
Abstract
A highly efficient flow cell for sequential electrolysis containing two complete electrochemical cells, capable of generating reactive species at the upstream working electrode and transporting them to the downstream working electrode, is demonstrated. Deconvolution of the intermixed electrode circuits is accomplished through analysis of the inherent resistance of the electrolyte, which allows for precise and independent control of the electrochemical potential at each electrode without altering concentrations of supporting or background electrolyte species. Sequential electrolysis involving oxidation of hydrogen and reduction of the generated protons downstream is demonstrated at nearly 100% efficiency on Pt-decorated dealloyed porous Nb catalysts. The conversion efficiency of the catalysts is discussed in terms of their geometries and active surface composition, elucidating strategies for use of sequential electrolysis cells for fundamental and applied studies.
Export citation and abstract BibTeX RIS
Several widely studied electrochemical reactions critical to environmental applications such as the hydrogen oxidation and evolution reactions (HOR and HER), oxygen reduction and evolution reactions (ORR and OER), reduction of CO2 (CO2RR), and synthesis of ammonia (NH3) are sensitive to local electrochemical environments including pH and concentrations of supporting species in the electrolytes.1–15 One kind of persistent problem in CO2RR and ammonia reduction is competitive hydrogen evolution at the thermodynamically required reducing potentials. In this study we take a step toward solving this kind of problem by demonstrating a new technique to dynamically vary the local pH at an electrode of interest without altering the concentration of the supporting species or bulk electrolyte using a sequential electrolysis flow cell with dealloyed porous electrodes. As a trend in electrocatalysis studies, this work follows on foundational work of Gurudayal et al. who used sequential planar electrocatalysts to separate the steps of the CO2RR reaction, controlling the flow of CO from a first to a second electrode.16 In contrast, our work here focuses on controlling proton flux, and in addition introduces a new dealloyed electrode with highly tortuous porosity that increased overall conversion efficiency.
The HOR and HER reactions were studied as models to demonstrate the sequential flow cell process capabilities. Figure 1 shows the flow cell and its schematic cross section. One notable feature relative to other sequential flow cells is the order of electrodes, which allows for precise and variable control of the pH at the downstream HER working electrode (the cell denoted HER) by delivering only products from the HOR working electrode (the cell denoted HOR) to the HER working electrode. Keeping the counter electrode of the HOR cell downstream of the HER working electrode prevents H2 and OH− species generated at the HOR counter electrode by reduction of water from flowing downstream and recombining with H+ products we wish to efficiently employ as reactants at the downstream electrode. In this way, the flow cell here controls the H+ concentration over nearly two orders of magnitude at the HER electrode through oxidation of dissolved H2 at the upstream HOR working electrode.

Figure 1. Image of the assembled flow cell (top) and cross-sectional schematic of the flow cell with the orientation and position of the porous electrodes (bottom).
Download figure:
Standard image High-resolution imageThe intermixed order of the electrodes complicates analysis of the voltammetry data because the working electrodes do not operate entirely independently of each other due to the location of the HER working and reference electrodes between the HOR working and counter electrodes. We observe minimal effects at the HOR working electrode, which was operated only in galvanostatic mode and exhibits only minor variation in the applied potential on the order of tens of mV as a function of the HER cell operation. In contrast, operation of the HOR cell dramatically alters the state of the HER cell by imposing current flow through the HER cell (a side-effect of the electrode order) and altering the local pH at the HER working electrode. A straightforward method to account for the effects of imposed current flow through the HER cell employs a simple model of the electric circuits of the electrochemical cells (Fig. 2). Because the entirety of the current generated in the HOR cell necessarily passed through the electrolyte (and its associated solution resistance) between the HER reference and working electrodes, the model predicts that the measured potential at the HER working electrode shifts linearly with the applied HOR current. This model is analogous to the uncompensated resistance potential drop in a conventional electrochemical cell; we confirmed this model experimentally (see Supplemental Information is available online at stacks.iop.org/JES/167/106510/mmedia).
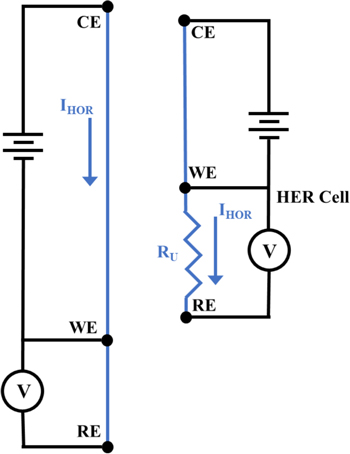
Figure 2. Circuit diagram of the flow cell electrodes with portions in blue indicating paths of ionic charge flow in the electrolyte.
Download figure:
Standard image High-resolution imageElectrode materials for sequential flow cells must satisfy a number of stringent requirements. First, they must contain relatively small scale and open porosity. However, the average pore size  cannot be too small, because the pressure required to maintain the desired flow increases as
cannot be too small, because the pressure required to maintain the desired flow increases as  17 Most high surface metals, for instance those made by electrochemical dealloying such as nanoporous gold, have pore sizes below 100 nm and are too mechanically fragile to maintain a high pressure drop. Second, it is advantageous that the electrode porosity be highly tortuous in order to force contact between species in the electrolyte and the surface. In this study, we used a new electrode material—porous niobium structurally reinforced by niobium silicide plates co-formed during a liquid metal dealloying process used to make electrode materials.18 This conductive material has highly random pores with characteristic diameter of order 1 micron (Fig. 3a) and can be easily machined into disks of order 1 mm thick and integrated into the flow cell. In addition, the surface of the material can be easily modified chemically; here, for instance, we decorated the surface of porous niobium with platinum nanoparticles. In total, three materials were examined compared and contrasted in detail as HER working electrodes in the flow cell: 0.78 mm thick silicide reinforced porous niobium electrodes created by liquid metal dealloying, both bare (denoted Nb) and coated with Pt nanoparticles (denoted NbPt) and a 1.4 mm thick stack of carbon cloth coated with Pt nanoparticles (denoted CCPt) (Fig. 3b).
17 Most high surface metals, for instance those made by electrochemical dealloying such as nanoporous gold, have pore sizes below 100 nm and are too mechanically fragile to maintain a high pressure drop. Second, it is advantageous that the electrode porosity be highly tortuous in order to force contact between species in the electrolyte and the surface. In this study, we used a new electrode material—porous niobium structurally reinforced by niobium silicide plates co-formed during a liquid metal dealloying process used to make electrode materials.18 This conductive material has highly random pores with characteristic diameter of order 1 micron (Fig. 3a) and can be easily machined into disks of order 1 mm thick and integrated into the flow cell. In addition, the surface of the material can be easily modified chemically; here, for instance, we decorated the surface of porous niobium with platinum nanoparticles. In total, three materials were examined compared and contrasted in detail as HER working electrodes in the flow cell: 0.78 mm thick silicide reinforced porous niobium electrodes created by liquid metal dealloying, both bare (denoted Nb) and coated with Pt nanoparticles (denoted NbPt) and a 1.4 mm thick stack of carbon cloth coated with Pt nanoparticles (denoted CCPt) (Fig. 3b).

Figure 3. Plan-view SEM micrographs of Pt functionalized porous Nb (top) and Pt functionalized carbon cloth (bottom). The inset micrographs show micron scale Pt particles distributed across the catalyst surfaces.
Download figure:
Standard image High-resolution imageResults
Wide range cyclic voltammetry (CV) measurements (Fig. 4) of the NbPt HER working electrode at various HOR current densities demonstrate the utility of shifting the HER data by the appropriate potential values. The water oxidation peaks (onset at 1.6 V vs SHE, not shown) of the CV curves align as expected when the applied potential data are shifted to account for HOR current density flowing across the solution resistance, confirming that the applied potential shifts were appropriate and allowed for detailed study of the catalytic properties of the HER working electrode.

Figure 4. (a) Open circuit potential as a function of time for a porous Nb electrode during application of galvanostatic pulses at the current densities (mA cm−2) as noted in red in the HOR cell; (b) instantaneous shifts in the open circuit potential as a function of the applied HOR current density as calculated from (a); (c) cyclic voltammetry of the NbPt HER working electrode at various applied HOR current densities prior to shifting the data to account for the effects of the HOR cell (50 mV sec−1 in H2-saturated 1M Na2SO4 at 10 ml min−1 volumetric flow rate). The low frequency noise is due to the peristaltic pump; (d) cyclic voltammetry data from (c) shifted according to the applied HOR current density.
Download figure:
Standard image High-resolution imageLinear sweep voltammetry (LSV) measurements were made at the HER working electrode at different fixed HOR current densities to produce differing fluxes of protons from the HOR electrode (Fig. 5). The data show that each HER working electrode converted the protons created upstream back to hydrogen at significant efficiencies in the potential regime associated with HER, because the reductive current clearly scaled with the HOR current density. Analysis of the data on the absolute SHE reference scale (left column in Fig. 5) is often employed for fundamental HER/HOR studies where the local reactant (H+) and product (H2) concentrations are constant, especially as the SHE scale is simply a constant offset from the reference electrode potential. In our case, however, the central volume between the HER and HOR electrodes is filled with protons generated at the HOR electrode, and thus has a different pH from the rest of the system. For this reason, the most common form of the Nernst equation in acids, where the H2 pressure is constant, cannot be used. To assess the HER electrode performance as a function of overpotential, it is better to use a pseudo RHE reference scale (hereafter referred to as the RHE scale). To align the LSV curves on the RHE scale, we used the facts that Pt is known to deviate from typical potential/pH behavior in neutral electrolytes9,11,19,20 and that the water concentration is fixed and large compared to any H+ or H2 concentration. It follows that we appropriately shift the SHE data to put the LSV data on the RHE scale by aligning the potential for H2O reduction at the CCPt electrode. This gives the correct offset for each HOR current density, and we shift the NbPt data and shifted the Nb data by these amounts (right column of Fig. 5).
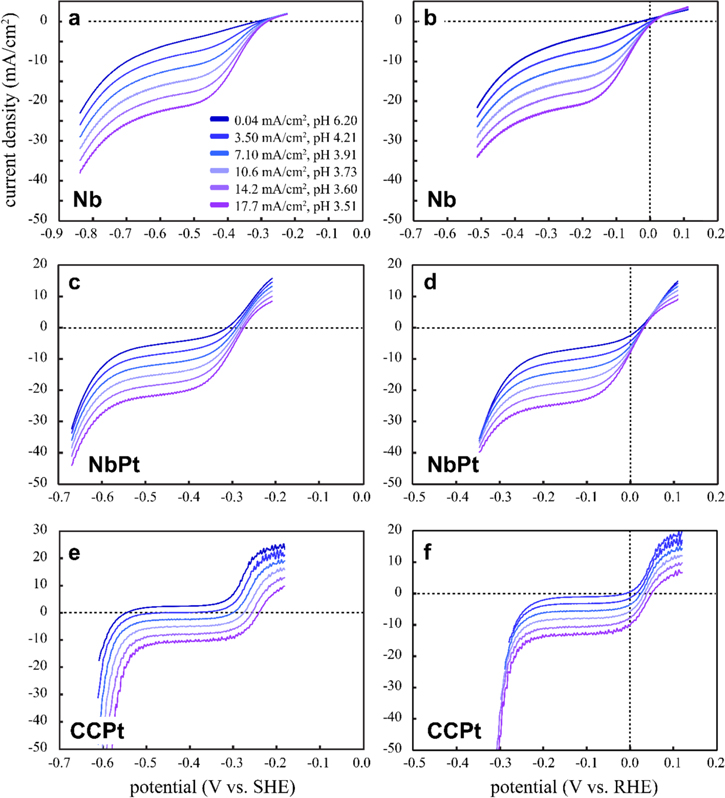
Figure 5. LSV data (5 mV s−1 positive to negative sweep, H2 saturated 1M Na2SO4, 10 ml min−1 volumetric flow rate) obtained with HER working electrodes and differing applied HOR current densities: Nb on the (a) SHE scale and (b) RHE scale, NbPt on the (c) SHE scale and (d) RHE scale, and CCPt on the (e) SHE scale and (f) RHE scale. Due to noise caused by the cyclic pumping, each curve is the average of 6 LSV for Nb and NbPt and 4 LSV for CCPt.
Download figure:
Standard image High-resolution imageThe baseline LSV curve was subtracted from each of the other LSV curves at larger HOR current densities to yield the excess current density due to proton reduction as a function of potential at the HER electrode and then allow calculation of the HER conversion efficiency. Figure 6 shows the proton conversion efficiency as calculated as a function of potential by dividing the excess HER current density data by the appropriate excess HOR current density. The Nb electrode exhibits broadly peaked behavior at the onset of diffusion limited proton reduction, with the peak values ranging from just above 100% for the lowest upstream HOR current density to 98% for the largest upstream HOR current density. The conversion efficiencies then decrease linearly with increasing overpotential to a minimum of ∼70% efficiency just prior to the onset of H2O reduction. Conversion efficiency on NbPt curves was nearly constant over the diffusion limited potential range before decaying at the onset of H2O reduction, with a maximum of 94% conversion efficiency. The CCPt electrode exhibits lower peak efficiencies in the diffusion limited region between 64% and 73% with the efficiency increasing slightly with increasing HOR current density. These single pass conversion efficiencies compare well with state-of-the-art gas diffusion electrode systems for CO reduction, which have been demonstrated in the range of greater than 70%.21

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 6. HER conversion efficiencies (%) calculated from Fig. 5 on the RHE scale (a) Nb, (b) NbPt, (c) CCPt.
Download figure:
Standard image High-resolution image{kind=link}
Discussion
The data presented in Fig. 2 provide substantial evidence in support of the proposed model based on passing HOR generated current through the solution resistance between the HER reference and working electrodes. The shifts followed a strictly linear, highly repeatable trend, where different electrochemical reactions were occurring upstream at the HOR working electrode. The oxidative current densities used during the HOR pulses ranged from current densities where only H2 was directly oxidized to H+ to potentials where water was oxidized. The reductive HOR pulses reduced water to H2 and OH–. The measured solution resistance was also similar among the varying HER and HOR working electrodes, as well as with the reference electrodes, further indicating that the electrolyte caused the instantaneous potential shifts, rather than the electrodes. The model and experimental results matched previous studies of a multiple working electrode microfluidic fuel cell where potential shifts at working electrodes within externally imposed electric fields scaled linearly with current density flowing between the external electrodes.22
Three primary electrochemical reactions govern the form of measured HER voltammetry curves: reduction of protons (Eq. 1, E0 = 0.0 V), reduction of water to (Eq. 2, E0 = −0.829 V), and reduction of Nb2O5 oxide to a hydride (Eq. 3, E0 ≅ 0.0 V23):
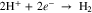
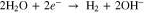
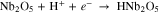
In further discussion below, we primarily focus on reduction of protons, and the potential ranges where this occurs on each electrode are −260 to −650 mV vs SHE for Nb, −235 to −500 mV for NbPt and −190 to −450 mV for CCPt, except when noted. Functionalization of the Nb electrode with Pt dramatically affected the HER behavior of the porous Nb. The diffusion limited H+ reduction plateau was 150 mV shorter compared to bare Nb, and the CCPt electrode exhibited a slightly lower overpotential for H+ reduction relative to the NbPt electrode with similar potential range for diffusion limited H+ reduction. The Pt functionalized electrodes exhibited significant HOR current density with the magnitude of HOR current density inversely scaling with the upstream HOR current densities due to the upstream oxidation of H2 limiting its availability. On pure Pt electrocatalysts, the combined effects of Eqs. 1 and 2 have been successfully modeled to match the experimental HER voltammetry curves over a wide range of pH values.11,19,20 The behavior of the porous electrodes containing Pt used in this study follow those trends observed in previous studies for Pt electrodes under neutral pH conditions demonstrating that observed differences in voltammetry were effects of local pH changes.
The additional complexity of HER conversion on Nb based catalysts is attributed to the concurrent reduction of the Nb oxide according to Eq. 3 at HER active potentials. The reduction of Nb2O5 to a hydride is reversible and mass transport limited by diffusion of protons through the transforming oxide layer (as observed in previous studies of cathodically polarized Nb oxide films23) and observed repeatedly in the LSV data. The reversibility was further evidenced by the repeatability of individual LSV curves at a given HOR current density (Fig. S3 in Supplemental Information). The onset potential of the Nb2O5 reduction reaction is pH dependent as required by Eq. 3, but does not exhibit the same pH/potential dependence as Pt. That is, different potential offsets are required to align the CCPt- and Nb-based LSV data with their baselines on pseudo RHE scales. The HER conversion curves of the Nb based catalysts also exhibit aligned onset waves on the SHE scale, rather than the RHE as observed for CCPt, further evidencing subtle differences in pH/potential behavior of Pt and Nb.
Protons reaching the surface of the polarized Nb based electrodes have two competing pathways towards eventual reduction: direct reduction to hydrogen or diffusion through the growing hydride to the Nb2O5 interface for reduction. The latter process requires a finite concentration of protons in the hydride and consequently changes the resistivity of the barrier layer. This adds complexity to the HER curve due to applied potential effects on the migration of protons and electrons through the outer layer of the Nb.23,24 The slope of the HER potential region for the Nb catalysts varied subtly, but remained relatively similar as a function of potential with varying HOR current density (Fig. 5). This suggests that a portion of the reductive current associated with Nb2O5 might not have scaled directly with the proton activity for HER and caused the observed decrease in conversion efficiencies on the RHE scale for the Nb electrode. The presence of the sloped diffusion limited plateau in the NbPt LSV data indicated that the exposed Nb2O5 surfaces were also electrochemically active on the NbPt catalyst during HER and caused the small decreases from 100% HER conversion. However, the significant HER conversion efficiencies on both scales still demonstrated that the Nb based materials were extremely efficient as flow-through catalysts. Furthermore, the increased efficiency of the NbPt catalyst relative to Nb as well as the ideal shape of the HER conversion curve demonstrated that the Pt particles were the primary active catalytic sites for proton reduction.
A useful figure of Merit (FOM) assessing the effectiveness of porous electrocatalysts has been proposed by Ateya and co-workers,25 with the FOM calculated as the ratio of the time a reactant species requires to diffuse radially to the electrode surface (which scales with the square of the pore size) to the time for the species to flow entirely through the pore based on its linear velocity (which scales linearly with the thickness of the catalyst layer). We estimated the figure of merit as 0.019 for the Nb based electrodes, where experimental values less than 0.5 predict complete conversion of reactant species passing through a sufficiently polarized porous electrode. Dimensional measurements, however, which are used to calculate the FOM don't accurately account for the non-uniform distribution of flow through the pores, leading to an overestimate of the FOM. That is, more electrolyte would be expected to flow through larger pores, and in these regions there would be poorer conversion for reactants passing through the catalyst. This effect is taken to the extreme for the carbon cloth. Considering just the micron scale porosity within the weave of the fibers, we calculate an FOM of 0.025 for the CCPt material. However, this does not account for the weave pattern of the carbon cloth, which had a broad pore size distribution with regularly spaced pores up to 100 μm × 100 μm.26 Not only did the carbon not reach high Faradaic efficiency for HER, but the catalyst membrane could not sustain a pressure drop, which would reflect good retention of the electrolyte in the pores. In contrast, based on the catalyst geometry, we calculated the pressure drop across Nb-based catalysts during operation of the cell based on Darcy's Law to be 5.8 kPa (0.84 psi).24
The agreement between the figure of Merit and trends in conversion efficiency confirms that the geometry of the electrode was the primary factor determining its ability to utilize reactants flowing through the catalyst. The small pore size of the Nb catalysts compared to the CCPt led to increased conversion efficiency in the Nb catalyst despite the CCPt having nearly eight times greater Pt surface area than the NbPt catalyst and two times more Pt surface area than the combined Nb and Pt surface area. The substantial tortuosity of the Nb pores compared to the CCPt pores also contributes to the increased efficiency of the Nb catalyst, as increasing tortuosity of a flow through electrode has been predicted to increase conversion efficiency for a given porosity,25 and the associated convection due to forced flow through a curved pore may aid in mass transport at the electrode surface.27
The sequential flow cell introduced here provides controlled fluxes of intermediate species or reactants to an electrocatalyst that can efficiently utilize them in order to decouple critical reaction steps in complex and challenging reactions such CO2RR and NH3 synthesis reactions through the use of multiple optimized catalysts joined by reactant flow, and by which concentrations of chemical intermediates can be controlled on-the-fly.28–30 This is a different electrocatalysis design strategy than the more common approach of using bimetallic and alloy catalysts that attempt to utilize effects of different atomic species at the catalyst surface to alter binding energies of intermediate species for improved performance over elemental catalysts.31–34 Our concept should be useful, for instance, in CO2RR, where, under favorable conditions, Cu catalysts can more efficiently catalyze formation of high value products when supplied with the critical reaction intermediate CO rather than CO2; indeed, in the course of preparing this manuscript, we became aware of work by Ager and co-workers doing this by controlling the flux of CO from one electrode to another in an analogous manner.16 Similarly, electrochemical NH3 synthesis is notably challenging due to the stability of the triple bonded N2 molecule and scaling of the binding energies of key intermediates, which means large overpotentials are required to drive the reaction.35 In aqueous electrolytes, hydrogen evolution tends to dominate at large cathodic overpotentials leading to low Faradaic efficiencies. Because active hydrogen is necessary for NH3 formation, we propose that well controlled and periodic (i.e., time-modulated) delivery of an activated hydrogen species from one electrode to another, one that is optimized for N2 adsorption, could be a possible route to more efficient electrochemical NH3 synthesis.
Conclusions
A novel electrochemical flow cell employing porous electrodes and two complete but intermixed electrochemical cells for highly efficient sequential electrolysis is introduced. We demonstrate that this cell can be well-controlled through operation of the upstream cell in galvanostatic modes, accounting for the potential shifts observed at the second electrode due to the HOR current flowing through the solution resistance between the second working electrode and its reference electrode. The flow cell exhibits highly efficient hydrogen oxidation and subsequent hydrogen reduction with the use of porous Nb, Pt-decorated porous Nb, and Pt-decorated carbon cloth as the downstream working electrode. HER conversion efficiency results show that the Nb and carbon cloth-based electrodes have geometric morphologies well-suited to efficient electrolysis in the flowing electrolyte. The large toruosity and small pore size of porous Nb lead to nearly perfect conversion efficiency, especially when functionalized with Pt. Our approach opens the door for to a new approach to tackling important electrochemical reactions with complex reaction pathways and multiple reactants, as well as studies of fundamental aspects of important electrochemical reactions over a range of pH without the need to alter the composition of the supporting electrolyte.
Acknowledgments
This research was supported by the U.S. Department of Energy, Office of Basic Energy Sciences under grant number DE-SC000868.