction zone of 7 × 11 Å2. Strong affinity has been found between this narrow constriction region and antibiotics of a comparable size, reaching an acceptable dwell time at the millisecond level. 16 In addition, it is also noted that, within a pH range of 5–9, WT OmpF exhibits a constant cation selectivity. 139 The resulting cis-to-trans EOF under negative potentials has been attributed as the dominant driving force for the capture and transport of various kind of β-lactam 16 and quinolone 140 antibiotics.
OmpG
The X-ray 59,60 and NMR 61 structures of monomeric OmpG reveal that it comprises a 14-stranded β-barrel with a larger constriction of 12 × 15 Å2 than OmpF and a wider channel diameter of 20–22 Å. The pH-dependent flexibility of its loop 6 and β-strand 12 can induce spontaneous gating. A series of strategies have been developed to form a "quiet" OmpG pore for sensing, such as to cross-link the strands 12 and 13 with a disulfide bond (G231C/D262C), 17 to remove the loop 6 (ΔL6) 121 or pin it to the lip membrane by dodecylation of a cysteine residue in loop 6 (I226C), 120 and to delete an irregularly positioned aspartic acid residue near the loop 6 (ΔD215). 17,121 Intriguingly, the loop 6 dynamics can be exploited to detect analytes outside the OmpG nanopore, after functionalization with extra recog-nition probes. 117
FhuA
The monomeric, 22-stranded FhuA (ferric hydroxamate uptake protein component A) forms a huge β-barrel of 69 Å in length and 46 × 39 Å2 in cross section. 62,63 A globular cork domain consisting four β-strands obstructs almost the entire pore lumen. Therefore, removal of the cork and several other extracellular loops that partially occlude the pore lumen (L3/L4/L5/L11 or L10) can form a stable, rigid truncated FhuA (t-FhuA) with huge conductance. 119 Since the wide pore geometry of t-FhuA is unfavorable to prolong the analyte residence time, a flexible peptide tether/probe has also been functionalized to the cis entry of t-FhuA for the detection of proteins outside the nanopore. 71
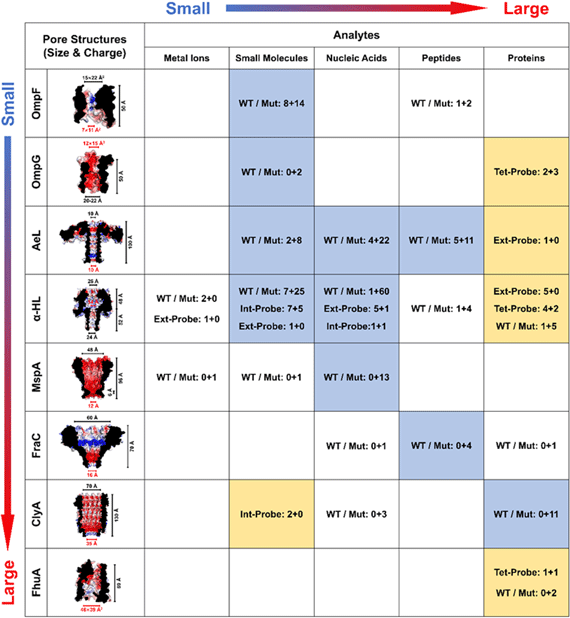
A summary of the above eight protein nanopores is given in Table I, in combination with their applications in single-molecule sensing and analysis. Pores and analytes are put in the order of their size, e.g., the diameter of nanopore constriction and entry/exit. It is noted that the detection strategies can be broadly divided into two categories:
- 1)the direct use of WT or mutant pores (highlighted in "light steel blue"), of which recognition sites are embedded in the nanopore cavity;
- 2)the additional use of probes (highlighted in "peach"), either externally or internally, to bind the target analytes.
Table I. Summary of the common protein nanopores (OmpF, OmpG, AeL, α-HL, MspA, FraC, ClyA, and FhuA), five major kinds of analytes (metal ions, small molecules, nucleic acids, peptides, and proteins), and the number of publications for each type of detection strategy ("WT/Mut": WT/mutant pores, or "Ext-, Int- Tet-Probe": external/internal/tethered probes). Pores and analytes are arranged according to their sizes. The diameters or cross sections at the nanopore constriction (highlighted in red), entry and exit are calculated based on the X-ray or cryo-EM structures, as well as the length of the pore. 54–59,62,122 The electrostatic potential of the inner surface of "WT" pores (except for FhuA, of which the cork is removed) is calculated by APBS 141–143 in 0.15 M NaCl, and is displayed in PyMOL (blue: +5kT/e, and red: −5kT/e). For analytes, polyethylene glycol and other polymers are considered as small molecules, and unfolded proteins are classified into peptides. The number of publications is shown as "A+B," in which "A" is the number for single molecule sensing, and "B" represents the number for non-sensing applications. A pore-analyte combination that applies mainly the "WT or Mutant" strategies is shown in light steel blue, while those adopting the "Probe" strategies are highlighted in peach. Only a selection of high-impact applications is counted for α-HL, but for the rest of protein pores, the number of publications is comprehensive.
|
When the "WT or Mutant" strategy is applied, a positive correlation can be observed between the size of pore and analyte, suggesting that the steric hindrance at nanopore entry and exit is crucial to the precise control of capture rate and dwell time, and should always be the primary consideration in the development of nanopore SMS. By contrast, when there is too great a difference between the size of pore and analyte, e.g., the folded proteins for OmpG, 117 α-HL, 144 and AeL, 145 or small molecules for ClyA, 111 the detection can only be realized with the aid of an extra probe. In particular, if the external probe is adopted, the molecular recognition will be extended to the outside of a nanopore, and therefore, decoupled from the signal transduction region inside. Such a strategy has been proven to be particularly effective in the reduction of detection limit for SMS. 1
Analytes and Applications
In the following sections, progress on the development of nanopore SMS over the last two decades is discussed, based on the type of analytes. As shown in Table I, the number of previously published sensing applications, i.e., those include at least a concentration-dependent measurement, is only 30% of the non-sensing counterpart. One challenge that has been discussed above is the dilemma between response time and the minimum number of signals for statistical significance, which defines the lower concentration limit of detection for nanopore SMS, in spite of their single-molecule sensitivity. 10 Moreover, the selective and simultaneous determination of multiple analytes in complex environments is another challenge. To achieve an efficient and specific capture, as well as to improve the detection limit, a series of "external probe" strategies 144,146,147 have been proposed, which will be elaborated in detail in the following sections.
Metal ions
The first prototype of nanopore SMS was pioneered by Hagan Bayley in 1997 for the detection of metal ions. 148 Four histidine residues were introduced at the trans exit of a α-HL pore(N123H/T125H/G133H/L135H) in one of its seven subunits (WT64H1), creating a binding site that was able to detect nanomolar level of divalent metal ions, such as Zn2+. Later in 2000, the same system was utilized to quantify multiple metal ions (Zn2+, Co2+, and Cd2+) simultaneously, since the 4H binding site interacted with each kind of metal ions in a mutually exclusive manner (Fig. 4a). 6 An almost identical detection limit of ca. 50 nM was reached for Zn2+, both in the absence and the presence of a sub-micromolar to tens-of-micromolar level of Co2+ interference.
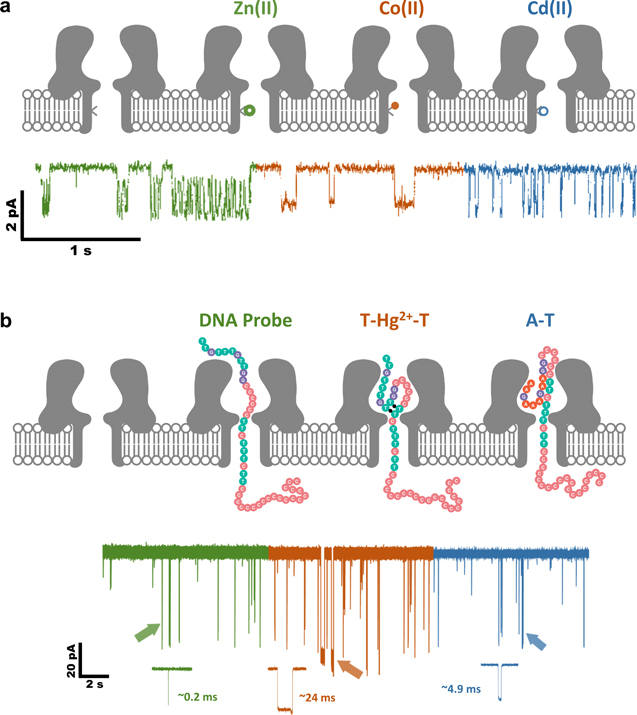
Figure 4. Nanopore SMS for metal ions. (a) Simultaneous detection of Zn2+, Co2+ and Cd2+ by a mutant α-HL pore. The metal ion binding site is composed of four histidine residues (N123H/T125H/G133H/L135H) and is located only in one of the seven subunits of α-HL heptamer (WT64H1). (Adapted with permission from Ref. 6. Copyright 2000 Springer Nature.) (b) Detection of Hg2+ by WT α-HL with the aid of a T-rich ssDNA probe, which upon the binding with Hg2+ ions, can form a unique T-Hg2+-T structure to prolong the residence time of probe in the α-HL lumen. (Adapted with permission from Ref. 146. Copyright 2011 American Chemical Society).
Download figure:
Standard image High-resolution imageDespite its early success, the diffusion limited capture of metal ions implies that the limit of detection can hardly go below a nanomolar concentration, which is not competitive to other established electrochemical 149 or optical 150 sensors. In order to solve this inherent limitation, Yuliang Zhao and Hai-Chen Wu came up with a "DNA probe" strategy. 146,151,152 The probe mainly has two functions: one is of course to bind metal ions in solution, and the other is to enhance capture rate via the additional EPF on DNA. Inspired by the specific binding of Hg2+ to thymine-thymine base pairs (T-Hg2+-T), 153 T-rich ssDNA probes were designed, which could turn into stable hairpin structures upon the interaction with Hg2+. 146 The resulting T-Hg2+-T complex exhibited a distinct current response, compared to the ssDNA probes (Fig. 4b). Probability of the T-Hg2+-T hairpin signal was positively associated with the concentration of Hg2+. Integrating this DNA probe approach with a salt gradient (0.15Mcis/3Mtrans) led to a detection limit of 7 nM Hg2+, which could be further lowered down to 0.5 nM when using an optimized DNA hybrid probe. 151 Such a metal ion mediated duplex formation strategy was also applied to the simultaneous detection of Pb2+ and Ba2+, which pro-moted the folding of guanine-rich DNA probes into different G-quadruplex structures. A sub-nanomolar limit of detection was realized for both metal ions. 152 Xiyun Guan and Xiaofeng Kang also investigated the binding of Hg2+ with DNA hairpin loop, instead of the stem, achieving a detection limit of 25 nM. 154
In addition, the concept of metal chelation induced signal change was extended to polypeptide probes by Guan. 155–158 In a typical example, as low as 40 nM Cu2+ was detected by (M113F)7 α-HL, with the help of a HHHHHHHHHH probe. 155 Note that the exact value of the current blockade by metal chelates cannot provide additional information on the type of metal ions detected, an "on-off" sensing strategy may be more efficient than a ratiometric or "off-on" method. In this sense, a superior sub-nanomolar level of detection limit was reached for UO2 2+ (0.1 nM) 156 or Th4+ (0.45 nM) 157 using a longer HHHHHHKHHHYHHH or YEVHHQKDDPDD probe.
Apart from the above, Kang also studied the possibility of using modified βCD as an internal recognition probe for the detection of Cu2+. 159,160 The long dwell time of βCD in α-HL enabled the interaction of Cu2+ with the functional groups on it. An estimated detection limit of 50 nM was reached with carboxymethyl-β-cyclodextrin (CMβCD), and a slightly improved value of 12 nM was realized when using heptakis-(6-deoxy-6-amino)-β-cyclodextrin (am7 βCD), possibly due to the almost permanent residence of the latter in α-HL. Besides, 5,10,15,20-tetrakis(4-sulfonatophenyl)-porphyrin 161 was applied as another type of probe, and a detection limit of 16 nM Cu2+ was obtained with α-HL. Note that βCD or porphyrin cannot offer additional EPF for the capture of metal ions, the resulting detection limits are thus not comparable to the DNA or peptide probes.
Small molecules
Although a wide range of antibiotics, e.g., ampicillin, 16 enrofloxacin, 162 cefpirome, 163 have been directly quantified by a narrow WT OmpF pore, its diffusion-limited capture suggests that the detection limit can only reach a sub-millimolar level. By contrast, other pores with a wider constriction do not exhibit strong enough affinity with small molecules, ending up with too rapid translocation to be detected. In order to solve this issue, all three types of strategies have been implemented, i.e., 1) to introduce a stronger binding site in nanopore; 2) to embed an internal adapter; and 3) to incorporate an external probe.
Regarding the first approach, Bayley proposed to include covalent interactions inside α-HL so as to enhance its binding with small molecules. 164 Proof-of-concept experiments were demonstrated through the detection of organoarsenic species. A thiol group was introduced to one of the seven subunits of α-HL (T117C), which could form a reversible covalent bond (−S-As-) upon the reaction with arsenic compounds. However, the ultralong duration for bound analytes as well as the slow association rate indicated that a practical limit of detection could only reach the sub-millimolar level. To solve this issue, supplementary cleavage agents, such as dithiothreitol (DTT), were adopted to reduce the lifetime of those highly stable or even "irreversible" covalent bonds. 165 Since the capture of DTT was also diffusion controlled, the enhancement in detection limit was therefore very limited.
In spite of the undesirable detection limit, the advantage of "covalent interaction" strategy lies in its discriminability to tiny structural differences, including the chirality of small molecules. 75 For example, using a Cu2+ probe bound to the covalently attached phenanthroline group in α-HL, Bayley successfully determined six neurotransmitters 166 and four pairs of amino acid enantiomers. 167 Moreover, another tethered 3-(maleimide)phenylboronic probe was used to quantify the cyclic isomers of monosaccharides as well as a range of diols. 77 Interestingly, it was noted that the L- and D-amino acids were discernible from their current blockade, 167 but the cyclic isomers of D-glucose and D-fructose could only be distinguished by their dwell time in α-HL. 77
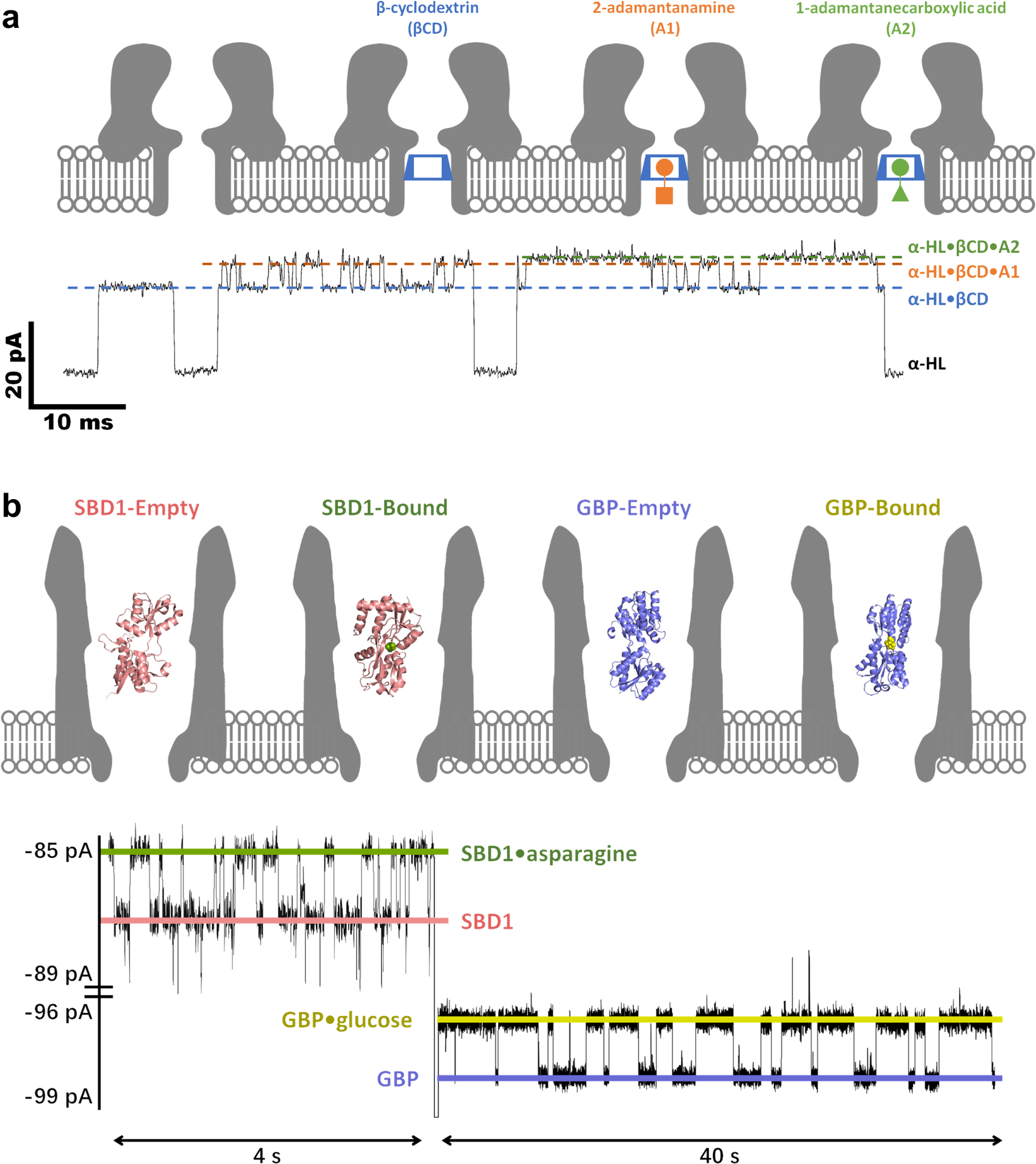
The second "internal adapter" strategy was also proposed by Bayley initially, who equipped the α-HL nanopore with a βCD probe for the simultaneous detection of multiple adamantane derivatives (Fig. 5a) or imipramine/promethazine. 7 The reduced-volume and hydrophobic cavity of βCD could significantly enhance the host-guest interactions with organic compounds, so as to prolong their residence time in the adapter and facilitate the resolution of structurally similar species. Following this lead, Kang, Guan and Bayley further applied the βCD embedded α-HL to determinate drug enantiomers, reaching a sub-micromolar level of detection limit for ibuprofen and thalidomide. 168
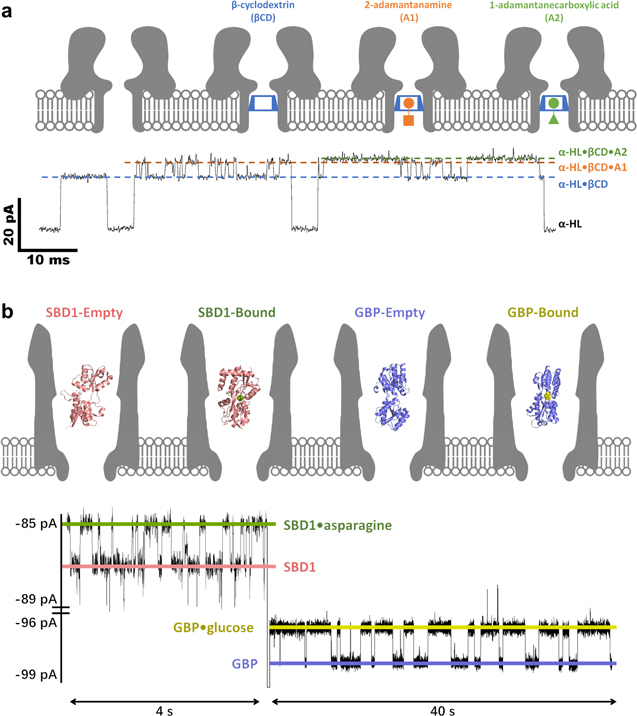
Figure 5. Nanopore SMS for small molecules. (a) Simultaneous detection of 2-adamantanamine (A1) and 1-adamantanecarboxylic acid (A2) by a βCD embedded WT α-HL nanopore. The signals of α-HL·βCD·A1 and α-HL·βCD·A2 only appear when a βCD is temporally trapped in α-HL. (Adapted with permission from Ref. 7. Copyright 1999 Springer Nature.) (b) Simultaneous detection of glucose and asparagine by a GBP (glucose-binding protein) or SBD1 (a substrate-binding domain analogous to GBP) confined ClyA-AS nanopore. The signals of GBP·glucose and SBD1·asparagine appear only when the internal probes are trapped in ClyA. (Adapted with permission from Ref. 111. Open Access Copyright under a Creative Commons Attribution 4.0 International License).
Download figure:
Standard image High-resolution imageInstead of using one internal adapter to detect various analytes, Giovanni Maglia proposed to equip ClyA with different ligand-binding proteins successively, which was able to remain functional when trapped in the nanopore. 111,136 Note that there is a wealth of proteins evolved to recognize small substrate molecules with ultra-high sensitivity and selectivity, integration of multiple protein probes can guarantee the precise determination of each target. As demonstrated in Fig. 5b, as low as 50 nM glucose and sub-micromolar asparagine could be simultaneously quantified by ClyA when it was incorporated with GBP (glucose-binding protein) and SBD1 (a substrate-binding domain analogous to GBP).
The third "external probe" strategy was attempted only one time by Ryuji Kawano and Shoji Takeuchi. 169 A cocaine-binding aptamer was purposely designed, which upon the recognition of target molecules, could turn into a Y-shaped three-way junction with a 30-mer polycytosine tag. Such a structure prevented its translocation but facilitated the capture, and so as low as 1 μM cocaine was detectable.
Nucleic acids
Although the nanopore based sequencing of a long DNA strand has already been experimentally validated years ago, 69 the quantitative determination of short DNA or RNA fragments by nanopore remains challenging to date, and is not competitive to the nucleic acid amplification tests. 170 Note that nucleic acids exhibit strong and uniformly distributed negative charge, their capture should be in most cases EPF driven, which is distinct from the sensing of metal ions and small molecules. The enhanced driving force also indicates that a stronger binding affinity is required so as to trap analytes long enough in the pore. It was recently shown by Yi-Tao Long that WT AeL was able to effectively discriminate short oligonucleotides of different length 109 or single nucleobase variations, 171 which were hardly detectable by α-HL, possibly due to the more favorable steric and electrostatic interactions between AeL and nucleic acids. Owing to its narrow pore entry, the capture efficiency of DNA by AeL was however low, restricting the practical detection limit at 1 μM for short 4-mer DNA fragments. 171 When using a lower pH to enhance EOF and capture rate, the detection limit for a longer 16-mer DNA could be pushed down to 0.4 μM. 134
Compared with the AeL nanopore, α-HL featured a higher capture rate of oligonucleotides but much shorter residence time. To solve this limitation, a 3'-terminal polyuridylic acid leader strand was designed by Bayley which, after linking to the target DNA, could significant increase the dwell time by two orders of magnitude. 172 Another approach initiated by Howorka and Bayley was to functionalize the pore entry of α-HL with a complementary probe of the target DNA. 173,174 Embedding the recognition site at the cis opening of α-HL guaranteed not only strong binding but efficient capture of the target analytes as well. An optimal detection limit of 60 nM was therefore realized for an 8-mer DNA. 174
In addition to the above, Li-Qun Gu developed a series of DNA external probes to determine lung cancer-associated and tumor suppressor miRNAs. 126,147,175 The first proof-of-concept design was shown in Fig. 6a, where the ternary oligonucleotide probe consisted of a central recognition domain fully complementary to the miR-155, and a poly(dC)30 signaling tag on both ends. After binding, the complex could be rapidly captured by the α-HL nanopore and was trapped inside until unzipping. The following short-lived residence of miR-155 and its fast translocation caused two additional current responses (levels 2 and 3), which was distinct from the trapping signal (level 1). Although sub-picomolar miR-155 was detectable under an asymmetric salt gradient, the practical detection limit using this probe and WT α-HL pore should be at the sub-micromolar level. 147
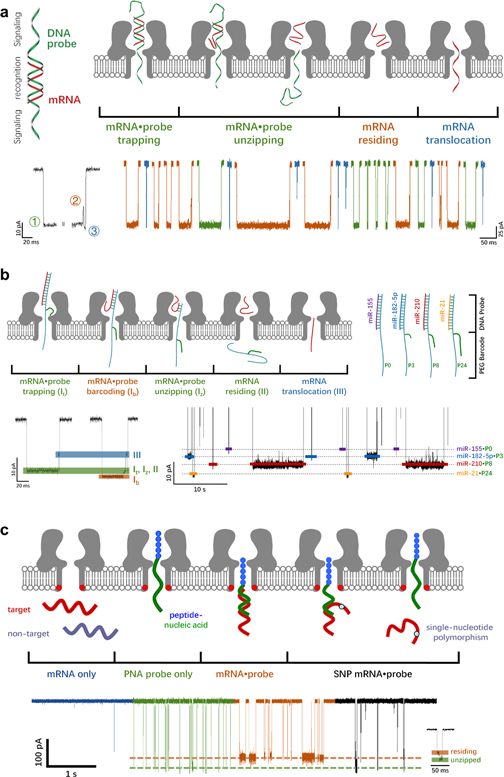
Figure 6. Nanopore SMS for nucleic acids. (a) Determination of lung cancer-associated miRNAs, miR-155 (red), by WT α-HL with the help of an external ssDNA probe (green), which is composed of two signaling tags and a central recognition domain. A typical current trace for a mRNA·probe hybrid includes three stages: level 1 refers to the trapping and/or unzipping of mRNA·probe, while levels 2 and 3 are the residing and translocation of mRNA. Presence of level 2 current confirms the unzipping of a mRNA·probe complex (brown), while the absence of level 2 in a long blockade (blue) suggests that the complex may return back to the cis side. Short current blockade (blue) refers to the rapid translocation of mRNA or probe. (Adapted with permission from Ref. 147. Copyright 2011 Springer Nature.) (b) Simultaneous detection of miR-155, miR-182–5p, miR-210, and miR-21 by α-HL with the aid of DNA probes. Concerning the probes for miR-182–5p, miR-210, and miR-21, an additional 3-, 8- or 24-mer PEG barcode is tethered to the signaling tag, which thus generates an increasingly greater barcoding current (shown as orange in a typical current trace) when the mRNA·probe complex is trapped in α-HL. (Adapted with permission from Ref. 175 Open Access Copyright under an ACS AuthorChoice License.) (c) Highly selective detection of tumor suppressor miRNAs, let-7b (target), by a mutant α-HL nanopore (K131D7) with the help of a polycationic probe, in the presence of miR-155 and miR-21 interference (non-target). The probe consists of a cationic peptide leader (blue) and a peptide nucleic acid (green) that can specifically bind to the let-7b. Both mRNAs and probes are added in the trans side of α-HL at positive applied voltages. The residence/unzipping time of mRNA·probe is able to reveal single-nucleotide polymorphisms in mRNAs (brown and black). (Adapted with permission from Ref. 126. Copyright 2013 American Chemical Society).
Download figure:
Standard image High-resolution imageWith regard to the simultaneous determination of multiple miRNAs, Gu proposed a barcoding strategy, in which the PEG tags of various lengths were attached to the analyte-specific probes (Fig. 6b), thus generating well separated current responses for various kind of miR-NAs. 175 More specifically, four DNA probes were designed, each consisting of a signaling tag and the respective recognition site for miR-155, miR-182–5p, miR-210, or miR-21. Meanwhile, an extra 3-, 8- or 24-mer azide-terminated PEG barcode was conjugated to the probes for miR-182-5p (P3), miR-210 (P8), or miR-21 (P24), respectively, while no PEG barcode was tethered to the probe for miR-155 (P0). When an encoded complex was trapped in WT α-HL, the PEG barcode could thread completely into the β-barrel, giving rise to a greater (and unique) current blockade than that of the miR-155·P8. It was further noticed that the capture rate of a particular type of miRNA·probe was not affected by the presence of others. Therefore, sub-micromolar level of detection limit may be reached for all of the four miRNAs in a mixture.
Apart from the programmable barcode for multiplex detection, the external probe strategy may also be used to promote the rapid and selective capture of target miRNAs from the mixture, as displayed in Fig. 6c. 126 In a typical test, a polycationic probe was rationally designed, which compromised of a positively charged leader peptide and a peptide nucleic acids recognition site. Only the target let-7b could be bound to the polycationic carrier and trapped by the WT α-HL nanopore, while the non-target miRNAs were unlikely to be captured, due to the unfavorable EPF driving force and the electrostatic repulsion barrier at the pore entry. More importantly, it was further found that the peptide-PNA conjugate exhibited a superior capture rate to the previously mentioned DNA probes, leading to an excellent limit of detection below 50 nM, even without using the salt gradient. Besides, it was also noted that the single-nucleotide poly-morphisms in miRNAs were readily resolved by the dwell time of miRNA·probe complex in α-HL.
Peptides and proteins
Determination of peptides or unfolded proteins can be realized by measuring their translocation through a narrow nanopore. For example, OmpF was used to quantify the analogue of an antimicrobial peptide, HP(2–20), with a sub-micromolar level of detection limit. 176 Similarly, AeL was adopted to detect the chemically or thermally unfolded maltose-binding pro-teins, 177,178 the passenger domain of pertactin, 179 and an 18-mer analogue of the C-terminal fragment of synaptobrevin 2, Sb2(76–93). 180 The detection limit was about 300 nM for the former two, and could surprisingly get down to 20 nM for the third one, possibly due to the efficient EOF-driven capture at a high bias.
The detection of folded proteins, however, is not easy, because the narrow entry of most protein nanopores prevents the capture of analytes, except for ClyA or FraC. In these two pores, the integration of a wide cis opening and a much narrower trans exit suggests that the exit barrier is higher than the entry one, which is crucial to decouple the interval and residence time. As a result, the shape of ClyA or FraC is not only able to facilitate the capture of protein analytes but to prolong their residence time as well. Note that both of these two nanopores are cation-selective, the cis-to-trans EOF at a negative bias has been commonly employed as the driving force in order to capture the proteins analytes with diverse charge density and distribution. 13,113 For instance, as shown in Fig. 7a, the WT FraC was used to quantify five distinct peptides and proteins (Fig. 7a), of which the isoelectric point ranged from 4.2 to 8.8 and the atomic mass from 1.2 to 25 kDa. 113 The huge difference in the current blockade of each kind of protein was also closely related to their various positions in the funnel-shaped FraC nanopore, enabling the simultaneous sensing, and the limit of detection could reach 20–30 nM under a favorable EOF.

Figure 7. Nanopore SMS for proteins. (a) Direct determination of chymotrypsin (25 kDa, PI 8.8), β2-microglobulin (11.6 kDa, PI 5.6), EGF human (6.2 kDa, PI 4.5), Endothelin 1 (2.5 kDa, PI 4.2), and Angiotensin I (1.3 kDa, PI 7.9) by WT FraC. (Adapted with permission from Ref. 113. Open Access Copyright under a Creative Commons Attribution 4.0 International License.) (b) Quantification of the site-specific phosphorylation in thioredoxins by WT α-HL with the aid of a chemically linked oligonucleotide leader. A four-step translocational unfolding pathway is demonstrated with a typical current trace. The amplitude and standard deviation of the level 3 current blockade can be used to discriminate various unphosphorylated (-P) and phosphorylated (+P) thioredoxins (TrX_S112, TrX_S017, and TrX_S95). (Adapted with permission from Ref. 127. Copyright 2013 Springer Nature.) (Adapted with permission from Ref. 181. Copyright 2014 Springer Nature.) (c) Simultaneous determination of mouse anti-biotin (mAb) and polyclonal anti-biotin (pAb) by an OmpG nanopore functionalized with a PEG-biotin probe. Two distinct binding conformations are found for pAb (brown for polyAb.1 and green for polyAb.2). (Adapted with permission from Ref. 117. Copyright 2015 American Chemical Society).
Download figure:
Standard image High-resolution imageAs for those proteins, of which the sizes are greater than the nanopore opening, three other types of sensing strategies have been proposed. The first one was to unfold the target analytes. Instead of using unfoldases, David Rodriguez-Larrea and Bayley designed a chemically linked oligonucleotide leader to initiate the protein unfolding. 127,181–183 More specifically, a poly(dC)30 tag was tethered to the C-terminal of thioredoxin through a hexamethylene linker (Fig. 7b). After the DNA·protein complex was trapped in α-HL, three levels of current blockades were observed, which were assigned to the residence of DNA strand (level 2) or the partially unfolded thioredoxin (level 3) in α-HL, and the translocational unfolding of the remaining protein (level 4). 127 It was further found that connecting the DNA leader to the N-terminal of thioredoxin resulted in a similar pattern of three current stages, but a much faster transition from level 2 to level 3, indicating that the unfolding kinetics of protein may be revealed from the characteristics of current blockade. 182 Based on this concept, the site-specific phosphorylation of thioredoxins was quantified at a (sub-)micromolar level. 181
The second widely applicable strategy is to utilize a tethered probe for the recognition of large-size folded protein analytes outside a variety of nanopores (α-HL, 8,184,185 ClyA, 13 OmpG, 117,186 and FhuA 71 ). The first demonstration of this approach was given by Liviu Movileanu, Howorka, and Bayley, in which a maleimide-PEG3400-biotin probe was linked at the S106C position in one of the seven subunits of α-HL for the detection of WT streptavidin and a lower-affinity variant, as well as the mouse anti-biotin (mAb). 8 The simultaneous determination of multiple protein analytes using a tethered probe was later realized by Min Chen with OmpG (Fig. 7c). 117 Herein, a PEG-biotin probe was functionalized to the flexible loop 6 of OmpG, which upon the binding with mAb and polyclonal anti-biotin, caused three distinct types of current response, compared to the original current fluctuations caused by nanopore gating. Although efficient capture was achieved in both measurements, the ultra-strong binding of the biotin·protein complex implied that it was almost impossible to reach the statistical significance within a reasonable timeframe.
Very recently, the simultaneous detection of WT barstar (Bs) and a lower-binding-affinity variant, D39A Bs, was further illustrated by Movileanu using a ribonuclease barnase (Bn, the receptor of Bs) tethered t-FhuA nanopore. 71 The Bn recognition domain was fused to the truncated FhuA via a (GGS)2 linker and was equipped with an additional peptide signaling tag (O). A surprisingly stable open-pore conductance was observed for the OBn(GGS)2t-FhuA nanopore, and was slightly lower than that of the Bn(GGS)2t-FhuA, suggesting that the signaling tag partially blocked the through-pore ionic current flow at a constant position. When a Bs molecule was bound to the Bn receptor, the peptide adaptor was driven away from the entrance of t-FhuA, leading to a step increase in the current state. Such an "on-off" type of current response, analogue to the common nanopore stochastics sensing, 7 could simplify the identification of various analytes. The discrimination of WT and D39A Bs in a mixture, however, was only possible based on their dwell time, while no difference was found in the binding induced blockade. In addition, it is noted that the binding affinity of the tethered probe with protein analytes can influence both the capture rate and dwell time—in other words, it is not easy to design a suitable recognition element to show both a high association and a high dissociation rate for the target protein.
The third strategy for the nanopore sensing of folded proteins is to adopt external probes. Rather than determine the protein targets, this method decides to measure the probes or protein·probe complex. A proof-of-concept experiment was first demonstrated by John J. Kasianowicz in 2001, in which two biotinylated oligonucleotide probes of different length, bT-poly(dA)10 and bT-poly(dA)50, were designed for the detection of avidin by α-HL. 187 After binding, the signal frequency of the shorted probes decreased, as the resulting avidin·bT-poly(dA)10 complex was extremely difficult to be captured by α-HL. By contrast, the longer DNA leader of avidin·bT-poly(dA)50 was able to thread into the lumen of α-HL, and to trap the complex at the nanopore entrance for an extended period, generating a new current blockade of long dwell time. Hence, integration of multiple external probes allowed the simultaneous detection of various proteins.
To further put this external probe strategy into practice, Wu redesigned the DNA probe approach by incorporating an "analyte capture induced probe release" scheme. 144,188,189 As shown in the first example (Fig. 8a), a ferrocene@cucurbit[7]uril (Fc@CB[7]) decorated ssDNA signaling probe was hybridized with a 25-mer DNA aptamer that could specifically bind to the analyte, vascular endothelial growth factor 121 (VEGF121). 144 Sequence of the signaling probe was complementary to that of the aptamer, except for the site to link Fc@CB[7] barcode, and/or several other mismatched bases to facilitate the competitive binding by protein targets. After the incubation with analyte, streptavidin-modified magnetic beads were used to remove the biotinylated aptamers that were either hybridized with protein or probe, leaving only the released DNA signaling probes for the susequent nanopore quantification. Owing to the presence of Fc@CB[7] barcode, the probe signals included a characteristic current oscillation (level 2–2'), which was attributed to the collision of a dissociated CB[7] within the vestibule of α-HL. A linear detection range of 5–500 pM was achieved for VEGF121, despite the extremely low signal frequency. The beauty of this scheme lay in that the incubation (molecular recognition) and the following nanopore detection (signal transduction) were carried out in two separate chambers. In this regard, it was now possible to incorporate a broad variety of signal amplification methods to push the detectable concentration towards fM level. 188–191

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Nanopore SMS for proteins by an external probe strategy. (a) Detection of vascular endothelial growth factor 121 (VEGF121) by α-HL with the help of a ferrocene@cucurbit[7]uril (Fc@CB[7]) modified DNA signaling probe and DNA aptamer hybrid. Upon the binding of VEGF121 with DNA aptamer, the DNA-Fc@CB[7] probe is released for nanopore analysis. A characteristic CB collision induced current oscillation (level 2–2') can be used for the accurate identification of signaling probes. (Adapted with permission from Ref. 144. Copyright 2015 John Wiley and Sons.) (b) Five Fc@CB[7] barcoded DNA signaling probes with distinct current response. DNA1 is 21-mer, including an alkyne-modified thymine (T*) in the middle to tether Fc@CB[7]; DNA2 is also 21-mer, but with a shorter amine-modified thymine (T'); DNA3 is 11-mer with a T* at the center; DNA4 is 21-mer, with a 5'-terminal modified Fc@CB[7]; and DNA5 is 21-mer, with a 5'-terminal modified Fc@CB[7] and a PEG-6 spacer. (Adapted with permission from Ref. 188. Copyright 2018 John Wiley and Sons.) (c) Recognition of divalent and monovalent antibodies through a triplex molecular beacon (tMB) strategy. The tMB is composed of two ssDNA strands, an antigen-terminated loop-forming one (black) for molecular recognition and a Fc@CB-barcoded stem-forming one (red) for signal transduction, which are bound via the Watson-Crick and Hoogsteen base pairing. Upon the binding with antibody targets, the respective signaling probes are released for the subsequent nanopore detection. (Adapted with permission from Ref 189. Copyright 2018 John Wiley and Sons).
Download figure:
Standard image High-resolution image{kind=link}
Similar to the aforementioned external probes for nucleic acids, the simultaneous detection of various proteins is also readily achievable. 145,188,189,192 On one hand, it is required to encode the signaling probes. As shown in Fig. 8b, a group of five Fc@CB modified oligonucleotide probes were rationally designed, by changing the length of the linker (DNA2 and DNA5), the length of the DNA strand (DNA3), or the position to tether the Fc@CB barcode (DNA4 and DNA5); each individual signaling probe was able to exhibit a unique current signature. 188 Different lengths of ssDNA strands 145 and ssDNA/dsDNA hybrids 192 were recently applied for the encoded signaling probes as well. On the other hand, the specific recognition domain on each probe needs to be adapted to the respective protein analyte. To enable this process, a series of triplex molecular beacon (tMB) was designed, which was composed of a loop-forming antigen-terminated ssDNA strand (molecular recognition) and another stem-forming Fc@CB-encoded DNA signaling probe (Fig. 8c). 189 The stem of a tMB was formed by the Watson-Crick and Hoogsteen base pairing between two strands, and could be opened upon the binding of antibody targets, so as to release the signaling probes for nanopore determination. The selectivity for a particular protein target, therefore, can be easily accomplished by replacing the recognition sites with, e.g. other antigens or aptamers, 145 at the end of tMB stems.
Conclusions
In summary, by going through the nanopore sensing principle, the typical kind of protein pores, and the major types of analytes or SMS applications, this review attempts to answer two key questions:
- a)is there an intrinsic structure-function relationship between the analyte and nanopore? In other words, for a given protein pore, is it possible to predict the (most) suitable applications, or vice versa?
- b)what is the unique selling proposition of nanopore SMS, i.e., for which applications or type of analytes do they have a competitive advantage over other sensors?
Starting with the fundamental theory of nanopore SMS, it is noted that a full cycle of signal in any current trace can be simplified into four parameters, e.g., the open-pore current (I0 ), the interval time (toff ), the current blockade (ΔI), and the dwell time (ton ). Among them, I0 and ΔI are determined by the size and charge of the nanopore and/or analyte, as well as the position of analyte in nanopore; while toff is inversely proportional to the analyte concentration, but ton is independent of it. As discussed throughout this review, the minimum number of signals to be recorded within the required response time defines the higher limit for the sum of toff and ton, and furthermore, the lower detection limit of nanopore SMS. For instance, assuming that at least 1,000 signals need to be detected in 10 min, the maximum value for the sum of ton and toff should therefore be 600 ms, on average. Assuming that ton is 100 ms, which as we know is independent of the analyte concentration, the LOD will be reached when toff increases to 500 ms. Clearly, a high capture rate (1/toff ) is pivotal to the development of ultra-sensitive nanopore SMS, which is diffusion-limited and/or entry-barrier-dependent.
Theoretically speaking, there is an "energy barrier" on both sides of nanopore. In order to accelerate the analyte capture while maintaining a sufficiently long dwell time in nanopore, it is crucial to ensure that the entry barrier is smaller than the exit one, and thus toff and ton can be decided by two different structural domains of a protein pore, allowing a precise tuning of each parameter. Taking into consideration of the size of analytes, those nanopores that have a "wide" opening and a "narrow" exit may fulfill the above requirements, such as to capture βCD from the trans side of α-HL, 129 cationic peptides from the cis side of AeL, 131 and unfolded proteins from the cis side of ClyA 103 and FraC. 113
According to the published literature, it is found that the (di-)electrophoresis-driven capture rate of highly charged DNA 144 or peptide 126 targets/leaders can reach a maximum of 107−108 M−1 s−1. Hence, with the help of asymmetric salt gradient or other signal amplification methods, the (probe-assisted) detection limit may be pushed to a picomolar level. Such an estimated value is comparable/superior to the electrochemical 149 or optical 150 detection of metal ions; is comparable/superior to the liquid chromatograph-mass spectrometry (LC-MS) 193,194 for the environmentally relevant or biologically active small molecules; is far below the isothermal amplification 170 for nucleic acids; and is inferior to the immunoassay 195 for proteins.
Bear in mind that nanopore SMS are more than just another ultra-sensitive chemical or biosensors. A low limit of detection is important, but definitely not the (only) feature. Instead, the competitive advantage should lie in the exceptionally high spatiotemporal resolution of nanopore transducers, which offers sub-angstrom precision with sub-millisecond accuracy, 196 enabling a high-throughput detection of analytes with tiny structural variations7 and even stereoisomers. 168 To maximize the current resolution of nanopore SMS, the signal transduction volume sometimes needs to be reduced, so that the peak separation between two different current states is increased. 168 Alternatively, it is also possible to introduce covalent binding sites into nanopore. In this regard, the residence time of analytes is prolonged, decreasing the respective FWHM of current blockade. 75 Importantly, both of these two approaches are readily achievable with protein pores, leveraging their well-defined structural basis and the versatility and precision of protein engineering.
Note that a prerequisite to realize such a resolution is that the target molecules have to enter the pore. For folded proteins that are too large to be trapped, even though they can be recognized outside nanopore through external probes, it is unlikely to transduce their structural feature by current blockade. On the other hand, with regard to the determination of metal ions, there seems little structural information that needs to be resolved. In comparison, the broad range of isomers of small molecules, which are hardly discriminable by LC-MS, may be the ideal targets for nanopore SMS. Here, the critical conceptual advance is indeed to observe those tiny structural differences directly by transducing them, rather than recognize them via a label, and in particular, in a single-molecule fashion.
So, finally, back to the above two questions. A simple answer to the first one is "yes." The most suitable protein nanopore for a particular kind of analytes must have a proper size and charge distribution, including a wide entry and narrow exit, and/or an electrostatic attraction and trap, as well as a controllable EOF, so as to allow the rapid capture and extended residence of the targets. In addition, based on the structures of protein pores, oligonucleotides and charged peptides seem to be the most appropriate types of analytes (leaders). A high capture rate and long dwell time of DNA or peptide are essential to the development of efficient and reliable nanopore SMS.
Regarding the second question, the answer is still open to debate, but as mentioned previously, one possibility may be the precision sensing of those extremely low concentration isomers of, e.g., per- and poly-fluoroalkyl substances 197 or antibiotic drugs. Determination of those species remains challenging even for the state-of-the-art analytical techniques. Therefore, the high resolution of nanopore SMS is supposed to play a role in the accurate identification of these structurally similar compounds. Clearly, a lot more research efforts are needed to improve the detection limit, 198 especially via the probe-directed capture and trapping strategies. Apart from that, it is also noted that many new nanopore applications are booming in almost every aspect of research, including but not limited to the medical diagnosis and treatment, 199–201 bionanotechnology, 202,203 environmental monitoring, 158 and even energy storage. 204 Since nanopore technology features many other unique advantages, including the ease of use, portability, reasonably low cost, and parallelization possibility, the authors envision that the new era of nanopore SMS will be coming soon, as long as the concentration limit can be properly addressed.
Acknowledgments
This work was supported by National Natural Science Foundation of China (grant no. 21972041 and 22006037), Shanghai Sailing Program (grant no. 19YF1410400), and State Key Laboratory of Pollution Control and Resource Reuse Foundation (grant no. PCRRF20001).