

Abstract
Non-biodegradable pharmaceuticals are a group of emerging contaminants that have gained much attention because of their potential risk of inducing adverse ecological and health effects. An efficient Ti/SnO2-Sb2O5-IrO2-RuO2 electrode was prepared by Pechini method and employed as the anode for electrochemical degradation of selected pharmaceuticals (i.e., ibuprofen, atenolol, and carbamazepine). Compared its counterparts, Ti/SnO2-Sb2O5-IrO2-RuO2 exhibited higher oxygen evolution potential, lower chlorine evolution potential, larger active area, and higher stability. Unlike the negligible effect of solution pH from 4.0 to 10.0, increasing current density favored the target pollutants and COD removal. The removal of IBU was mainly attributed to the reaction with •OH, the contribution of which to IBU removal was enhanced with increasing current density. In contrast to IBU, the removal of ATEN and CBZ was mainly ascribed to the reaction with selective radical chlorine species (RCS) mainly in the form of Cl•, which was produced from direct oxidation of Cl− or indirect oxidation by •OH. It was found that the removal of ATEN and CBZ was less affected by dissolved organic matter (DOM) than that of IBU in municipal effluent. This study demonstrates that Ti/SnO2-Sb-RuO2-IrO2 is an attractive electrode for the destruction of pharmaceutical contaminants.
Export citation and abstract BibTeX RIS
The increasing utilization of pharmaceuticals has induced the discharge of massive pharmaceutical contaminants into the natural environments from pharmaceutical industries, livestock runoff and municipal wastewater treatment plants.1 Despite the extremely low concentration residual in natural water bodies, ranging from ng l−1 to μg l−1, a wide range of pharmaceuticals and their transformation products may still cause certain adverse ecotoxicological effects to the organisms after long-term exposure.2 Consequently, the natural and engineered waters contaminated by pharmaceuticals have been emerging as a matter of considerable concern in the past several decades.3
In recent decades, it is therefore an urgent task to develop efficient technologies to attain the appreciable decontamination of pharmaceutical contaminants present in water. Some pharmaceutical contaminants are resistant to the conventional water treatment technologies, e.g., biological process, coagulation, and filtration.4 Electrochemical advanced oxidation processes (EAOPs) are capable to degrade any organic compounds until complete mineralization with reaction rate constant of 108 ∼ 1010 M−1 s−1 since non-selective •OH (E0(•OH/H2O) = 2.80 V/SHE) can be generated in situ.5,6 Thus, EAOPs are increasingly recognized as promising next-generation technologies for the treatment of pharmaceuticals in water.7,8 The performance of EAOPs fundamentally depends on the properties of anode materials. Normally, the boron-doped diamond (BDD), PbO2 and Sb-SnO2 with relatively high oxygen evolution overpotentials are known as the typical non-active anode material for •OH production via the one-electron oxidation of H2O.9–12 More importantly, the powerful oxidant •OH is relatively weakly adsorbed on these anode surface, so that it is easily available for the non-selective oxidation of pollutants. Worth mentioning, the industrial application of these anode materials is seriously impeded by the high cost, release of toxic metal ions or low electrochemical stability, etc. Recently, sub-stoichiometric titanium oxides, mainly Ti4O7 have been demonstrated promising anode materials as alternative to BDD due to its low fabrication cost, but it suffered from the deactivation owing to the transformation of Ti4O7 to TiO2 after the electrolysis process.13
As compared with the above non-active anodes, the active anodes, especially dimensionally stable anode (DSA) with a thin mixed metal oxide layer on Ti substrate, are more attractive for wastewater treatment because of their excellent electrocatalytic activity, mechanical stability and adequate service life.14–16 RuO2 is propitious to the electrochemical evolution of Cl2, but it is stable only under alkaline condition,17 and modest life is achieved in acidic media even if a stabilizing agent is used.18 IrO2 is the desirable electrocatalyst to enhance the production of heterogeneous •OH and facilitate the oxygen evolution reaction. In contrast to RuO2, IrO2 is stable under acid condition and moderate life at alkaline pH.17,19 SnO2 is utilized as a dispersing agent, while Sb is often introduced as a dopant.17 Thus, the use of ternary oxides like IrO2-SnO2-Sb2O5 or RuO2-SnO2-Sb2O5 as the anode materials cannot only enhance the electrode stability, but also exhibit relatively high electrocatalytic activity,20 which therefore have been applied to degrade various contaminants. As compared with the above ternary oxides electrodes, the quaternary oxides electrode, i.e., Ti/SnO2-Sb2O5-IrO2-RuO2, probably has more merits such as electrochemical activity and stability. Although a handful of studies paid attention to the electrochemical or physicochemical properties of this electrode,21 it is still necessary to inspect its electrochemical performance for specific pharmaceutical contaminants removal.
Similar with inert anodes, the electrolysis of H2O at DSA anode surface can also generate the heterogeneous •OH. However, the formed •OH is chemisorbed on the DSA anode surface and more readily transforms to oxygen due to its remarkably lower oxygen evolution overpotential. Instead, DSA(•OH) can be also consumed for the formation of radical chlorine species (RCS), including Cl•, Cl2•− and ClO•, which can selectively oxidize some specific contaminants.22 Thus, it has been widely considered that DSA mediated EAOPs is a feasible strategy for Cl− containing wastewater treatment. Despite of this, the involvement of •OH in the decontamination of various pharmaceutical pollutants has been also unveiled in Cl− free electrolyte solution.20 In consideration of the different reactivities of •OH and RCS, EAOPs tend to be a complementary solution for the oxidation of many structurally diverse pharmaceutical compounds. Unfortunately, few study paid attention to quantitatively identity the contributions of •OH and RCS for the removal of pharmaceuticals with various chemical structure when employing DSA as the working anode.
To this end, Ti/SnO2-Sb2O5-IrO2-RuO2 quaternary oxides electrode was prepared by the Pechini method and was employed for the electrochemical degradation of the selected pharmaceuticals (i.e., ibuprofen (IBU), atenolol (ATEN) and carbamazepine (CBZ)). The electrochemical properties of the electrodes was evaluated based on oxygen evolution potential, chlorine evolution potential, linear cyclic voltammetry, cyclic voltammetry and electrochemical impedance spectroscopy. Additionally, the effects of solution pH and current density on the oxidation performance of Ti/SnO2-Sb2O5-IrO2-RuO2 electrode were also investigated. Finally, the contributions of •OH and RCS to the electrochemical oxidation process for the three selected pharmaceutical contaminants were investigated by establishing a mathematical model.
Materials and Methods
Chemicals and materials
Sodium sulfate (Na2SO4), tin(IV) chloride pentahydrate (SnCl4·5H2O), hexachloroiridic acid hexahydrate (H2IrCl6·H2O), antimony trichloride (SbCl3), citric acid (C6H8O7), ruthenium chloride (RuCl3), ethylene glycol (C2H6O2), nitric acid (HNO3), oxalic acid (C2H2O4), sulphuric acid (H2SO4) were purchased from Sinopharm Chemical Reagent Co. Ltd., China. Ibuprofen (C13H18O2, IBU, >98%), atenolol (C14H22N2O3, ATEN, >98%), carbamazepine (C15H12N2O, CBZ, >98%), potassium dihydrogen phosphate (KH2PO4) were provided by Shanghai Aladdin Bio-Chem Technology Co., Ltd., China. All obtained chemical reagents were analytical grade. Ultrapure water (≥18.2 MΩ cm) was utilized for all solution preparation.
Electrode preparation and experimental procedure
Titanium mesh (φ 10 cm × 20 cm) was degreased in 20% NaOH solution at 100 °C for 1 h and then etched in 10% oxalic acid solution at 100 °C for 3 h. Subsequently, the pre-treated titanium mesh was thoroughly washed with deionized water. Pechini method was applied to fabricate the target DSA electrode. The polymeric precursor solution was obtained by dissolving citric acid in ethylene glycol at 80 °C. Then, the metallic precursors (SnCl4·5H2O, SbCl3, RuCl3 and H2IrCl6·H2O) at certain molar ratio was dissolved in the above polymeric precursor solution at 80 °C with the concentration of 0.5 mol l−1. As mentioned above, RuO2 was favorable for the evolution of Cl2, while IrO2 was considered as the most effective component to enhance the stability of DSA electrode. However, Ru and Ir was the noble metals. Thus, to achieve the balance between electrode properties and cost, the content of Ir or Ru was both set at 5% and 10%, respectively, which were similar with the previous literatures.23–25 The molar ration of Sn/Sb was 9:1. To obtain Ti/SnO2-Sb2O5-RuO2 (molar ratio of 0.81:0.09:0.1), Ti/SnO2-Sb2O5-IrO2 (molar ratio of 0.855:0.095:0.05), Ti/SnO2-Sb2O5-RuO2-IrO2 (molar ratio of 0.765:0.085:0.1:0.05) electrodes, the precursor sol was painted onto the pre-treated titanium mesh and then was dried at 80 °C. Subsequently, it was sintered at 450 °C for 10 min to form the oxide film. These procedures were repeated 15 times and finally the electrode was sintered at 450 °C for 1 h.
The electrochemical experiments were conducted in a 1000 ml cylindrical vessel, in which cylindrical titanium mesh (φ 6 cm × 20 cm) and cylindrical Ti/SnO2-Sb2O5-RuO2-IrO2 mesh (φ 10 cm × 20 cm) were concentrically inserted in the vessel as the cathode and anode, respectively, with electrode distance of 20 mm. The solution was recirculated between the electrochemical vessel and the reservoir by a peristaltic pump at flow rate of 800 ml min−1. A constant cell current was applied to the electrode couple from a DC-power supply (DPS-18500). The electrolysis was sustained in 35 mM Na2SO4 + 20 mM NaCl solution. The typical working solution parameters were: [pharmaceutical]0 = 30 mg l−1, pH0 = 7.0 and initial volume of 2 l. The real wastewater without pretreatment was collected from the local WWTP (Qingdao, China). The main composition of the wastewater was provided as the following: [COD]0 = 62 mg l−1, [NO3−-N]0 = 6.45 mg l−1, [TN]0 = 6.6 mg l−1, [TOC]0 = 4.93 mg l−1, pH = 7.56, conductivity = 3520 μS cm−1, [Cl−] = 1300 mg l−1. During the electrolysis processes, 5 ml aqueous sample was extracted from the electrolytic cell for analysis at certain intervals.
Electrochemical analysis
The electrode properties were conducted in a standard three-electrode cell on an electrochemical workstation (PGSTAT302N, Metrohm). The working electrodes were Ti/SnO2-Sb2O5-RuO2, Ti/SnO2-Sb2O5-IrO2, Ti/SnO2-Sb2O5-RuO2-IrO2 anode (1 cm × 1 cm), a saturated calomel electrode (SCE) was used as the reference electrode, and the counter electrode was a platinum plate electrode (3 cm × 3 cm). The oxygen evolution potential (OEP) and chlorine evolution potential (CEP) were obtained based on linear cyclic voltammetry (LSV) at a scan rate of 10 mV s−1 in 0.5 M Na2SO4 and 3.5% NaCl solution, respectively. The electrochemically active surface area was calculated from the cyclic voltammetry (CV) in 0.5 M Na2SO4 solution. Electrochemical resistance and polarization resistance were measured by electrochemical impedance spectroscopy (EIS), the reaction was carried out in a 0.5 M Na2SO4 solution at a potential of 1.11 V/SCE. Frequency sweep from 10 kHz to 30 mHz with a sine wave amplitude of 10 mV/SCE. In order to evaluate the long-term stability of the as-prepared anode materials, accelerated service life tests were conducted in 3 M H2SO4 solution at current density of 1 A cm−2. Python was used to integrate the results over the simulation time.
Analysis method
The concentrations of ibuprofen (IBU), atenolol (ATEN) or carbamazepine (CBZ) were monitored by Agilent 1100 series high-performance liquid chromatography (HPLC) with Eclipse XDB-C18 column (4.6 mm × 150 mm, 5 μm). The mobile phase at a flow rate of 1.0 ml min−1 and the UV detector at wavelength of 235 nm, 235 nm and 225 nm were employed to determine the concentration of IBU, ATEN and CBZ, respectively. N,N-dimethyl-4-nitrosoaniline (20 μM) was employed as a chemical probe of •OH. The N,N-dimethyl-4-nitrosoaniline (RNO) solution was employed as the scavenger for quantitative determination of •OH yield, and the electrochemical bleaching of RNO was monitored by UV–vis spectrometer at wavelength of 440 nm.26 The chemical oxygen demand (COD) was detected using a COD analyzer (DGB-401, China).
Results and Discussion
Electrochemical property of DSA electrodes
In the electrochemical reaction system, the electrochemical properties such as OEP and CEP of anode material are of particular concern since they are the significant indexes of oxidants generation for target pollutants removal.27,28 Figure 1a shows that OEP value of the Ti/SnO2-Sb2O5-RuO2 and Ti/SnO2-Sb2O5-IrO2 electrodes were about 1.07 V/SCE and 1.08 V/SCE, which were less than that of Ti/SnO2-Sb2O5-RuO2-IrO2 (1.15 V/SCE). In addition, the CEP value of Ti/SnO2-Sb2O5-RuO2-IrO2 electrode was 1.11 V/SCE, which was slight lower than other two electrodes, i.e., 1.14 V/SCE for Ti/SnO2-Sb2O5-IrO2 and 1.12 V/SCE for Ti/SnO2-Sb2O5-RuO2 (Fig. 1b). The electrodes with higher OEP and lower CEP can lead to higher yield of •OH owing to the inhibited oxygen evolution reaction and better active chlorine production performance.20,29 This indicates that the as-prepared Ti/SnO2-Sb2O5-RuO2-IrO2 was more reactive to the oxidation of organic pollutants in comparison with its counterparts. The electrochemical active surface areas were also calculated according to CV tests collected between −0.2 and −1.2 V/SCE (see Fig. 2). From XRD analysis in Fig. S1 (available online at stacks.iop.org/JES/167/143503/mmedia), it can be found that SnO2 was the main component on Ti/SnO2-Sb2O5-RuO2-IrO2, and other three elements probably served as the dopants and marginally existed in the forms of metal oxides. From the previous literatures, we could infer that the high OEP and low CEP were probably ascribed to the doping of Sb2O5, RuO2 and IrO2 in SnO2, which might reduce the binding energy of •OH and increase the bind energy of Cl• on SnO2.30 The electrochemical active area surface was calculated as 3.8 × 10−4 cm2, 4.2 × 10−4 cm2 and 7.9 × 10−4 cm2 for Ti/SnO2-Sb2O5-RuO2, Ti/SnO2-Sb2O5-IrO2 and Ti/SnO2-Sb2O5-RuO2-IrO2, respectively, based on the calculation in Eq. 1. The Ti/SnO2-Sb2O5-RuO2-IrO2 electrode exhibits larger electroactive area than the other two electrodes due to the presence of both Ru and Ir oxide.
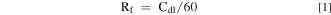
where Cdl is the double layer capacitance. Plot the current density and scan rate at a potential of 0.9 V to obtain a straight line, and Cdl value is the slope of the straight line in each inset in Fig. 2. The ideal smooth oxide surface electric double layer capacitance is 60 μF cm−2.

Figure 1. LSV curves for Ti/SnO2-Sb2O5-RuO2, Ti/SnO2-Sb2O5-IrO2 and Ti/SnO2-Sb2O5-RuO2-IrO2 electrodes in 0.5 M Na2SO4 electrolyte solution (a) and 3.5% NaCl electrolyte solution (b) at a scan rate of 50 mV s−1.
Download figure:
Standard image High-resolution image
Figure 2. Cyclic voltammetry curves of Ti/SnO2-Sb2O5-RuO2-IrO2 (a), Ti/SnO2-Sb2O5-RuO2 (b) and Ti/SnO2-Sb2O5-IrO2 (c) electrodes (Conditions: 0.5 M Na2SO4, scan potential: −0.2 V ∼ 1.2 V, scan rate: 20 mV s−1 ∼ 80 mV s−1); Inset in each figure represents the linear relationship between scan rate and current density.
Download figure:
Standard image High-resolution imageAs shown in Fig. 3, the Nyquist plots for Ti/SnO2-Sb2O5-RuO2, Ti/SnO2-Sb2O5-IrO2 and Ti/SnO2-Sb2O5-RuO2-IrO2 electrodes can be fitted with the equivalent circuit in the inset. Table I shows that the Rct value was 8.23 Ω, 12.21 Ω and 6.60 Ω for Ti/SnO2-Sb2O5-RuO2, Ti/SnO2-Sb2O5-IrO2 and Ti/SnO2-Sb2O5-RuO2-IrO2 electrode, respectively. This behavior indicates that both RuO2 and IrO2 doping enhanced electrode conductivity, which was conductive to the transfer of electrons from electrode surface to Ti substrate during electrochemical process. The capacitance value of Ti/SnO2-Sb2O5-RuO2-IrO2 electrode was 2.50 × 10−2 F cm−2, which was much higher than those of other two electrodes. As a result, Ti/SnO2-Sb2O5-RuO2-IrO2 electrode would exhibit more appreciable charge and discharge performance, which was favorable for the electrochemical wastewater treatment.31
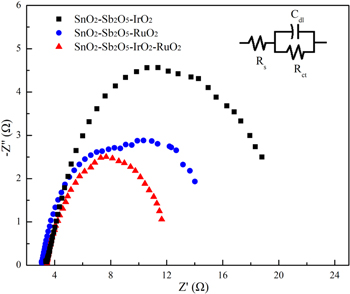
Figure 3. EIS Nyquist plots of Ti/SnO2-Sb2O5-RuO2, Ti/SnO2-Sb2O5-IrO2 and Ti/SnO2-Sb2O5-RuO2-IrO2 electrodes by applying an impedance amplitude of 10 mV with the frequency range from 10 kHz ∼ 30 mHz.
Download figure:
Standard image High-resolution imageTable I. Simulated data of each parameter (EIS) for different electrodes.
| Electrode | Rs/Ω | Rct/Ω | Cdl/F cm−2 |
|---|---|---|---|
| Ti/SnO2-Sb2O5-IrO2 | 3.78 | 12.21 | 1.29 × 10−2 |
| Ti/SnO2-Sb2O5-RuO2 | 3.35 | 8.23 | 1.42 × 10−2 |
| Ti/SnO2-Sb2O5-RuO2-IrO2 | 3.65 | 6.60 | 2.50 × 10−2 |
Figure 4 shows the results obtained for the accelerated life tests of the three electrodes. The accelerated service life of the anodes tested increased in the order: Ti/SnO2-Sb2O5-RuO2 (16 h) < Ti/SnO2-Sb2O5-IrO2 (28 h) < Ti/SnO2-Sb2O5-RuO2-IrO2 (68 h). IrO2 has a lifespan that is 20 times that of an equivalent amount of RuO2, and also can be used in acidic conditions. However, IrO2 is more expensive than RuO2, and its electrocatalytic activity is not as good as RuO2. In order to improve the coating performance, RuO2 and IrO2 were simultaneously doped into the Ti/SnO2-Sb electrode to enhance its oxygen and chlorine evolution capabilities. This can effectively improve both the stability and electrocatalytic activity of the electrode.32,33 Generally, the Ti/SnO2-Sb2O5-RuO2-IrO2 electrode has high electrochemical properties, suggesting that this electrode possessed an excellent performance during the electrochemical oxidation process.
Figure 4. The accelerated life tests of Ti/SnO2-Sb2O5-RuO2, Ti/SnO2-Sb2O5-IrO2 and Ti/SnO2-Sb2O5-RuO2-IrO2 electrodes. (Conditions: 3 mol l−1 H2SO4, current density = 1 A cm−2).
Download figure:
Standard image High-resolution imageElectrochemical treatment performance
It is known that the current density fundamentally governs the global effectiveness of the electrochemical treatment performance for pollutants removal because the generation of reactive species was positively related with the current density. However, it is much more pronounced for competition of oxygen evolution reaction with the production of reactive species (•OH and RCS) at higher current density, which is a waste energy side reaction during electrochemical process. Therefore, the electrochemical performance of Ti/SnO2-Sb2O5-RuO2-IrO2 electrode was evaluated at different current density by selecting IBU, ATEN and CBZ as the model pollutants. Figure 5 illustrates IBU, ATEN and CBZ removal efficiency and COD removal efficiency increased with elevating current density. At pH 7.0, the removal efficiency of IBU reached 74%, 93% and 98%, at 10, 20 and 30 mA cm−2, respectively, after 120 min electrochemical reaction, accompanying the enhanced COD removal efficiency from 22% to 68%. Unlike IBU, relatively low current density applied was enough for the removal of ATEN and CBZ. The removal efficiency of ATEN and COD removal efficiency reached 81.9% and 99.6%, 32% and 38% at 2 and 6 mA cm−2, respectively, within 60 min. And almost complete removal of ATEN was attained within 30 min at 10 mA cm−2, accompanying COD removal efficiency of 54%. The removal efficiency of CBZ reached 99.5% at 2 mA cm−2 after 60 min. The removal efficiency of ATEN reached 99.5% after 20 min of electrolysis when current density was 6 mA cm−2 and 10 mA cm−2. In this case, the COD removal efficiency increased from 54.5% to 61.4% at current density increasing from 6 to 10 mA cm−2.

Figure 5. The effect of current density on the removal efficiency and COD removal efficiency for IBU (a), (d), ATEN (b), (e) and CBZ (c), (f), respectively. (Conditions: [pharmaceutical]0 = 30 mg l−1, pH = 7.0).
Download figure:
Standard image High-resolution imageIt has been demonstrated that the influence of solution pH on pollutant removal is compound specific in UV/chlorine system,34 which was associated with the change in the radical chemistry for the pharmaceutical pollutants with different chemical structures. As solution pH increased from 4.7 to 8.4, the rate constants for metronidazole, nalidixic acid, diethyltoluamide and caffeine, decreased by more than 70%.34 However, in present electrochemical reaction system, negligible effect of solution pH on the oxidation of IBU, ATEN and CBZ over the pH range of 4.0–10.0 (see Fig. 6). These results indicate that the electrochemical removal of IBU, ATEN and CBZ mediated by Ti/SnO2-Sb2O5-RuO2-IrO2 electrode can be carried out in a wide pH range.
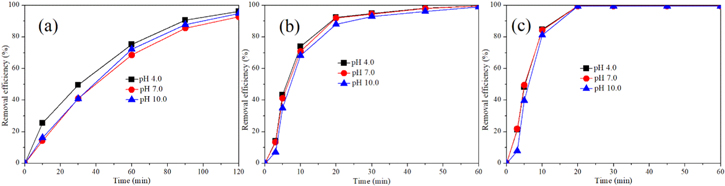
Figure 6. The effect of solution pH on the removal efficiency of IBU (a), ATEN (b) and CBZ (c) (Conditions: [pharmaceutical]0 = 30 mg l−1, (a) current density = 20 mA cm−2, (b) and (c) current density = 6 mA cm−2).
Download figure:
Standard image High-resolution imageRoles of reactive species
EAOPs relies on the production of reactive species, i.e., •OH (Eq. 2) and RCS (e.g., Cl•, Cl2−• and ClO•) in NaCl electrolyte solution. •OH was the effective oxidant for recalcitrant organic pollutants with electron-deficient moieties at nearly diffusion limited rates through electron transfer, H abstraction and addition reaction. The production of RCS is initiated by the direct oxidation of Cl− at the anode surface with the generation of Cl• following the reaction in Eq. 3. Cl• with standard reduction potentials of 2.47 V/SHE can be also formed via the indirect oxidation of Cl− mediated by •OH (Eq. 4). The selective oxidant Cl2•− (2.0 V/SHE) can be also produced via being in equilibrium with Cl• (Eq. 5). Because of the long lifetime of Cl2•− (microsecond) and the high reactivity of Cl•, the generated Cl2•− mainly decays through the equilibrium of Cl• and Cl2•− in Eq. 5 and shows activity towards micropollutants through Cl• chemistry at chloride concentrations lower than 100 mM.35 Thus, in present electrochemical reaction process, Cl2•− contributed negligibly to the oxidation of the selected pollutants since the consideration of Cl− was only 20 mM. Both •OH and Cl• can react with HOCl/OCl− to produce the more selective ClO• with standard reduction potentials of 1.5−1.8 V/SHE (Eqs. 6–8). Among the RCS, Cl• has the highest standard reduction potentials and favorably attacks the compounds containing amino and/or carboxylic groups at reaction rate constants ranging from 108 to 109 M−1 s−1,36–38 which therefore probably dominated the contribution of RCS to the electrochemical oxidation of pollutants.
Besides, it has been widely known that both RuO2 and IrO2 belong to the category of "active" anodes and are able to form the so-called higher oxide MOx+1. Thus, the formation of MOx+1 on Ti/RuO2-IrO2 electrode may contribute to the oxidation of pollutants. However, it has been demonstrated that there is an significant inhibition of MOx+1 and MOx( ) toward pollutant oxidation in the presence of Cl− ions, where Cl− can be oxidized with the electro-generation of active chlorine.39 On the other hand, only physisorbed MOx(·OH) species, can form on non-active anodes such as Ti/SnO2-Pt.40 Notably, in present study, negligible RuO2 and IrO2 was detected on Ti/SnO2-Sb2O5-RuO2-IrO2 based on XRD pattern in Fig. S1. From these, it can be reasonably inferred that contribution of MOx+1 to the oxidation of pollutants could be neglected during the present Ti/SnO2-Sb2O5-RuO2-IrO2 mediated electrochemical process with the presence of Cl−.
) toward pollutant oxidation in the presence of Cl− ions, where Cl− can be oxidized with the electro-generation of active chlorine.39 On the other hand, only physisorbed MOx(·OH) species, can form on non-active anodes such as Ti/SnO2-Pt.40 Notably, in present study, negligible RuO2 and IrO2 was detected on Ti/SnO2-Sb2O5-RuO2-IrO2 based on XRD pattern in Fig. S1. From these, it can be reasonably inferred that contribution of MOx+1 to the oxidation of pollutants could be neglected during the present Ti/SnO2-Sb2O5-RuO2-IrO2 mediated electrochemical process with the presence of Cl−.
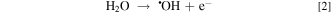
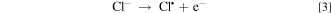
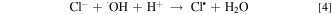
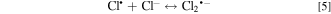
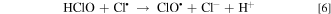
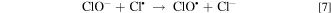
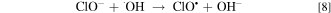
The kinetic expression of pollutant (P) in the electrochemical system is shown in Eqs. 9 and 10 without considering the contribution of HClO and Cl2.41
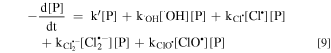
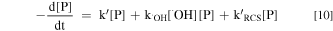
where k' is the rate constant of P removal by direct oxidation at anode surface; k•OH and k'RCS are the second-order rate constants for the reaction of P by •OH and RCS (Cl•, Cl2−• and ClO•), respectively.
The CV analysis in Fig. S2 shows that the direct oxidation of the three selected pollutants can be neglected. Then, the oxidation removal of IBU, ATEN and CBZ can be integrated following Eq. 11.
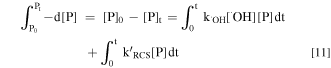
Then the removal efficiency (R) of P can be expressed as:
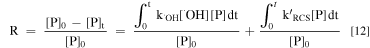
That is:
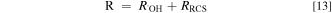
where [P]0 and [P]t represent the concentration of P at time 0 and t, respectively. R is the overall removal efficiency of P at time t, R•OH and RRCS represent the removal efficiency of P by •OH and RCS at time t, respectively.
The •OH concentration can be gotten by an indirect measurement of the depletion of RNO that was used as a •OH probe (Eqs. 14 and 15)26.
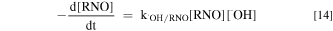

where k•OH/RNO was defined the second-order rate constant of RNO oxidation by •OH (1.25 × 1010 M−1 s−1). [•OH] is the steady state concentration of •OH.
k•OH/RNO[•OH] represents the steady state generation rate constant of •OH. From Fig. S3, we can conclude that the electrochemical generation rate constant of •OH was enhanced from 0.058 s−1 to 0.14 s−1 with increasing current density from 2 to 30 mA cm−2. R•OH can be obtained by •OH exposure (![$\displaystyle {\int }_{0}^{{\rm{t}}}\left[{}^{\cdot }{\rm{O}}{\rm{H}}\right]{\rm{dt}}$](https://content.cld.iop.org/journals/1945-7111/167/14/143503/revision2/jesabc30bieqn2.gif) ), target pollutant oxidation kinetics and k•OH (7.2 × 109 M−1 s−1 for IBU, 6.84 × 109 M−1 s−1 for ATEN and 8.8 × 109 M−1 s−1 for CBZ).41 In consideration of the unavailable rate constants of most pollutants oxidation by specific RCS, the overall contribution of RCS (
), target pollutant oxidation kinetics and k•OH (7.2 × 109 M−1 s−1 for IBU, 6.84 × 109 M−1 s−1 for ATEN and 8.8 × 109 M−1 s−1 for CBZ).41 In consideration of the unavailable rate constants of most pollutants oxidation by specific RCS, the overall contribution of RCS ( ) can be calculated by subtracting the contribution of •OH to pollutant removal (
) can be calculated by subtracting the contribution of •OH to pollutant removal ( ). The removal of IBU, ATEN and CBZ that are attributed to •OH and RCS as a function of time at pH 7.0 were presented in Figs. 7a–7c. The removal of IBU was mainly due to the reaction with •OH, which was consistent with the experimental results in previous studies.42 Taking the reaction for 60 min as an example, the overall removal rate of IBU was 69%, the contribution rate of •OH and RCS were 60% and 9%, respectively. It can be also observed that the contribution of •OH to IBU removal was enhanced with increasing current density, whereas the contribution of RCS was decreased. This indicates that higher current density facilitated the production of •OH during DSA mediated electrochemical process. In addition, the role of RCS in IBU removal was enhanced with the process of electrochemical reaction. In contrast to IBU, the removal of ATEN and CBZ was mainly ascribed to the reaction with RCS. Noticeably, the contribution of •OH changed slightly with extending the electrochemical reaction time. For example, at reaction time of 60 min, ATEN overall removal rate was 95%, •OH contribution rate was 17%, RCS contribution rate was 75%; CBZ overall removal rate was 99%, •OH contribution rate was 13%, RCS contribution rate was 86%.
). The removal of IBU, ATEN and CBZ that are attributed to •OH and RCS as a function of time at pH 7.0 were presented in Figs. 7a–7c. The removal of IBU was mainly due to the reaction with •OH, which was consistent with the experimental results in previous studies.42 Taking the reaction for 60 min as an example, the overall removal rate of IBU was 69%, the contribution rate of •OH and RCS were 60% and 9%, respectively. It can be also observed that the contribution of •OH to IBU removal was enhanced with increasing current density, whereas the contribution of RCS was decreased. This indicates that higher current density facilitated the production of •OH during DSA mediated electrochemical process. In addition, the role of RCS in IBU removal was enhanced with the process of electrochemical reaction. In contrast to IBU, the removal of ATEN and CBZ was mainly ascribed to the reaction with RCS. Noticeably, the contribution of •OH changed slightly with extending the electrochemical reaction time. For example, at reaction time of 60 min, ATEN overall removal rate was 95%, •OH contribution rate was 17%, RCS contribution rate was 75%; CBZ overall removal rate was 99%, •OH contribution rate was 13%, RCS contribution rate was 86%.

Figure 7. The contributions of •OH and RCS to the removal efficiency of IBU (a), ATEN (b) and CBZ (c) as a function of time. (Conditions: [pharmaceutical]0 = 30 mg l−1, (a) current density = 20 mA cm−2, pH = 7.0, (b) and (c) current density = 6 mA cm−2, pH = 7.0).
Download figure:
Standard image High-resolution imageThe structure of IBU, ATEN and CBZ are rather complex, which were constructed by pyrimidine and benzene rings, carbonyl, amine, alkyl and methoxyl groups. Consequently, the different contributions of •OH and RCS toward the electrochemical oxidation of IBU, ATEN and CBZ can be attributed to their compound specific reactivity, which have been extensively investigated in the UV/chlorine advanced oxidation process (AOP) involving the formations of •OH and RCS. As for IBU, the primary RCS for IBU oxidation removal was assumed to be Cl• since a carboxylic group was contained in IBU molecule. Thus, its degradation was initiated by •OH dominated hydroxylation and chlorine substitution marginally induced by Cl•, and sustained by decarboxylation, demethylation, chlorination and ring cleavage.42 The final degradation products, including carboxylatic acids and chlorine containing products, may be aliphatic compounds formed through the cleavage of the benzene ring. Despite negligible contribution of Cl•, many dichloro and monochloro products (i.e., trichloromethane, 1,1,1-trichloropropanone, chloral hydrate, 1,1-dichloro-2-propanone, dichloroacetic acid and trichloroacetic acid) were probably formed during the electrochemical degradation of IBU process.42 Considering the presence of amine moieties in ATEN molecule, the attack of Cl• at amine moiety was expected to initiate the degradation of ATEN and CBZ. The transformation of amine moiety can be enhanced by •OH in addition to the pathway mediated by Cl•,43,44 but the radical pathway is usually less efficient owing to its low selectivity. Then, the •OH and Cl• together led to the consecutive degradation of ATEN and CBZ via hydroxylation, hydrogen atom abstraction, acylamino cleavage and decarboxylation and chlorination reactions.44,45 Take CBZ as an example, intermediates (e.g., acridine, acridone and hydroxyl-CBZ) and some chlorine substituted products (e.g., C14H10ClNO2, C13H8ClNO2 and C13H8ClNO) were detected during the electrochemical degradation process.
Solution pH determines the dissociation of HOCl/OCl− (pKa = 7.5) and therefore influences the electrochemical radical production. From reactions in Eqs. 6–8, the formation of ClO• was associated with solution pH. Actually, solution pH posed negligible effect on target pollutant oxidation removal in Fig. 6. This can be interpreted by the fact that formation of •OH and Cl• was independent of solution pH (Eqs. 2 and 3), which were primarily responsible for the degradation of pollutants.
Electrochemical removal of target pharmaceuticals in municipal effluent
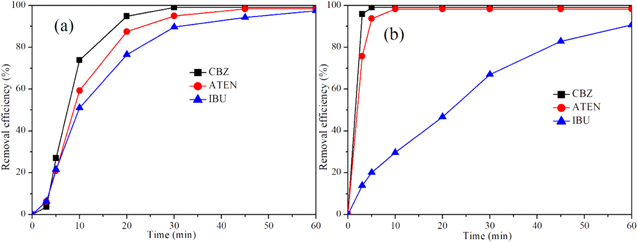
Water matrix components in the effluent such as dissolved organic matter (DOM) and Cl− were likely to affect the pharmaceuticals degradation. The presence of Cl− was favorable for the generation of RCS, promoting the oxidation of the pollutants. In contrast, DOM has been reported as a typical radical scavenger. The rate constants of DOM reacting with •OH and Cl• are 2.5 × 104 l (mg·s)−1,46 and 1.3 × 104 l (mg·s)−1,36 respectively. Figure 8a shows the electrochemical oxidation of target pollutants in the municipal tertiary effluent spiked with 5 mg l−1 IBU, ATEN or CBZ. The removal efficiency of IBU, ATEN and CBZ all reached more than 97% within 60 min. The removal rate of CBZ was faster than that of atenolol, and the removal rate of ibuprofen was the slowest. Figure 8b shows the electrochemical oxidation removal of IBU, ATEN and CBZ with total concentration of 5 mg l−1 in the municipal tertiary effluent at 20 mA cm−2. It was found that the removal efficiency of ATEN and CBZ reached 98% after 10 min, whereas reaction time of 60 min was required for the removal of 90% IBU. The oxidation removal of ATEN and CBZ was less affected by DOM than that of IBU. This indicates that the contribution of •OH dominated the electrochemical oxidation of IBU, whereas RSC was the main oxidants for the removal of ATEN and CBZ, which were consistent with the results in Fig. 7.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. The removal efficiency of IBU, ATEN and CBZ in the municipal effluent (Conditions: (a) [IBU]0 = [ATEN]0 = [CBZ]0 = 5 mg l−1, current density = 20 mA cm−2, 6 mA cm−2 and 6 mA cm−2 for IBU, ATEN and CBZ, respectively; (b) [IBU]0 = [ATEN]0 = [CBZ]0 = 5/3 mg l−1, current density = 20 mA cm−2).
Download figure:
Standard image High-resolution image{kind=link}
Conclusions
In this study, quaternary oxides DSA electrode, i.e., Ti/SnO2-Sb2O5-IrO2-RuO2, was prepared by the Pechini method and exhibited higher electrochemical properties in comparison with its ternary counterparts. Solution pH negligibly affected the electrochemical performance for the selected pharmaceuticals removal, whereas the degradation of pharmaceuticals was evidently enhanced with elevating current density. At pH 7.0, the degradation efficiency of IBU reached 74%, 93% and 98%, at 10, 20 and 30 mA cm−2, respectively, after 120 min electrochemical reaction. Unlike IBU, the degradation efficiency of 81.9%–100% could be attained for ATEN within 60 min even at current density of 2–10 mA cm−2, while almost complete removal of CBZ was achieved at current density of 2–10 mA cm−2 within 60 min. These results revealed that •OH played the most important role in IBU degradation, RCS played the most important role in ATEN and CBZ degradation. Besides, the degradation of ATEN and CBZ was less affected by DOM than that of IBU. In summary, the electrochemical treatment of pharmaceutical pollutants by the employment of Ti/SnO2-Sb2O5-RuO2-IrO2 as the anode is a feasible and attractive strategy.
Acknowledgments
This work is financially supported by the National Major Science and Technology Program for Water Pollution Control and Treatment (No. 2017ZX07101-006).