
Abstract
Viable anionic exchange membrane (AEM) electrolysis for H2 production requires highly active hydrogen evolution reaction (HER) catalysts that are also robust and cost-effective. Traditionally juxtaposition of two transition metals with one being more oxophilic is necessary for easier access to water in the inner Helmholtz plane as a source of protons in the crucial Volmer step for enhanced HER activity. However, they are prone to passivation at anodic potentials or deactivation via the formation of sub-surface metal hydrides at high cathodic overpotentials. Here we report a method of enabling close juxtaposition of Ni-Ni-oxide surfaces by functionalizing Ni-based catalysts via the careful thermal treatment of carbon-supported Ni nano-particles chelated with a select organic chelating compound referred to as cupferron. This functionalization results in an embedded Ni surface with a clear graphitic coating engendering long-term stability for passivation and subsurface hydride formation. The stability and activity of the functionalized Ni catalyst are demonstrated in half-cell RDE and full-cell AEM membrane-based hydrogen pump experiments. In the case of the latter, the functionalized Ni outperforms carbon-supported Pt.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Water electrolysis is a potentially cost-effective and carbon-neutral means of hydrogen gas production. In the United States, the targeted cost of hydrogen production, as set by the Department of Energy (DOE), is $2/kg by 2020. 1 The development of low-cost and reliable catalysts for the oxygen evolution reaction (OER) and the hydrogen evolution reaction (HER) is a key component in achieving this goal. The relatively recent introduction of anion exchange membranes has allowed for the substitution of platinum group metals(s) for cheaper transition metal catalysts in these reactors. 2–7 In alkaline media, HER presents a particular challenge: at high pH's HER is more sluggish because of the external energy needed to generate H+ from the catalytic dissociation of water, 8–10 whereas in acidic media H+ is freely available. 11,12
In alkaline media, three steps can be used to describe the HER mechanism. 13–15 Although there is ambiguity whether the HER mechanism undergoes either Volmer-Tafel or Volmer-Heyrovsky, it is generally accepted that the Volmer step occurs first, breaking the water molecule before the hydrogen recombination to form H2. However, some publications claim that a Volmer-Heyvrosky pathway is preferred in alkaline media 16,17 due to smaller energy barriers than Volmer-Tafel. Therefore, the Volmer-Heyvrosky pathway is the one considered throughout this work, see Eqs. 1 & 2. Also, the side product OH–, needs to be removed from the surface efficiently to complete the Volmer step.
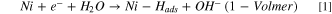
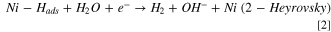
Based on the work by Markovic et al. 2 oxophillic sites facilitate the adsorption and dissociation of water into Hads and OHads according to the bi-functional mechanism. The oxophillic sites effectively weaken the H–OH bond and facilitate the transport of the resulting OHads away from the (Ni0) active site (Eq. 4). This mechanism is summarized in Fig. 1.
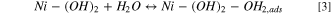
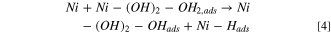

Figure 1. Schematic depicting a bifunctional mechanism using a functionalized mono-metallic surface using Ni. Note the use of oxophilic moieties for enabling water transport to the Ni0 surface for hydride formation.
Download figure:
Standard image High-resolution imagePreviously, bifunctional metal/metal oxides (MMOx) have been used in the water-gas shift reaction, 18,19 proton hopping, 20 carbon monoxide oxidation. 21 Even though the logic of bifunctional HER mechanism was well known for decades its true delineation was demonstrated by Markovic, et al. using a Pt surface decorated with Ni(OH)2 clusters, resulting in an increase of HER activity by a factor of eight. 8
Nickel, and other 3d transition metals, possess a 3d band spanning the Fermi level which satisfies the Sabatier Principle and therefore makes them attractive candidates for HER catalysts in an alkaline environment. Among these metals, Ni shows a hydrogen binding energy close to that of Pt despite having a lower activity towards HER. 22 In addition, Ni has the highest resistance to corrosion in an alkaline environment. These properties of Ni result in a close affinity for electrons to transfer from the adsorption site to proton. 23–25 Therefore, in terms of the rational design of non-Pt HER catalysts, a material comprised of Ni and NiOx would appear to be a promising candidate.
Wang et al., 26 Gong et al., 27 and Lovell, et al. 28 each reported the HER performance of Ni/NiOx catalysts either unsupported or loaded onto nanostructured carbon with the two latter studies reporting a performance close to that of Pt. However, the stability of these catalysts was not adequately addressed. Gong et al. reported a degradation rate of 2.4 mV hr−1 in a full electrolysis cell, however, this was conducted at 20 mA cm−2, a current density far too low to be comparable to actual operating conditions. The study by Wang et al. 26 highlighted the importance of creating a favorable Ni:NiOx ratio noting that above 1:2, had a detrimental impact on performance by not providing an adequate specific surface area of Ni for hydrogen adsorption and recombination. While the previous studies demonstrate the ability and importance of creating a catalyst with a favorable ratio of Ni:NiOx they do not show an ability to maintain it: exposing the catalyst to anodic potentials or even ambient conditions over time could create additional NiOx sites. In this work, we report a functionalized monometallic Ni-based catalyst where layers of graphene introduced during synthesis envelop the Ni0 particles making these moieties resistant to oxidative passivation and formation of deactivating metal hydride regions while maintaining a favorable ratio of Ni0 to NiOx. 16,29,30 Fundamental studies using rotating disk electrodes (RDE) show the anodic and cathodic stability of the functionalized Ni catalyst as opposed to a standard metal-metal oxide (MMOx) catalyst such as those represented by NiCrOx/C used in this study. In addition, the efficacy of this catalyst is demonstrated in the practical context of a full cell operated in a hydrogen pump configuration that exhibits superior alkaline HER activity and durability. These complementary characterizations show that unlike standard Ni or MMOx catalysts, whose surfaces are susceptible to poisoning through oxidation and hydride formation, 16,29,30 the surface of functionalized Ni catalyst maintains stable HER activity even after anodic and cathodic polarization. This study is further embellished by the use of synchrotron-based in situ X-ray absorption spectroscopy for probing durability of the Ni surfaces for passivation and hydride formation and also a kinetic analysis of the HER reaction as qualitative support of the electrochemical data represented by RDE studies.
Experimental
Catalyst synthesis
30% Ni metal on Ketjen Black-EC600JD (Akzo Nobel Polymer Chemicals abbreviated as K600) was synthesized using a standard reduction method. Briefly, carbon black was dispersed in 18.2 MΩ H2O (Millipore) and stirred overnight. The appropriate amount of NiCl2 6H2O dissolved in 10 ml H2O was added to the carbon dispersion and the reaction mixture was stirred in an ice bath for one hour under a constant stream of nitrogen. 3 molar equivalents (with respect to the metal) of the sodium borohydride (NaBH4) reducing agent solution (Sigma Aldrich) were added dropwise to the reaction mixture which remained in an ice bath for the remainder of the process. Once the exothermic reaction was completed, the mixture was vacuum filtered, washed three times with H2O, and dried overnight at 80 °C under reduced pressure. The catalyst was heat-treated at 700 °C for 3h in argon.
6H2O dissolved in 10 ml H2O was added to the carbon dispersion and the reaction mixture was stirred in an ice bath for one hour under a constant stream of nitrogen. 3 molar equivalents (with respect to the metal) of the sodium borohydride (NaBH4) reducing agent solution (Sigma Aldrich) were added dropwise to the reaction mixture which remained in an ice bath for the remainder of the process. Once the exothermic reaction was completed, the mixture was vacuum filtered, washed three times with H2O, and dried overnight at 80 °C under reduced pressure. The catalyst was heat-treated at 700 °C for 3h in argon.
Ni-Cr/K600 was prepared using the method described by Bates et al.
31
Briefly, K600 was dispersed in water followed by enough NiCl26H2O and Cr(NO3)39H2O (reagent grade, Sigma-Aldrich) to create a 1:1 ratio of Ni:Cr and a 60% wt. metal loading on the carbon support. The reaction mixture was stirred in an ice bath under a constant stream of nitrogen for one hour. 10 ml of freshly prepared NaBH4 solution (3 molar excess with respect to the metals) was added dropwise and the mixture was stirred for 24 h to ensure the reaction reached completion. The solid was subjected to the same workup as described above (filtered, dried, etc) and heat-treated at 500 °C for 6h in a 5% H2/95% Ar atmosphere to fully anneal the alloy.
Ni-functionalized/K600 (NFK) was prepared via the chelation of Ni2+ with cupferron (Alfa Aesar) in the presence of carbon support. 150 mg K600 was added to 20 ml of 18.2 MΩ H2O and stirred overnight. NiCl26H2O and enough cupferron to create a 1:2 molar ratio of Ni2+ to cupferron were dissolved in water separately. The solutions of NiCl26H2O and cupferron were concurrently added to the dispersion of K600 and the mixture was stirred for 48h. Excess solvent was removed with vacuum filtration and the product was dried overnight under reduced pressure at 80 °C. The complex was heat-treated at 700 °C in Ar for three hours to yield a 30% Ni catalyst. The Ni2+-cupferron complex was also prepared without carbon support or heat treatment for characterization purposes.
Electrochemical methods
Electrochemical measurements were conducted using Autolab potentiostat/galvanostat (PGSTAT30, Metrohm). All RDE experiments were conducted in a 3-electrode configuration using a fluorinated ethylene propylene (FEP) cell with a 50 °C 0.1M KOH solution (semiconductor grade 99.99% metal trace, Sigma Aldrich) as an electrolyte. A gold flag was used as a counter electrode and all potentials were measured against a reversible hydrogen electrode (RHE). Inks composed of 0.6 ml H2O, 1.39 ml 2-propanol, 10 μl of Nafion dispersion (5% wt., Alfa Aesar), and an appropriate amount of catalyst were drop-casted onto a 0.247 cm2 glassy carbon electrode. 10 μg cm–2 and 50 μg cm–2 were the target metal loadings of Pt/C and Ni-based catalysts, respectively. The electrolyte was purged with Ar and catalysts were conditioned prior to acquiring electrochemical data by cycling the potential from 0.05 VRHE to 1.05 VRHE ten times. Unless otherwise stated, RDE measurements were acquired with a scan rate of 50 mV s–1 with the working electrode rotating at a rate of 2500 rpm. Current densities for Ni-based catalysts were normalized to A cm–2 Ni based on the available Ni active sites taken from 0.03 VRHE to 0.35 VRHE. A more detailed explanation of how the ECSA was calculated can be found in the Supporting Information. All potentials reported were corrected for uncompensated resistance using electrochemical impedance spectroscopy (EIS). EIS boundaries were set at open circuit potential (OCP) while applying 10 kHz to 0.1 Hz with a 10 mV perturbation for 50 points.
The anodic stability of each catalyst was assessed after anodic and cathodic conditioning. After conditioning in Ar gas, the solution was purged with H2 gas, and three different upper limits (0 VRHE, 1.00 VRHE, and 1.05 VRHE) were obtained. After an initial sweep from OCP (typically + 0.02 VRHE) to –0.60 VRHE, the potential was cycled ten times from –0.60 VRHE to either 0 VRHE, 1.00 VRHE, or 1.05 VRHE. Chronoamperometry was taken from OCP to 0 VRHE to –0.02 VRHE to –0.20 VRHE in increments of 5 mV increment/step immediately after each CV.
The cathodic stability of the catalysts was also tested after the electrolyte was purged with H2 gas. The potential was cycled from OCP to –0.3 VRHE to 1.05 VRHE for a total of 9 complete scans. After the 9th scan, the potential was brought to –0.3 VRHE and held for 30 s after that, the potential was cycled from –0.3 VRHE to 1.05 VRHE 9 times. This procedure was repeated with a 10 min hold at –0.3 VRHE.
Hydrogen pump experiments were conducted using 5 cm2 gas diffusion electrodes (GDEs) comprised of Toray 90 carbon paper hot pressed to a novel polyaryl piperdine–triphenol methyl (PAP-TP-ME) based anion exchange membrane (AEM) (University of Delaware, W-7 Energy). This membrane exhibited an ion exchange capacity of 2.0 mili-equilvalent/g and ionic conductivity (104.1 mS @ 60 °C) and excellent thermal stability at 90 °C. All catalyst inks were prepared with a targeted metal loading of 3 mg cm−2. Isopropanol and water (1:1) were added to the catalyst yield a 3% wt. solution with respect to the total mass of the catalyst. Finally, a solubilized form of the ionomer (25% wt. with respect to the catalyst) was added to the ink and sonicated for one hour. Catalyst inks were applied to the GDL material via airbrush. In each experiment, Pt/C (47.2%, Tanaka) was used as an HOR catalyst, with one of the Ni-based used as the HER catalysts. All hydrogen pump experiments were conducted at 60 °C with humidified H2 (50 sccm) flowing across the anode and humidified N2 (200 sccm) flowing across the cathode. Backpressure was not applied to either gas feed. Galvanostatic polarization curves were obtained by collecting the potential at the end of a series of one-minute chrono-potentiometric holds from 0–500 mA cm−2 in 50 mA cm−2 increments. Potentials were corrected for interfacial resistance using high-frequency resistances obtained through impedance spectroscopy. Impedance spectra were collected between 100 kHz and 0.1 Hz with a signal amplitude of 0.01 VRMS.
Characterization
X-ray diffraction (XRD) patterns were obtained using an Ultima IV XRD (Rigaku) operated at 40 kV and 44 mA. The X-ray source was Cu Ka (λ = 1.541 A). The procedure was a function of time that uses 0.1° step size and 5 s hold per step. XRD patterns were analyzed with PDXL 2 (Rigaku) software.
X-ray photoelectron (XPS) spectra were acquired using a Kratos Axis Ultra DLD spectrometer using a monochromatic Al Ka power source at 125 W. Charge neutralization was not necessary. Three areas of each sample were analyzed to ensure consistency. Pass energies of 20 eV were used for the acquisition of high-resolution spectra. The fitting and quantifications were made obtained with Casa XPS software using the default sensitivities factors. High-resolution spectra were acquired for Ni 2p, C 1 s, O 1 s, and N 1 s. Acquisition times for the surveys were of 3 min, while for high-resolution spectra, surveys were acquired for 1 h.
Overall metal loading was determined by inductively coupled plasma optical emission spectroscopy (ICP-OES) performed by Robertson Microlit Laboratories (Ledgewood, NJ).
The preparation of the XAS electrodes can be found in our previous work. 32 The XAS experiments were conducted at room temperature in a previously described flow half-cell in which continuously H2-purged 0.1 M KOH was circulated at a flow rate of 0.4 cm3s−1. 5 Spectra were obtained at –0.1, 0, and 0.6 VRHE. The XAS spectra at the Ni edge were collected in the transmission mode, at the ISS 8–ID beamline of the National Synchrotron Light Source (NSLS) II, Brookhaven National Laboratory (BNL). Typical experimental procedures were utilized with details provided in our previous work. 33
High resolution–transmission electron microscopy (HR-TEM) and their Fast Fourier Transformations (FFT) and selected area diffraction (SAD) data were collected on an FEI Titan Themis 300 HR-TEM. Scanning electron microscope (SEM) images and energy dispersive spectra (EDS) were collected on an S-4800 FE (Hitachi).
TGA data of Ni-functionalized catalyst was collected on an SDT Q600 (V20.9 Build 20, TA instruments). The Ni2+-cupferron complex (i.e. without carbon support) was characterized using a 10 °C min–1 ramp to 700 °C, Ar flow (100 ml min–1).
Results and Discussion
Cupferron was ultimately chosen as a chelating agent in this study because of its ability to form complexes with nickel in aqueous solutions under mild conditions. Cupferron has been extensively used for chelation–based extraction of trace metals and its complexes have been characterized, however, this is the first instance where it has been used for the preparation of electrocatalysts.
34
In an inert atmosphere, metal-organic complexes such as Ni(Cup)22H2O thermally decompose to form metal or metal oxide particles dispersed in amorphous carbon.
35–37
The structure and composition of the metal-organic complex as well as the heat treatment process determine the structure and composition of the final products, therefore a thorough mechanistic understanding of the decomposition reactions is essential to improve the performance of these types of catalysts. The decomposition mechanism generally involves
1
rupture of metal-ligand bonds to nucleate nanoparticles;
2
particle growth via catalytic reaction at particle surface;
3
carbonization of organic residues and reduction of metal oxides.
The decomposition process of Ni(Cup)22H2O was characterized by TGA/DSC analysis combined with SEM/EDS and XRD. First, dehydration of the complex occurs at ≈100 °C forming the anhydrous complex followed by the melting of the complex at ≈200 °C. The TGA profile of the nickel-cupferron complex (Fig S1 (available online at stacks.iop.org/JES/168/084501/mmedia)) shows a ∼50% loss of mass beginning just below 200 °C. Bottei and Schneggenburger reported the decomposition of cupferron at this temperature which resulted in the evolution NO and NO2.
38
In the case of the nickel-cupferron complex, it is reasonable to assume that the thermal degradation of the complex yields NiO in addition to nitrogenous gases. Carbonization of the carbon residues begins at ≈400 °C and the NiO is reduced to Ni0. Above 650 °C, Ni has the ability to graphitize the carbon backbone.
39,40
The chelation and decomposition processes are summarized in Fig. 2.

Figure 2. Schematic of chelating process and heat–treatment method together with SEM of the final catalyst.
Download figure:
Standard image High-resolution imageXRD patterns of both Ni functionalized and non-functionalized on carbon show one phase of Ni metallic (Ni0) (Supporting information; Fig. S2) having a similar crystallite size of about 11 nm which matches TEM images showing an average particle size range was 10–12 nm. SEM images show high surface area nanomaterials embedded on carbon for both catalysts (Fig. 2). The overall Ni loading of the Ni-functionalized was determined by ICP-OES and corroborated with TGA and EDS. The systematic approach used to optimize catalyst synthesis (e.g. metal loading, heat treatment time, etc.) is discussed in the supporting information.
Electrochemical results
Three catalysts: Ni/K600, Ni-Cr/K600, and nickel-functionalized/K600 (NFK) are used in this study as representative examples of non–functionalized monometallic, bifunctional mixed metal oxide (MMOx), and functionalized catalysts, respectively. Figure 3 compares the polarization curves of these three catalysts to that of Pt/C in the HER region (0 to –0.6 VRHE). Although Pt/C still shows superior HER activity in alkaline pH, the non-Pt group catalysts show promising activity, in particular the NFK. The HER performance trend among the non-Pt catalysts was: NFK > Ni-Cr > Ni agreeing with the proposed bifunctional mechanism and demonstrating the efficacy of NFK relative to its mixed-metal and non-functionalized counterparts.

Figure 3. HER comparison for Pt/C (LD: 10 μg metal cm–2) vs non-Pt (LD: 50 μg metal cm–2) from 0 V to –0.6 VRHE in H2 saturated 0.1M KOH.
Download figure:
Standard image High-resolution imageA study of the anodic upper limit was conducted (Fig. 4) to provide a comparison of oxidative passivation activity among the three classes of catalysts. An anodic study may seem unnecessary in the development of HER catalysts which, generally operate in negative potentials. However, in the practical context of an electrolysis cell, potentials can switch polarization during start-up and shut down, exposing the HER catalyst to anodic potentials.

Figure 4. A comparison of the steady-state (SS) chronoamperometry curves of NFK (A), Ni/K600 (B) and, Ni-Cr/K600 (C) after various cycling to different upper anodic limits. Conditions: 0.1M KOH, 50 °C, H2 purged, 2500 rpm.
Download figure:
Standard image High-resolution imageFigure 4 shows that cycling NFK to higher anodic potentials yields better HER performance with the best performance being achieved when the catalyst is cycled to 1.05 VRHE. The improved performance is likely due to the in situ formation of additional NiOx sites forming a favorable ratio of NiOx and Ni0. In contrast, the lower HER activity of the catalyst prior to anodic voltage threshold conditioning suggests that the NFK initially had insufficient NiOx sites. According to Hall et al., 29 the zero-valent, metallic Ni is oxidized to NiOx above 0.20 VRHE and Ni(OH)2 layers start to form and thicken right after the reaction of Ni → NiOx is complete, forming some nickel hydroxides irreversibly onto the surface. As shown, anodic cycling beyond a voltage threshold of 1.00 VRHE, improves the NFK's HER activity, presumably due to an increase in the number of surface oxides. When the catalyst is exposed to an oxidizing potential of 1.05 VRHE, NFK exhibited the best performance. As anticipated, Ni/K600 and Ni-Cr/K600 have the opposite trend compared to NFK because they lack protection against passivation in the anodic region.
Furthermore, these catalysts were tested for cathodic stability by holding the potential at –0.3 VRHE for different lengths of time. Figure 5 presents the CVs showing the anodic vs cathodic behavior of Ni/K600 vs NFK, initially without –0.3 VRHE—hold (0 s) followed by various hold times (30 s, and 10 m). The performance at the 9th scan was noted as it was the most stable after the cathodic hold Fig. 5.

Figure 5. Cathodic stability studies @ –0.3 VRHE (before iR comp) of (a) NFK, (b) Ni/K600 and (c) Ni-Cr/K600. HER activity plots are taken from the CVs, (0 VRHE to –0.3 VRHE). Insets show a complete CV, to focus on the Ni/NiOx redox peaks. Condition: 0.1M KOH, 50 °C, H2 purged, 2500 rpm.
Download figure:
Standard image High-resolution imageBesides suffering from oxidative passivation, unprotected Ni materials can be poisoned by hydride (Hx) formation on the surface resulting in a drop in HER catalytic activity. The NFK was stable in the cathodic region within 10 scans while the HER activity of both Ni/K600 and Ni-Cr/K600 degraded drastically within a couple of cycles. Based on evidence from Ni–hydride studies by Soares, 41 Hall et al. were able to show that the formation of hydride in the Ni decreased the HER activity. 29
Evidence of protection for Ni–functionalized/K600
HR–TEM/SAD/FFT
Figure 6a shows an HR-TEM image of NFK with a Ni particle encapsulated by five graphite layers. The Fast Fourier Transform (FFT) (Fig 6a, inset) and the selected area diffraction (SAD) (Fig. 6b) confirmed the carbonaceous material covering the nickel particle was graphite due to the presence of a G-002 lattice plane in both images. Figures 6b and 6d represent the difference in SAD patterns between 30% NFK and 30% Ni /K600, respectively, where the functionalized Ni had a graphite plane (G–002) while the non-functionalized Ni did not.

Figure 6. HR-TEM images of (a) 30% NFK and (b) 70% Ni/K600. (c) and (d): SAD patterns of NFK vs Ni/K600 taken at 100 nm areas.
Download figure:
Standard image High-resolution imageEx situ XPS
XPS was conducted using two samples: Ni/K600, and NFK with K600 as a control. The surveys (Supporting Information, S4) show that no Ni was present on the K600 sample while peaks associated with Ni0 were detected on both Ni samples. 42 Oxygen and carbon contributions are detected to roughly the same extent (comparable peak areas) for all three samples.
Figure 7 shows the deconvoluted peaks of the Ni 2p region XPS spectra of NFK (Fig. 7A) and Ni/K600 (Fig. 7B). According to the literature, the Ni0 peak is located at 852.5 ± 0.3 eV, 42–46 while nickel monoxide (NiO) can be found is centered at 854 ± 0.3 eV 42,44,46,47 and nickel hydroxide (Ni(OH)2) is centered at 855.5 ± 0.3 eV. 42,44,46 Degenerate peaks can also be detected due to the plasmon effect 48 at 858.5 eV for Ni0 and 861.2 eV for NiOx. 49 The XPS spectrum of Ni/K600 (Fig. 7B) indicates that the surface is comprised of Ni(OH)2 (main phase) and NiO. Peaks associated with Ni0 are absent in Ni/K600 due to oxidation in ambient conditions. Conversely, Ni0 was the main contribution in the spectrum of NFK (Fig. 7A and Table I), indicating that the encapsulation of the Ni, formed during the preparation, limits passivation of its surface under ambient conditions, as shown in Figs. 6a and 6b.

Figure 7. Ni 2p contribution for (A) NFK and (B) Ni/K600.
Download figure:
Standard image High-resolution imageTable I. XPS quantitative results for Ni 2p survey.
| Binding Energy (eV) | Bonds | Ni-functionalized /K600 | Ni/K600 |
|---|---|---|---|
| 852.6 | Ni metallic | 46% | 0.0% |
| 853.5 | NiO | 15% | 14% |
| 855.2 | Ni(OH)2 | 40% | 76% |
| 858.5 | Unknown | 0% | 10% |
As the two catalysts undergo comparable heat treatments and considering that NFK exhibits greater long-term stability under cathodic potentials (unlike the Ni/K600, Fig. 5), it can be inferred that this is due to the preservation of the Ni0 active sites. TEM images are consistent with this hypothesis as they show graphite layers encapsulating the nickel nanoparticles for the NFK (Fig. 6a), whereas no graphite layers could be detected for the Ni/K600 (Fig. 6c). Based on the XPS spectra of these two catalysts, it is reasonable to assert that in the case of NFK, the graphite layers prevent severe passivation of the nickel surface, while no such protection is available for Ni/K600. Also, since both metallic nickel and nickel hydroxide sites are necessary for HER kinetics (i.e., via the bi-functional mechanism), 50 the improved performance of NFK relative to Ni/K600 can be explained by a mixture of Ni0 and NiOx on the surface of the former whereas in the case of the latter the surface is comprised entirely of Ni(OH)2 (Table I). C 1s XPS spectra also showed evidence of C-C bonds on the surface of the catalysts (Supplementary Information, S7), but the C–C bonds from the K600 and the one encapsulating the Ni particles are not differentiable.
ex situ and in situ XAS
An in situ XAS study was conducted on Ni/K600 and NFK at the Ni K-edge in an H2-saturated 0.1 M KOH electrolyte. Spectra were collected as a function of applied potentials to monitor the active Ni/NiOx redox behavior of the two samples during catalysis. Figure 8a shows that the Fourier Transform of the extended X-ray absorption fine structure (FT–EXAFS) of the Ni/K600 at 0.6 VRHE has two peaks (1.5 Å and 2.8 Å) that coincide with the peaks of the Ni(OH)2 standard with reduced peak intensity because of the nanoscale particle size. In addition, Ni/K600 has a small peak around 2 Å that overlaps that of the Ni reference foil. Meanwhile, the intensity of the X-ray absorption near edge structure (XANES) at 0.6 VRHE is even higher for that of Ni(OH)2. These results indicate that the nanoscale Ni(OH)2 is the dominant Ni-species at 0.6 VRHE. As potentials gradually decrease to –0.1 VRHE, the XANES intensity gradually drops; meanwhile, the intensity of the FT-EXAFS peaks of Ni(OH)2 reduces, accompanied by the dramatic increase of the intensity of the FT-EXAFS peak of Ni0. These trends clearly indicate the transition from Ni(OH)2 to Ni0 triggered by lowering the potential.

Figure 8. Ni K edge FT-EXAS (left spectra) and XANES (right spectra) spectra were taken both in situ and ex-situ conditions. In situ data measured with 0.1 M KOH with H2 saturated electrolyte with potentials vs RHE. A&B represents data for surface-functionalized Ni (NFK) and C&D represent Ni/K600.
Download figure:
Standard image High-resolution imageConversely, both the XANES and FT-EXAFS spectra of the NFK closely resemble those of the Ni foil and remain nearly unchanged within the potential range of –0.1 to 0.6 VRHE. The spectrum of Ni(OH)2 is undetectable. These results agree with the XPS data and provide in situ evidence that the functionalized carbon prevents the Ni surface from forming a passivating oxide layer. Note that the lack of peaks associated with Ni(OH)2 in spectra shown in Fig. 8 does not necessarily indicate the absence of Ni(OH)2 but rather the spectra are overwhelmed by the spectrum of the bulk Ni0. This is a particularly reasonable deduction given that most of the Ni in the particles with an average particle size of 10 nm is electrochemically inactive and in the form of Ni0. Therefore, the in situ XAS provides clear evidence that the surface Ni in the NFK catalyst is protected by the graphitic layers and largely retains the Ni0 state.
Kinetic Study
Accurate normalization of the polarization curve to the ECSA of each catalyst ensures that the performance differences between NFK, Ni/K600, and Ni-Cr/K600 are unambiguously kinetics-based. Therefore, a kinetic model based on previous studies 16,51 was used to fit the Faradaic current of each catalyst to better understand how NFK outclasses the two other catalysts. Oshchepkov et al. described the reduction mechanism, which is considered when the electrode is polarized negatively. 51 Data used for the fitting were the forward scan of the ninth cycle from Fig. 5, where performance differences arise between the three catalysts. These data points were specifically selected to propose kinetic-based explanations of the observed different behaviors.
It must be emphasized that the kinetic model described here below aims at describing how hydride formation on the surface can impede, from a mechanistic point of view, the HER. This model is obviously using a simplified mechanism and the determined kinetic rate constants values must not be taken as absolute. The chemical and electrochemical steps considered are presented here below.
Sites considered in the model are:
- NiOx moieties responsible for water attraction
NiOx moieties responsible for water attraction
- Adsorbed water on the NiOx moieties
Adsorbed water on the NiOx moieties
- Metallic (Ni) site
Metallic (Ni) site
- Adsorbed hydroxyl on the NiOx moieties
Adsorbed hydroxyl on the NiOx moieties
- adsorbed hydrogen atom on the metallic moieties
adsorbed hydrogen atom on the metallic moieties
- surface hydride
surface hydride
The proposed bi-functionalized water adsorption mechanism is modeled by the following reactions:
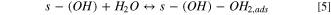
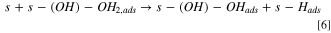
The Volmer–Heyrovsky mechanism was considered for the HER, as it was postulated that it is most likely this consecutive step mechanism that is occurring for the HER than a Volmer–Tafel. 16 Also, considering those consecutive steps enables a simple mathematic hypothesis, allowing the writing of a simple faradaic current expression (see below).
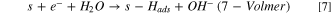
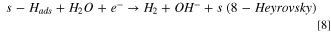
Also, it was important to take into account the formation of hydrides, as it is very well known that Ni0 tends to form hydrides when polarized negatively 16,29,30,52
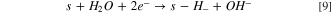
Finally, as hydrides are unstable in water solution, it is natural to consider the evolution of hydrogen from hydrides species:
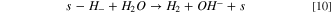
The kinetic model was developed assuming Butler–Volmer behavior, 53,54 and a Langmuir isotherm to simplify the system. The development of the Kinetic Model (See supporting information) has led to the following faradaic current density expression:
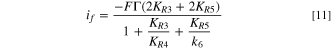
With:
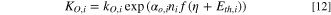
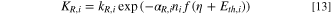
And:
αO,i: symmetry factor for the oxidation of the elementary step i
– αR,i: symmetry factor for the reduction of the elementary step i (and αO,i + αR,I = 1)
–f = F/RT
–kO,i: rate constant of the oxidation direction, for the elementary step i
–kR,i: rate constant of the reduction direction, for the elementary step i
–E: Electrode potential
–Γ: the concentration of the surface-active sites (mol cm–2)
The experimental data from Fig. 4 was fitted to the model using a non-linear least-square method program. The correlation between the experimental and the calculated Faradaic current density is presented in Fig. 9 (left).

Figure 9. (Left) Faradaic current fitting for cathodic curve after 10 m hold at –0.3V. Experimental data were taken from Fig. 5. (A)—NFK. (B)—Ni/K600. (C)—Ni/K600. Right) Surface occupancy of the five surface sites for. (A)—NFK. (B)—Ni/K600. (C)—Ni-Cr/K600.
Download figure:
Standard image High-resolution imageFigure 9 (left) shows a consistent fitting for each material, with sufficient R2 values. To keep a sensible fitting of the data, the rate constant for the Volmer step (easiest one) was constrained to the same order of magnitude for the three materials (as in the end, the barrier energy for the binding of H to a nickel active site should be the same for the three catalysts). The surface occupancy of 
 and
and  are not appearing here as the mathematical simplifications led to conclude that they are null. However, this does not imply that those sites do not exist on the surface of the catalyst, it just means they do not participate in the ECSA (thus they are not electrochemically active). Therefore, the coverage of those species cannot be calculated/assumed from the faradaic current. The fitted kinetic parameters are displayed in Table II.
are not appearing here as the mathematical simplifications led to conclude that they are null. However, this does not imply that those sites do not exist on the surface of the catalyst, it just means they do not participate in the ECSA (thus they are not electrochemically active). Therefore, the coverage of those species cannot be calculated/assumed from the faradaic current. The fitted kinetic parameters are displayed in Table II.
Table II. Fitted parameters value, for the 10 m cathodic hold at –0.3 VRHE experiment for NFK, Ni/K600, and Ni-Cr/K600.
| NFK | Ni/K600 | Ni–Cr /K600 | Reaction | |
|---|---|---|---|---|
| kr,3 (cm s–1) | 5,97E+00 | 3,10E+00 | 3,24E+00 |

|
| αr,3 | 5,00E-01 | 1,00E+00 | 1,00E+00 | |
| kr,4 (cm s–1) | 2,82E-02 | 2,10E-04 | 1,81E-04 |
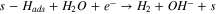
|
| αr,4 | 5,00E-01 | 5,75E-01 | 6,48E-01 | |
| kr,5 (cm s–1) | 8,02E-05 | 7,69E-02 | 7,91E-02 |
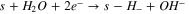
|
| αr,5 | 7,57E-01 | 5,00E-01 | 5,00E-01 | |
| k6 (cm s–1) | 2,84E+00 | 3,93E-03 | 3,24E-03 |
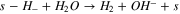
|
| kr,3/kr,4 | 211,99 | 14777,97 | 17940,77 | |
| k6/kr,5 | 35397,66 | 0,05 | 0,04 |
The fitting shows that the fastest step for the HER is the Volmer step, thus implying that Heyrovsky is the rate-determining step for the HER. However, this is not as simple: when looking at the kr3/kr4 ratio, it is evident that the latter is by far lower for the NFK than for the Ni/K600 and Ni-Cr/K600, showing a faster Heyrovsky mechanism for the NFK. Another relevant metric is the kr6/kr5 ratio that depicts the speed of oxidation of the hydrides. In both Ni/K600 and Ni-Cr/K600, this ratio is very low, implying that the hydride species are accumulating on the electrode surface. On the other hand, for the NFK, the ratio is very high, implying that the hydrogen evolution from the hydrides is a very fast process. Also, it must be noted that the value of kr5 (hydride formation on the electrode) is much slower for NFK than for the other two catalysts, implying that very few hydrides compounds are formed on the surface. In order to better grasp the role of the hydride poisoning of the surface, the surface occupancy of each active site versus the potential is depicted in Fig. 9 (right) (See supporting information for the mathematical expression of each active site occupancy).
Figure 9 (right) shows a surface poisoning/accumulation of NiHx for Ni/K600 (B) and Ni-Cr/K600 (C), while the surface of NFK (A) is nearly free of NiHx. The impact of surface hydrides on the HER is evident when looking at Fig. 5, as a high surface concentration of Hads is crucial for a fast proceeding of the Heyrovsky step, and the worst materials (Ni/K600 and Ni-Cr/K600) are clearly depicting that the greater the polarization to a negative potential, the lower the amount of Hads. It is thus highly probable that the graphite shell, surrounding the Ni nanoparticle is playing a key role in the deactivation of the hydride formation of Ni, while not affecting the HER performance in the NFK catalyst. Similar effects have been reported for Ni embedded in reduced graphene oxides. 33
H2 pump and its application
Hydrogen pump fuel cell experiments were performed to assess the performance and stability of Ni-based HER catalysts in a practical context. Full cell tests were run in a hydrogen pump configuration, as opposed to a water electrolysis configuration, in order to decouple the overall cell performance from the sluggish kinetics of the OER. In addition, since the kinetics of hydrogen oxidation on Pt are so fast, it is reasonable to assume that the HER catalyst is the sole contributor to the cell's overpotential. The reactions in a hydrogen pump fuel cell are:
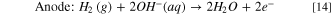
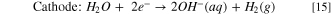
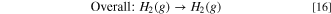
While hydrogen pump cells do have practical applications such as electrochemical hydrogen purification, here it is used only to demonstrate how these HER catalysts perform in a fuel cell. To the best of our knowledge, this is the first instance of anion exchange membrane hydrogen pump fuel cell performance data being reported in the literature. It should be noted that the several orders of magnitude higher overpotentials relative to analogous proton exchange membrane-based cells are not unexpected as similar disparities in exchange current densities have been observed in RDE studies of HER in acidic and alkaline electrolytes. 14 Figure 10A compares the polarization curves of three hydrogen pump cells, each equipped with one of the three Ni-based HER catalysts, these are compared with a cell containing Pt/C HER electrode. In all four cells, a Pt/C electrode was used for HOR. Surprisingly, the chronopotentiometric polarization curves in Fig. 10A contradict several trends in the curves obtained in RDE experiments shown in Fig. 3. At every point of the curve, NFK required a lower overpotential than the other HER catalysts to attain the same current density. Compared to the polarization curve obtained from the RDE study shown in Fig. 3, where NFK does not surpass the current density of Pt/C until ∼ –520 mV. NFK does appear to be preferable to Pt/C in a hydrogen pump cell: at 500 mA cm–2, a current density used to benchmark electrolysis cells, outperforming Pt/C by 154 mV. Differences in the performance between Pt/C vs NFK in fundamental and practical contexts will be examined in the subsequent studies. Figure 10A also contradicts Fig. 5 in that Ni/K600 outperforms Ni-Cr/K600 as higher cathodic currents are applied. However, it is important to remember Ni-Cr/K600 is more susceptible to hydride poisoning (Fig. 5C) and it is likely that the catalyst was deactivated during MEA conditioning.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. (A) Polarization curves of hydrogen pump fuel cells equipped with NFK, Ni-Cr/K600, Ni/K600 or Pt/C HER catalysts. (B) Chronopotentiometric stability curves of the three non-Pt catalysts obtained at 500 mA cm−2. The cells operated at 60 °C and used an anion-exchange membrane referred to in the experimental section as PAP-TP-ME from W-7 Energy.
Download figure:
Standard image High-resolution image{kind=link}
Steady–state testing of the NFK and Ni/K600 catalysts, collected at 500 mA cm–2 over a period of three hours were used to assess the stability of these catalysts (Fig. 10B). The cathodic stability trends shown in Fig. 5 match those collected in the H2 pump cell. Ni/K600 and Ni-Cr/K600 both required increasing potentials to maintain the current density at rates of 4.84 mV h–1 and 3.80 mV h–1 as compared to NFK catalysts which exhibited a loss of 5 μV h−1. The hydrogen pump experiments demonstrate the high-performance and robust nature of NFK catalysts relative to other classes of Ni-based HER catalysts in a practical context.
Conclusions
A functionalized monometallic nickel-based HER catalyst was prepared such that several layers of graphene encapsulate the metallic particles. This functionalized Ni catalyst demonstrates improved HER activity and both anodic and cathodic stability relative to unfunctionalized Ni and NiCr, a mixed metal-oxide catalyst. The encapsulation was visually confirmed by HR-TEM while the graphitic nature of the encapsulating material was determined by XPS and SAD. In-situ XAS studies of NFK show that the catalyst remained mostly metallic under anodic potentials. It appears that the graphitic shield limits anodic passivation of NFK thus maintaining a favorable ratio of Ni and oxophilic NiOx where the latter increases performance by providing a surface that favors water adsorption as proposed by the bifunctional mechanism. This theory is supported by RDE studies that show an improved HER performance by NFK after repeated anodic cycling to 1.05 V. For traditional Ni-based catalysts, prolonged HER performance is hampered by the formation of hydrides that form on the surface and the bulk of the catalyst. Greater stability of HER performance was observed with NFK relative to the other catalysts after holding at a cathodic potential. Kinetic modeling of experimentally obtained polarization curves suggests that the improved performance of NFK is linked to resistance toward hydride formation on the surface, which Ni and NiCr do not exhibit. Lastly, the performance of NFK was assessed in the practical context of an anion-exchange membrane hydrogen pump cell where the HER performance trends were confirmed and where NFK still demonstrated superior HER performance compared to other catalysts.
This work was sponsored under an arpa.e grant (DE-AR000688) and a grant from the Department of Energy under the HydroGen initiative (DE-EE0008082. We would like to acknowledge the valuable input from Bob Allen, Kevin Scanlan, Thomas Stracensky, Ryan Pavicek, Ershuai Liu and Kieran Halligan. This research used beamline 8-ID (ISS) of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704.