
Abstract
The electrochemical reduction mechanisms of diprotonated tetraphenylporphyrin (H2TPP) and mono- and diprotonated octaethylporphyrin (H2OEP) were studied in tetrabutylammonium perchlorate/benzonitrile. The diprotonated forms of both porphyrins undergo two one-electron reversible reduction processes forming isophlorin. Contrastingly, monoprotonated H2OEP is reduced in a single process involving a two-electron one-proton transfer that yields two types of short-lived intermediates, isophlorin and neutral phlorin. The existence of intermolecular proton transfer reactions, from the parent protonated porphyrin to the isophlorin or neutral phlorin, to form phlorin cation species (isophlorin protonated at the meso-position) was demonstrated. In-situ UV–vis spectroelectrochemical experiments allowed us to identify the absorption of the isophlorin species of H2TPP but not of H2OEP. These results show that the lack of phenyl substituents increases the rate of protonation at the meso-position. Finally, it was demonstrated that the protonation of the porphyrin macrocycle not only lowers the reduction potentials but also increases the reactivity of the electrogenerated species.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
The porphyrin ring structure has four nitrogen atoms, two of which are of pyrrolenine (=N−) type and have free electron pairs. Since they act as Brønsted bases, they can undergo protonation, yielding the mono- and diprotonated forms. Combining protonated porphyrins with anionic species can lead to self-assembled supramolecular structures.1–3 Since one of the effects of protonation is lowering the reduction potentials,4,5 protonated porphyrins are attractive for the construction of donor-acceptor assemblies, where the porphyrin moiety acts as an electron-acceptor.
The addition of one or two protons on the pyrrolenine nitrogens results in steric hindrance because of the N-H protons from the pyrrolic ring. This leads to the loss of planarity in the macrocycle.6 If the addition of the first proton induces a great distortion from the planar structure of the porphyrin free base, then the two protonation steps occur simultaneously, leading directly to the diprotonated form. However, if the conformational change is not that remarkable, there would be two protonation steps and the monoprotonated species can be observed.7–10
Moreover, the distortion induced by protonation leads to changes in the HOMO-LUMO energy levels,4,11 which in turn modifies the optical,4,11–14 and electrochemical4,5,15 properties. Thorough studies exist about the effects of protonation of porphyrins on the reduction/oxidation potentials based on the stabilization/destabilization of HOMO/LUMO orbitals.4 However, less is known about the reactivity of the reduced species and the chemical reactions coupled to the electron transfer. Only three early works by Wilson et al.16–18 for tetrapyridylporphins in acidic aqueous media, and a recent study by Kadish and col.15 of different meso- and β-substituted porphyrins, show that the reduction of diprotonated porphyrins leads to phlorin species. Even when there are examples of porphyrins capable to form stable monoprotonated species, including octaethylporphyrin (H2OEP),19 dodecaphenyl-porphyrin (H2DPP),20,21 deutero-, hemato-, meso-, and proto-porphyrin IX dimethyl esters;22 there are no studies on the reduction mechanisms of monoprotonated porphyrins.
In this work, we report the reduction mechanism of a monoprotonated porphyrin, H3OEP+. Experimental results indicate that the reduction occurs through a sequence of parallel reactions that produce short-lived intermediates, such as isophlorin H4OEP and neutral phlorin H3OEFH (Scheme
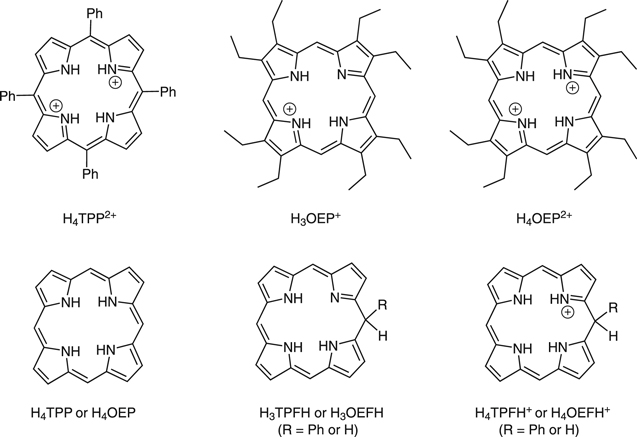
Scheme 1. Diprotonated meso-tetraphenylporphyrin (H4TPP2+) and mono- and diprotonated octaethylporphyrin (H3OEP+ and H4OEP2+). Their reduction products: isophlorin (H4TPP or H4OEP), neutral phlorin (H3TPFH or H3OEFH), and phlorin cation (H4TPFH+ or H4OEFH+). FH indicates phlorin species. Substituents in the porphyrin periphery for the reduced species are omitted for clarity.
Download figure:
Standard image High-resolution imageExperimental
Materials
2,3,7,8,12,13,17,18-octaethylporphyrin (H2OEP 95%) from Sigma-Aldrich was purified by chromatography using silica gel with toluene/ethyl acetate. Pyrrole, benzaldehyde and propionic acid were purchased from Sigma-Aldrich, in the case of pyrrole it was distilled prior to use. Electrochemical grade tetrabutylammonium perchlorate (Bu4NClO4 ≥ 99%) was purchased from Aldrich and dried in a vacuum oven for 48 h prior to use. Perchloric acid, used to protonate the free base porphyrin, H2TPP and H2OEP, was purchased from Aldrich (HClO4 ACS reagent 69 −72). Previous to use, this solution was standardized using sodium carbonate (Na2CO3 ≥ 99.5%, Sigma-Aldrich); the actual concentration was 11.5 M (69.44% purity). An aqueous tetrabutylammonium hydroxide solution from Sigma-Aldrich (40%, Bu4NOH ∼ 1.5 M) was also standardized with phthalic acid monopotassium salt from Merck (KHP 99.8%) yielding a 1.9 M concentration. The standards KHP and Na2CO3 were dried for 4 h at 120 °C prior to use. 1,8-diazabicyclo[5.4.0] undec-7-ene (DBU 98%, Sigma-Aldrich) was used as received. Benzonitrile (PhCN 99% extra pure, ACROSS Organics) was distilled under reduced pressure with phosphorous pentoxide (P2O5 ≥ 98%, Aldrich). The solutions for electrochemical experiments were bubbled with ultra-high purity nitrogen (99.999%, Infra).
NMR spectra were recorded using a Bruker ARX 300 spectrometer, the experiments were done with concentrations between 15 and 20 mg ml−1 at 25 °C. Chemical shifts (ppm) are relative to Si(CH3)4 for 1H and 13C (77.23 ppm) in CDCl3, all coupling constants are reported in Hertz.
Synthesis
meso-Tetraphenylporphyrin was prepared by placing 200 ml of propionic acid in a round-bottomed flask equipped with a condenser and a magnetic stirrer, followed by heating under reflux, and simultaneous addition of 5.5 g (5.3 ml, 51.8 mmol) of pyrrole and 3.5 g (3.6 ml, 32.9 mmol) of benzaldehyde. The reaction mixture was refluxed for 30 min and allowed to cool.23,24 The product was filtered over celite and purified by flash chromatography column using a mixture of hexane/CHCl3 (60/40). Finally, the product was washed with methanol. The macrocycle H2TPP was obtained as a purple solid, 2.7 g (4.40 mmol, 34%). P.f. > 300 °C. 1H NMR (CDCl3, 300 MHz): δ 8.85 (s, 8H, H-β), 8.22 (dt, 8H, J = 7.8, 1.7 Hz, Ho-Ph), 7.76 (m, 12H, Hm-Ph and Hp-Ph), −2.77 (s, 2H, N–H) ppm. 13C NMR (CDCl3, 75.5 MHz) δ: 142.3 (Cipso-Ph), 134.7 (Co-Ph), 131.2 (C-β), 127.8 (Cm-Ph), 126.8 (Cp-Ph), 120.3 (C-meso) ppm. Spectral data for H2TPP is in agreement with those previously reported.25,26
Electrochemistry
An Autolab PGSTAT 302 potentiostat controlled by the GPES software was used for the voltammetry and coulometry experiments. For the spectroelectrochemical measurements, a BASi Epsilon potentiostat-galvanostat was coupled to a UV–vis Agilent 8453 spectrophotometer. A three-electrode jacketed cell was used for the cyclic voltammetry and controlled potential coulometry. The counter and reference electrode were a Pt wire and the Ag/AgNO3 system (a silver wire immersed in 0.10 M Bu4NClO4/0.01 M silver nitrate/acetonitrile, separated from the main solution by a porous Vycor frit from Bioanalytical Systems). For the coulometry experiments, the Pt counter electrode was placed inside a fritted glass chamber isolated from the main solution. A glassy-carbon disc (0.3 and 0.1 cm diameter) and a porous carbon mesh (0.3 × 0.2 cm) were used as working electrodes for the cyclic voltammetry and coulometry, respectively. A thin-layer quartz electrochemical cell with a three-electrode arrangement (BASi) was employed for the spectroelectrochemistry measurements. Depending on the concentration of the solutions, two different path lengths where used, 0.5 and 1.0 mm. The working electrode consisted of an optically transparent gold minigrid, the counter electrode was a Pt wire, and the Ag/AgNO3 system as reference.
The redox potential of the ferrocenium/ferrocene couple (Fc+/Fc) was regularly measured, all potential values in this work are referred to the Fc+/Fc couple. All the experiments were performed in Bu4NClO4 0.1 M/PhCN at 25 °C; except for the spectroelectrochemistry that was carried out at room temperature. Prior to every experiment, the solutions were bubbled with N2 for approximately 50 min Also, the working electrode was polished with alumina paste (0.05 μm, Buehler) before each measurement.
The total uncompensated resistance in Bu4NClO4 0.1 M/PhCN at 25 °C (880 and 2800 Ω for the 0.3- and 0.1-cm diameter electrode) was determined as previously described.27 Approximately 80% of the total resistance was electronically compensated during the experiment. For the voltammetry simulation, the remaining ∼20% resistance was added during the simulation (DigiElch 8, http://digielch.de).28
Results and Discussion
UV–vis spectrometric titration of H2TPP and H2OEP
Free base H2TPP shows the typical UV–vis porphyrin spectrum: an intense Soret band at 422 nm, and four Q bands of lower intensity at 517, 551, 592 and 648 nm. When adding increasing amounts of HClO4 to the H2TPP benzonitrile solution, the Soret band gradually decreases, and a new signal appears and increases at 442 nm. Additionally, the bands at 517 and 551 nm gradually decrease until they disappear, and that at 648 nm increases and shows a 10-nm bathochromic shift (Fig. S1, Supplementary material is available online at stacks.iop.org/JES/167/155507/mmedia). The reduction from four to two Q bands is due to a symmetry change from D2h to D2d.14,29 These characteristics are typical of the direct formation of the diprotonated porphyrin, H4TPP2+, i.e., without the formation of the monoprotonated species.30,31
The free base H2OEP spectrum also presents the characteristic pattern, with the Soret band at 402 nm and four Q bands at 498, 532, 568 and 622 nm (Fig. 1, black line). Unlike H2TPP, the addition of acid induces spectral changes that are consistent with the two-step protonation where both the mono- and diprotonated species (H3OEP+ and H4OEP2+) are observed. When adding 0.2–1.0 HClO4 equivalents (Fig. 1 top), there is a slight hypsochromic shift of the Soret band (7 nm), and the shoulder at 372 nm disappears. In addition, Q-IV (498 nm) gradually decreases until it is no longer observed, and Q-II increases and is hypsochromically shifted (13 nm). At 622 nm, Q-I decreases while a new band gradually grows at 602 nm. Band Q-III, 532 nm, shows only very slight changes during this first protonation step. It is worth noting that the formation of the monoprotonated species is characterized by two isosbestic points at 515 and 610 nm. A similar behavior has been observed for other porphyrins.8,22,32,33 When adding between 1.2 and 2.0 HClO4 equiv (Fig. 1 bottom), a Soret band appears at 407 nm. Also, Q-III (532 nm) decreases and then disappears when two equivalents of acid have been added; the band at 571 nm decreases while that at 555 nm increases and shifts to the blue. Band Q-I has a similar behavior to that observed during monoprotonation, i.e., the signal at 602 nm decreases and one at 594 nm rises. The spectra measured in-between the mono- and diprotonaded species (those with 1.2–1.8 acid equivalents) show three isosbestic points at 400, 541 and 584 nm. Based on the spectral pattern for the acid titration of H2TPP and H2OEP, the following equilibria were determined:
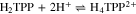
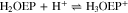
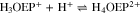

Figure 1. Changes in absorption spectra during acid titration of 0.14 mM H2OEP with HClO4 in PhCN. Top: conversion of neutral H2OEP to monoprotonated H3OEP+ species. Bottom: conversion of monoprotonated H3OEP+ to diprotonated H3OEP+ species. Path length 0.1 mm (Soret bands) and 10.0 mm (Q bands).
Download figure:
Standard image High-resolution imageElectrochemical behavior of diprotonated meso-tetraphenylporphyrin
The reduction of H2TPP in dry benzonitrile (PhCN) shows the typical porphyrin behavior; two reversible one-electron reductions, E1/2 = –1.65 and –2.03 V (Fig. 2a, black curve), corresponding to the formation of the radical anion and the dianion.34–39 In both processes, the plot of peak current vs square root of the sweep rate shows a linear relation with the intercept at zero, indicating the electron transfers are diffusion-controlled.
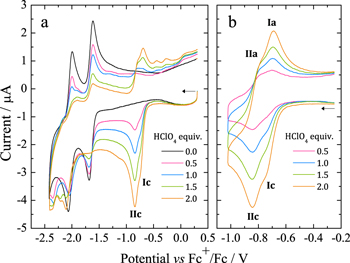
Figure 2. Cyclic voltammograms of H2TPP 0.25 mM in Bu4NClO4 0.1 M/ PhCN at 25 °C with increasing HClO4. (a) Eλ = –2.42 V and (b) Eλ = –1.02 V. Scan rate: 0.1 V s−1.
Download figure:
Standard image High-resolution imageIncreasing additions of HClO4 induces the rise of two new contiguous reduction processes, Ic and IIc, that gradually increase. Simultaneously, the current intensity of the neutral H2TPP reduction process decreases (Fig. 2a). When the potential is switched right after IIc, we observe the oxidation processes Ia and IIa, which are coupled to Ic and IIc (Fig. 2b). This indicates that the reduced species are stable enough to allow for their oxidation at the scan rates used during these experiments. Based on the results of section 2.1, the addition of 2 equiv of HClO4 provokes the full disappearance of the H2TPP bands, indicating that the equilibrium of reaction 1 is mostly shifted toward the H4TPP2+ species, and almost no free protons are in solution. Therefore, Ic/Ia and IIc/IIa must correspond to one-electron reduction processes of H4TPP2+ that yield the radical cation H4TPP•+ and isophlorin H4TPP, reactions 4 and 5. The direct reduction of free protons at these potentials was ruled out by recording voltammograms of HClO4 in the electrolytic media. A similar behavior has been reported for the reduction of meso-tetra(4-N-methyl-pyridyl)porphyrin diacid in aqueous HCl 1.0 M.17

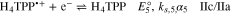
Digital simulation was performed for the cyclic voltammograms obtained for H4TPP2+ at three concentrations (0.27, 0.47 and 0.73 mM), and scan rates from 0.1 to 5.0 V s−1. For the simulation, reactions 4 and 5 with  = –0.72 V and
= –0.72 V and  = –0.82 V, and ks,4 and ks,5 = 0.04 cm s−1 were used. Examples of the fit are shown in Fig. 3. At low scan rates, 0.1 V s−1, the current intensity of Ia and IIa is slightly larger in the simulated voltammogram than that observed in the experiments. This indicates that the isophlorin, H4TPP, formed at IIc undergoes some kind of chemical reactions.
= –0.82 V, and ks,4 and ks,5 = 0.04 cm s−1 were used. Examples of the fit are shown in Fig. 3. At low scan rates, 0.1 V s−1, the current intensity of Ia and IIa is slightly larger in the simulated voltammogram than that observed in the experiments. This indicates that the isophlorin, H4TPP, formed at IIc undergoes some kind of chemical reactions.
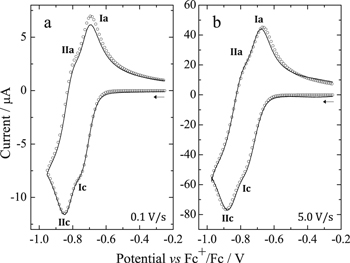
Figure 3. Cyclic voltammograms of 0.73 mM H4TPP2+ in Bu4NClO4 0.1 M/PhCN at 25 °C. Solid line: background-corrected experimental voltammograms. Open circles: simulation using E°4 = –0.72 V and E°5 = –0.82 V with ks,4 and ks,5 = 0.04 cm s−1, α = 0.5, and D = 2.7 × 10−6 cm s−1.
Download figure:
Standard image High-resolution imageCoulometry of H4TPP2+ at –0.89 V (process IIc) consumed 1.4 ± 0.15 e− per H4TPP2+ molecule in approximately 50 min (the experiment was performed in triplicate). The voltammetric analysis of the solution after electrolysis demonstrates that Ic and IIc completely disappeared, indicating that H4TPP2+ was depleted (Fig. 4a, green line). The voltammogram also shows a new peak, IIIc, and the partial recovery of the reduction peaks of free base H2TPP. In addition, the anodic sweep (blue line) does not show the oxidation processes of isophlorin (Ia and IIa, reverse of reactions 4 and 5), but a new oxidation process is observed at 0.07 V, peak IVa. The presence of IIIc and IVa confirms the existence of chemical reactions coupled to the formation of H4TPP.

Figure 4. Voltammograms of (a) H4TPP2+ and b) H4TPP2+ + 1 equiv of HClO4 in Bu4NClO4 0.1 M/PhCN at 0.1 V s−1, before (cathodic—pink; anodic—black) and after (cathodic—green; anodic—blue) electroreduction at E = –0.89 V (peak IIc). Voltammograms of H2TPP are included for comparison (dotted grey line).
Download figure:
Standard image High-resolution imageThe electroreduced solution was further analyzed by UV–vis spectroscopy (Fig. 5, blue line). Compared to the solution before electrolysis (pink line), a new band rises at 773 nm. This agrees with previous reports for phlorin species, which present absorption bands in the 700–850 nm region.40–47 It is worth noting that the H2TPP Q bands at 517 and 551 nm before electrolysis (pink line) had disappeared but they partially reappear after electroreduction (blue line).
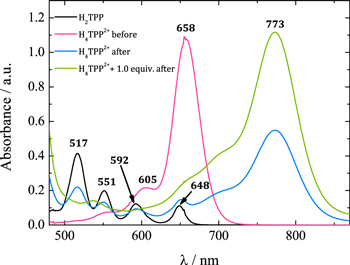
Figure 5. Absorption spectra of H4TPP2+ and H4TPP2+ + 1 equiv HClO4 in Bu4NClO4 0.1 M/PhCN, before and after electroreduction at E = –0.89 V (peak IIc). Spectrum of H2TPP is included for comparison. No additional bands are observed in the region between 850 and 1000 nm.
Download figure:
Standard image High-resolution imagePhlorins are two-electron reduced porphyrin species that have been protonated at one of the meso- positions of the main ring (Scheme
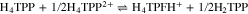
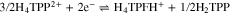
To further confirm the protonation of isophlorin H4TPP, cyclic voltammetry was carried out for H4TPP2+ solutions with 0.5 and 1.0 equiv of HClO4. Figure 6a shows that the presence of free protons results in the loss of reversibility for Ic/Ia and IIc/IIa, and also induces a new oxidation process IVa. Additionally, when the inversion potential is –1.30 V (Fig. 6b), a new reversible reduction process, IIIc/IIIa, appears. This matches the observations for the electroreduced solution (Fig. 4), and indicates the formation of phlorin cation H4TPFH+, which is feasible because of the presence of free protons from HClO4, reaction 8. The formation of phlorins has also been reported during the reduction of metallated porphyrins in aprotic media and in the presence of proton-donating species.43,53–55
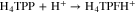
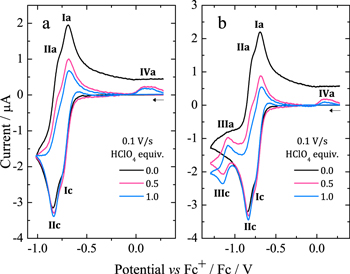
Figure 6. Voltammograms of H4TPP2+ 0.25 mM in Bu4NClO4 0.1 M/PhCN at 25 °C with 0.5 and 1.0 equiv of HClO4. (a) Eλ = –1.02 V and (b) Eλ = –1.33 V.
Download figure:
Standard image High-resolution imageA process similar to IIIc/IIIa has been observed during the reduction of H2TPP in CH2Cl2 with TFA, which was attributed to the one-electron reduction of phlorin species, reaction 9.15 Moreover, the experimental results show that the oxidation peak IVa corresponds to the two-electron oxidation of phlorin cation, reverse of reaction 10 (section A.1, Figs. S2 and S3, Supplementary material)
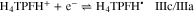
Based on the above and to unequivocally confirm the existence of the self-protonation reaction, it was assumed that the presence of 1 equivalent of HClO4 during the electroreduction of H4TPP2+ should suppress self-protonation, reaction 6. This, in turn, would result in a two-electrons global process, reaction 10, encompassing reactions 4, 5 and 8.
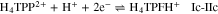
Exhaustive electrolysis of a H4TPP2+ + 1 equiv HClO4 solution at –0.89 V (process IIc), consumed 2.1 e–/H4TPP2+, which corresponds to what is expected from the global process 10. Similar to the electroreduction of H4TPP2+ with no HClO4 excess (Fig. 4a), the voltammograms of the electrolyzed solution (Fig. 4b) show the reduction (IIIc) and oxidation (IVa) of the phlorin cation species. In addition to the main oxidation peak, IVa, the anodic sweep (Fig. 4b, blue line) presents a small oxidation peak Va that was barely present in Fig. 4a. The addition of Bu4NOH to the electroreduced H4TPP2+ solution proves that IVa and Va are related to the oxidation of less protonated phlorin species, e.g. the neutral phlorin H3TPFH (Scheme
To obtain experimental evidence of intermediate species, such as H4TPP•+ and H4TPP, UV–vis spectroelectrochemistry was carried out. We acquired the absorption spectra of 0.26 mM H4TPP2+ at different reduction potentials (from –0.10 to –1.25 V) and in presence and absence of 1 additional equiv of HClO4 (Figs. 7b and 7c). It can be observed that, as the potential reaches Ic and IIc, the Q band at 658 nm decreases while two bands at 773 and 859 nm gradually increase. It is worth noting that the band at 859 nm appears before that at 773 nm, the former is barely perceptible at −0.74 V (Ic) but becomes clearer as the potential reaches that of IIc (−0.87 V, Fig. 7b). This band was not observed in the spectra of electroreduced H4TPP2+ (Fig. 5) and should thus correspond to short-lived species, such as isophlorin H4TPP, formed during the second electroreduction process and proven to be unstable in the time-scale of the electrolysis experiment. However, there were no bands that could be assigned to H4TPP•+.

Figure 7. (a) Cyclic voltammograms at 0.02 V s−1 in the quartz thin-layer electrochemical cell, path length: 1.0 mm. Potential-dependent spectral changes of (b) 0.26 mM H4TPP2+ and (c) 0.25 mM H4TPP2+ + 1 equiv of HClO4.
Download figure:
Standard image High-resolution imageThe spectra at Fig. 7b show that as the potential departs from that of IIc, the band at 773 nm increases and that at 859 nm decreases. This agrees with the fact that more H4TPP is consumed as time increases in the self-protonation reaction 6, producing more phlorin cation (which is responsible for the band at 773 nm). When the sweep direction is switched, the band at 859 nm drastically decreases at –0.06 V because at that point all the unreacted H4TPP is oxidized back to H4TPP2+ (reverse of reactions 4 and 5). After IVa (0.60 V), phlorin cation is oxidized, regenerating the diprotonated porphyrin, reverse reaction 10, as evidenced by the decrease in the phlorin cation band at 773 nm, and the increase of the H4TPP2+ Q band at 653 nm.
Spectroelectrochemistry experiments were performed for the solution of H4TPP2+ + 1 equiv of HClO4. A similar behavior was observed, however the increase of the band at 773 nm is more evident than when there are no excess protons, and the isophlorin band at 859 nm is smaller at all the potentials analyzed (Fig. 7c). These results agree with reaction 8, where the electrogenerated H4TPP is promptly consumed by the reaction in presence of free protons.
Electrochemical behavior of mono- and diprotonated octaethylporphyrin
In acid-free media, H2OEP is reduced via two consecutive one-electron processes, E1/2 = –1.91 and –2.34 V, that correspond to the formation of the radical anion and the dianion (Fig. 8a, black line).19,36 When adding HClO4 to the H2OEP solution, the behavior is different from that of H2TPP. Increasing amounts of HClO4 (from 0 to 1 equiv) lead to the appearance and increase of a reduction process at –1.06 V, peak Ic' (Fig. 8a). Simultaneously, the processes corresponding to the non-protonated H2OEP decrease. Based on the spectrophotometric titration from Fig. 1, which shows that with 1 equiv of HClO4 only the monoprotonated species H3OEP+ is present, peak Ic' can be assigned to the reduction of H3OEP+.
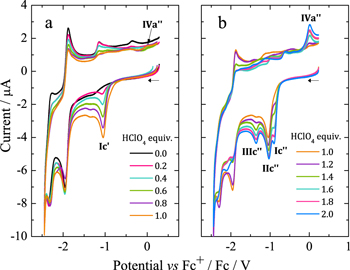
Figure 8. H2OEP cyclic voltammograms (0.50 mM in Bu4NClO4 0.1 M/ PhCN) at 25 °C with increasing amounts of HClO4: (a) 0–1 equiv and (b) 1–2 equiv. Scan rate: 0.1 V s−1.
Download figure:
Standard image High-resolution imageThe addition of 1.2 and 2.0 equiv of HClO4 (Fig. 8b), leads to three reduction processes (Ic'', IIc'' and IIIc'') that increase in intensity as the acid concentration increases. Peak IIc'' seems to be located at the same potential than Ic', suggesting the presence of the monoprotonated species H3OEP+, e.g. at 2.0 equiv of HClO4. However, voltammograms of H3OEP+ and H4OEP2+ solutions obtained at different scan rates (Fig. 9), demonstrate that Ic' and IIc'' are actually different: at 0.1 V s−1 Ic' (H3OEP+) and IIc'' (H4OEP2+) overlap; at 5.0 V s−1, Ic' is cathodically shifted by 0.131 V from the value obtained at 0.1 V s−1 and IIc'' barely shifted (0.049 V). A detailed analysis of the H3OEP+ voltammograms is provided below.
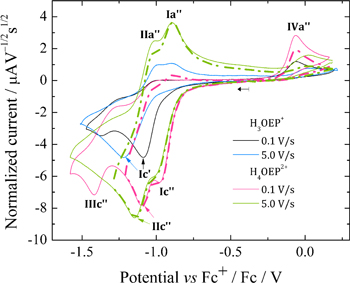
Figure 9. Scan rate normalized cyclic voltammograms of H3OEP+ and H4OEP2+ 0.8 mM in Bu4NClO4 0.1 M / PhCN at 25 °C.
Download figure:
Standard image High-resolution imageIn the H4OEP2+ voltammograms obtained when the potential is switched right after the second reduction process (IIc''), coupled oxidation peaks can be observed for both Ic'' and IIc'' (Fig. 9, dashed-dotted lines, peaks Ia'' and IIa''). Similar to the behavior of H4TPP2+, Ic''/Ia'' and IIc''/IIa'' can be assigned to consecutive one-electron transfers that yield the radical cation and isophlorin, reactions 11 and 12. It is worth noting that at low scan rates (Fig. 9, pink line), the reversibility of these processes decreases and peaks IIIc'' and IVa'', due to the phlorin cation H4OEFH+, become evident. This contrasts with the H4TPP2+ behavior, for which the formation of phlorin cation H4TPFH+ is only observed in longer timescale experiments, i.e., coulometry, or when excess HClO4 is added (Fig. 6). Moreover, the presence of IVa'' at 5.0 V s−1 (Fig. 9, green line), proves that the H2OEP isophlorin is more reactive than the H2TPP isophlorin, which is why self-protonation with the initial porphyrin occurs even at short timescales, reaction 13. Also, when comparing the experimental voltammogram at 5.0 V s−1 with a simulation based on reactions 11 and 12, the loss of reversibility of processes Ic''/Ia'' and IIc''/IIa'' is evident (Section A.3, Fig. S5, Supplementary material).
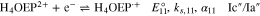
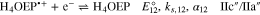
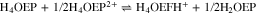
The charge consumed after exhaustive electrolysis of a H4OEP2+ solution corresponds to 1.35 e–/H4OEP2+ molecule. Voltammograms obtained after the electrolysis show signals of phlorin cation H4OEFH+, i.e., oxidation IVa'' during the anodic sweep and reduction IIIc'' during the cathodic sweep (Fig. S6a, Supplementary material). The UV–vis absorption spectrum of the electroreduced solution also presents signals from phlorin cation (band at 745 nm), and bands assigned to the formation of H2OEP at 498 and 622 nm (Fig. 10, orange line).
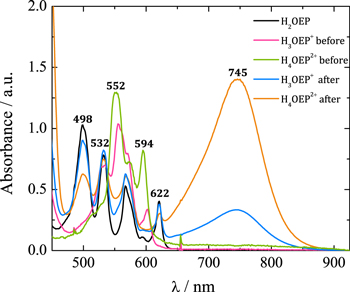
Figure 10. UV–vis absorption spectra of H4OEP2+ and H3OEP+ in Bu4NClO4 0.1 M/PhCN, before and after electroreduction at E = –1.11 V. H2OEP spectrum is included for comparison. No additional bands are observed in the region between 900 and 1000 nm.
Download figure:
Standard image High-resolution imageThese results agree with the global reaction 14, which is obtained from the combination of reactions 11–13, demonstrating that the reduction of H4OEP2+ follows the same behavior than H4TPP2+.
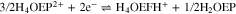
The reduction of H3OEP+ was also studied by performing an exhaustive electrolysis of H2OEP with 1.0 equiv of HClO4 at –1.11 V (Ic') for 1 h. The charge consumed was equivalent to 0.7 e–/H3OEP+ molecule. Both the UV–vis spectrum and the voltammograms before electrolysis confirm the presence of H3OEP+ with no interference from the H2OEP and H4OEP2+ species. After electroreduction, the spectrum presents bands at 745 nm and at 498 and 622 nm, corresponding to phlorin cation H4OEFH+ and to H2OEP as products (Fig. 10, blue line). The voltammograms show the same behavior than that of electroreduced H4OEP2+, i.e., peaks corresponding to phlorin cation (Figs. S6, S7 and S8, Supplementary material). These results can be explained by the global reaction 15. Unlike H4OEP2+ reduction, where H4OEFH+ and H2OEP are obtained in a ratio 1:0.5 (reaction 14); during H3OEP+ reduction, the ratio becomes 0.5:1 (reaction 15). This can be qualitatively observed in the spectra of the electrolyzed solutions (Fig. 10): the H4OEFH+ band at 745 nm is larger than that of H2OEP at 498 nm for the H4OEP2+ solution (orange line), and the opposite occurs in the H3OEP+ solution (blue line).
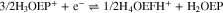
To better explain the mechanism involved in the H3OEP+ reduction, the voltammetric behavior was analyzed for different H3OEP+ concentrations and scan rates. Representative curves at two different concentrations (Fig. 11) show that the reduction process Ic' cathodically shifts as the scan rate increases, indicating the existence of coupled chemical reactions. Looking first at the 0.61 mM voltammogram, it is evident that, in addition to the cathodic shift, the current of Ic' changes. This is consistent with the presence of a coupled chemical reaction that yields a species whose reduction potential is similar to that of H3OEP+. Also, at 5 and 10 V s−1, two-reduction processes with similar potential values are observed (Fig. 11a). For the 1.85 mM solution, the current and shape of Ic' remain unmodified with increasing scan rates (Fig. 11b). Since the current of Ic' depends on the scan rate at low (0.61 mM) but not at higher concentrations (1.85 mM), it can be inferred that the coupled chemical reaction is of bimolecular nature. This behavior is characteristic of organic molecules with acid protons in their structure and is due to typical father-son50 or self-protonation49,56 reactions. Such process is feasible because the product of the first electronic transfer is more electronegative than the starting molecule.
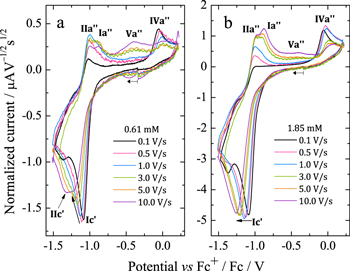
Figure 11. Scan rate normalized voltammograms for H3OEP+ in Bu4NClO4 0.1 M/PhCN at 25 °C: (a) 0.61 and (b) 1.85 mM.
Download figure:
Standard image High-resolution imageBased on the above, the reduction of H3OEP+ likely proceeds via a one-electron transfer, yielding H3OEP• (reaction 16) that is protonated in presence of the initial H3OEP+. For H3OEP•, there are two sites where protonation can occur: the free electron pair of the pyrrolic nitrogen (yielding H4OEP•+, reaction 17a) or in one of the meso- positions of the macrocycle54,55 (yielding the phlorin radical cation H3OEFH•+, reaction 17b), Scheme
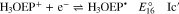
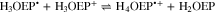
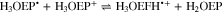
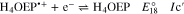
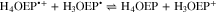
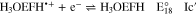
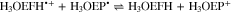
Voltammograms at 5 and 10 V s−1 for H3OEP+ 0.61 mM (Fig. 11a) indicate that the rate of the H3OEP• protonation can be partially overcome so that a second reduction process is observed as a small shoulder (peak IIc') at a slightly more negative potential than that of Ic', and is attributed to the reduction of the remaining H3OEP• that did not react with H3OEP+, reaction 19. The oxidation peaks observed during the reverse sweep of H3OEP+ coincide with peaks Ia'', IIa'' and IVa'' from Fig. 9, which were attributed to the oxidation of isophlorin H4OEP (IIa'' and Ia''; reverse reactions 12 and 11) and of phlorin cation H4OEFH+ (IVa''; reverse reaction 10). Therefore, it can be inferred that the protonation of the neutral radical H3OEP• occurs at the pyrrolic nitrogen electron free pair, yielding H4OEP•+ that is then reduced at Ic' to produce H4OEP (Scheme
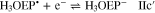
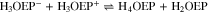
It is worth noting that, as the scan rate increases, the current of IVa'' decreases and a new oxidation process is observed, Va''. This is even more remarkable in the voltammograms obtained for the 0.61 mM solution than for that at 1.85 mM (Fig. 11). According to the acid-base behavior of the meso-tetraphenylporphyrin phlorin cation (Fig. S4, Supplementary material), it can be assumed that Va'' corresponds to the oxidation of neutral phlorin species H3OEFH, which was confirmed by experiments where DBU was added to a solution of H4OEFH+. The presence of neutral phlorin H3OEFH as intermediate in the mechanism for the formation of phlorin cation H4OEFH+ suggests that the protonation of H3OEP• occurs not only in the pyrrolic nitrogen, but also in the meso-position of the macrocycle so that H3OEFH•+ is obtained and immediately reduced to the neutral phlorin H3OEFH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scheme 2. Simplified diagram showing the electrochemical formation of phlorin cation H4OEFH+ from H3OEP+. It is considered that all the protonation steps are carried out by another H3OEP+ molecule. Ethyl substituents are omitted for clarity. In brackets is the number of the corresponding reaction.
Download figure:
Standard image High-resolution image{kind=link}
In summary, experimental results indicate that both of the reaction routes shown in Scheme
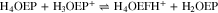
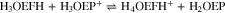
Spectroelectrochemical measurements, similar to those performed for H4TPP2+ (Fig. 7), were carried out for H3OEP+ and H4OEP2+. As the potential moved towards negative values, a band corresponding to phlorin cation was observed at 745 nm; however, no isophlorin bands were detected. This agrees with the voltammetry results in which the existence of oxidation and reduction processes of phlorin cation indicate the isophlorin H4OEP is a highly reactive species.
Conclusions
Acid titration of meso-tetraphenylporphyrin H2TPP and octaethylporphyrin H2OEP with HClO4 in PhCN shows that, for H2OEP, two protons are added in discrete steps forming the mono- and di-protonated species, H3OEP+ and H4OEP2+. However, for H2TPP, diprotonation occurs simultaneously to yield H4TPP2+ species. Since the monoprotonated form of H2OEP was stable in PhCN, it was possible for us to compare the electroreduction mechanism of the dipropotonated species, H4TPP2+ and H4OEP2+, with that of H3OEP+. Both H4TPP2+ and H4OEP2+ are two-electron reduced to phlorin cation species in an electrochemical process that involves a self-protonation reaction of the isophlorin intermediate. The results indicate that the absence of substituents at the meso-positions of the porphyrin ring increases the reactivity of the isophlorin. Thus, the rate of the self-protonation reaction between H4OEP and the starting protonated compound is higher than that of H4TPP. For H3OEP+, two parallel electrochemical reduction pathways were identified for the formation of the phlorin cation H4OEFH+: one involving the protonation of the one-electron transfer product, H3OEP•, at the pyrrolenine nitrogen atom, followed by a further one-electron reduction of the protonated radical to yield an isophlorin intermediate H4OEP; and the other via the protonation of H3OEP• at a meso-position and its further reduction providing a neutral phlorin intermediate, H3OEFH. Voltammograms at different scan rates and H3OEP+ concentrations suggest that both trajectories simultaneously occur.
In summary, the results of this work show that the protonation does lower the reduction potentials of porphyrin macrocycles, which can be useful for certain applications. However, it should be kept in mind that the electrochemical reduction of protonated porphyrins leads to shorter-lived intermediates than those from free base or metallated porphyrins. This is mainly because electroreduced species have basic properties, so they cannot coexist with the starting protonated porphyrin.
Acknowledgments
We thank to the Dirección General de Apoyo al Personal Académico (DGAPA-UNAM, PAPIIT IN212518) for the financial support of this research. A-A, L.R and C-C, H.M are grateful to CONACyT for their master and PhD grants, and to the Programa de Maestría y Doctorado en Ciencias Químicas, UNAM