
Abstract
This paper reviews the battery electrolyte technologies involving Hückel-type salts as a major electrolyte component. The concept was initially proposed by M. Armand in 1995 and then explored by several research groups. In the present review studies on the optimization of the electrolyte composition starting from molecular modeling through enhancing the yield of the salt synthesis to structural characterization and electrochemical performance are described. Furthermore, the use of the optimized electrolytes in a variety of lithium-ion and post-lithium batteries is presented and discussed. Finally, the commercialization of the up to date technology by Arkema is discussed as well as the performance of the present Hückel anion based electrolytes as compared to other marketed electrolyte technologies.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
The lithium-ion battery (LIB) market is developing at the fastest rate among chemical energy storage technologies. LIBs now dominate over all alternative power sources for consumer electronics and electric vehicles. As the LIB technology advances, they are also introduced to military and medical equipment, previously relying on lead-acid and nickel-cadmium batteries.
Electrolytes for LIBs are basically based on a salt of a lithium cation and a weakly coordinating anion (WCA) dissolved in an aprotic solvent.1 The most popular and frequently used LIB electrolyte is LiPF6 dissolved in a mixture of organic carbonates sold commercially as LP30®. Over the past 20 years many new lithium salts with new WCAs have been proposed, but very quickly their disadvantages have been found, finally preventing their practical use in LIBs. The most frequently encountered weaknesses are: i) a too low ionic conductivity of their carbonate-based electrolytes, the target being 1 mS cm−1 at room temperature, ii) anodic dissolution of aluminum (current collector) at the voltage range of the cells by electrolytes comprising selected salts, and iii) the contribution of the lithium cations to the ionic conductivity is too small, the rest is transferred by the anions, which are blocked by electrodes. The usable ionic conductivity is only that of lithium cations (σLi+), which can be calculated as the product of total ionic conductivity and the lithium cation transference number.
The electrolyte salts are also required to be thermally stable, to not limit the whole cell. In terms of operation temperature the organic carbonate solvents that boil at ca. 90 °C, they are usually are the limiting factor. Furthermore, the electrolyte also has to be stable in the full voltage range of the cell, i.e. from 0 up to 4.6 V vs Li+/Li (or another metal) and against all other cell components. The main issue is often the stability against the electrode active materials, but also against the current collectors—copper and aluminum. Unfortunately, most of the salts and solvents react with the electrode materials in the range of the LIBs cell's operation potential. Most electrolytes are only usable if they form a stable solid electrolyte interphase (SEI) at the anode, which is permeable for lithium cations, but not to electrons. This is achieved by the salt anion reaction at electrodes and/or adjunction of various electrolyte additives which reduce at a higher potential than the decomposition of the salt anion and solvents, and form stable SEI layer. All salts should also be safe in both handling and manufacturing and be inexpensive to manufacture. Salts that are stable to moisture present an added advantage for storage and handling.
Most salts tested for application in LIB cells do not satisfy the abovementioned requirements and have been rejected after failing stability and compatibility tests against standard electrode materials and solvents.
Today only eight lithium salts are commercially available and used by the LIB industry for cell manufacturing or at least for research purposes. A quick review of the properties of these salts shows that some do not fulfill every industrial requirement. For instance, LiClO4-based electrolytes may become explosive in the presence of electrodes containing transition metals or acetylene black, both components of most cathodes.2 The salt itself also has a low thermal stability.3 Electrolytes based on carbonate solvents and containing LiBF4 have lower ionic conductivities than analogous LiPF6-based electrolytes4 and form SEI layers that are very poor lithium cation conductors,5 especially at temperatures above 60 °C.6
In contrast, LiPF6-based electrolytes have very high ionic conductivity, up to 10 mS cm−1 and also form stable SEI layers with many different anodes. Unfortunately, LiPF6 is thermally unstable, it decomposes at ca. 70 °C in a solvent-based electrolyte (pure and dry it is stable up to ca. 200 °C).7–10 In addition, it always contains at least traces of hydrogen fluoride due to the auto-decomposition: LiPF6 → LiF + PF5 and then hydrolysis with traces of water: PF5 + H2O → POF3 + 2HF.11,12 Hydrogen fluoride is toxic and corrosive and can damage other cell components inside the cell, such as leaching out transition metals (Mn, Co...) from the positive electrode. Also POF3 is a toxic compound.13 This is why LIB cell manufacturers often must use moisture and HF scavenger additives and all components before cell assembly must be very dry. In addition, LiPF6 is metastable when cycled below 2.5 or above 4.2 V vs Li+/Li in cells containing organic carbonates, which encompasses every modern LIB cell.14 Simultaneously, LiPF6 is very demanding in terms of synthesis—apart from the moisture-less environment, preferably inert gas, it also requires very pure, dry and dangerous precursors, anhydrous HF and PCl5. All of the above requirements substantially increase the manufacturing cost of LIBs due to the special requirements for materials and reactors used, as well as security measures against environment pollution.15,16
Moving to other popular LIB salts, LiTf (Li[SO3CF3]), LiFSI (Li[N(SO2F)2]), LiTFSI (Li[N(SO2CF3)2]) and LiBETI (Li[N(SO2CF2CF3)2]) all do not protect aluminum current collector against corrosion.17,18 They can therefore only be used either in cells containing solid polymer electrolytes (SPE), at potentials below 3.9 V vs Li or with additives that inhibit aluminum corrosion. The latter, however, increase the cost of cell manufacturing.19 The less common LiFAP (Li[PF3(CF2CF3)3]) salt has proven to be too expensive in manufacturing cells for the consumer market.20
Only a few salts remain and are used in research laboratories—most commonly LiPF6, but also LiFSI, LiTFSI and LiBF4. In primary, non-rechargeable cells also LiBF4 and LiClO4 salts are used, while the salt used in electrolytes for rechargeable consumer market LIBs is almost exclusively LiPF6.
In late 1990s and early 2000s new lithium salts were searched by multiple teams all over the world, urged by the need of new lithium salt design motivated by the:
- –virtual market monopoly of one lithium salt—LiPF6;
- –well-known numerous disadvantages of LiPF6, particularly its instability;
- –failure of compatibility with new generation of electrode materials (in particular Si-based).
There has been a number of unsuccessful attempts to substitute the LiPF6 salt already and the field was abandoned by the end of the first decade of 21st century by most research teams. However, it was rational to develop new technology both from business and technological points of view and over 150 different salts were proposed:
- –
- –
- –
- –
- –a few perfluoroalkoxyaluminates, perfluorosulfonylmethides, and imidazolates.37
Many of the abovementioned salts turned out to be unstable either chemically (in contact with moisture, air or other cell components), thermally or electrochemically. Others were discontinued at an early stage by the researchers themselves due to low conductivity of electrolytes, problems with solubility in typical battery solvents, or inability to lower the manufacturing costs. One successful salt design that emerged in the mid-1990s was based on anions following the Hückel rule—and this review tells the story of how the Hückel salts as a major electrolyte component has become a possible battery technology.
Molecular Modeling Studies as a Useful Tool for Designing New Hückel Anions
Modeling has been an integral part of the development of new Hückel anions for battery application for nearly two decades. It started from very simple ab initio computational approaches to, together with IR and Raman spectroscopy, determine the dominant ion-ion interactions,38,39 and evolved to independently suggest new anion designs, mainly by DFT,40–42 and then reverted back to more complex models and approaches, including both implicit and explicit solvation, to real electrolytes of an existing anion.43 The studies have since been expanded in very different directions; using the knowledge gained to create all fluorine-free electrolytes,44 using the Hückel family anions as part of the computational benchmarking studies (CCSD(T)/CBS) of electrochemical stability windows (ESWs),45 and using one Hückel anion as a probe for solvate ionic liquid (SIL) or disproportionation-based electrolytes via DFT and MD simulations.46
The starting point for the first modeling efforts of a Hückel based anion, the 4,5-dicyano-1,2,3-triazole (DCTA or TADC) anion, performed from 2002 and onwards was that its lithium salt had been used already in 1995 in Grenoble to create solid polymer electrolytes (SPEs).47 The result was an electrolyte without any fluorine at all and the conductivities for the PEO-based SPE were on par with those employing LiTf. While the TADC anion itself was synthesized as early as in the 1920's48 it had not been attracting any attention since—and little was known about how it interacted with the lithium cation once dissolved in a medium. Hence a joint study between Univ. Montreal and Chalmers University of Technology was set up in late 2001 using small amounts of LiTADC dissolved in acetonitrile and record the Raman spectra for solutions ranging from 0.1–0.5 M and at the same time applying simple computational models positioning the lithium cation at different negatively charged sites of the anion. The latter was mainly used in two ways: i) to directly calculate the relative stabilities of different ion-pairs, as previously done for other anions of battery electrolyte interest,49 and ii) to calculate the Raman spectra for the anion itself and the different ion-pairs, including both depolarized and polarized spectra (as were recorded experimentally). While the computational level employed (HF/6–31+G*) is far from impressive now, it allowed to suggest that a bidentate coordination of the lithium cation to two adjacent ring nitrogen atoms clearly dominated energetically vs bidentate coordination to two nitrile nitrogen atoms and monodentate coordination to the apex nitrogen atom (Fig. 1). The latter was in fact found by the vibrational analysis to be a transition state and not a minimum in the energy landscape. The resulting ion-pair could not really be understood only from looking at the atomic charges of the TADC anion, but furthermore it was quantitatively a weaker ion-ion interaction than any other anion studied at the time including both PF6− and TFSI.49

Figure 1. The different ion-pairs located in Ref. 38.
Download figure:
Standard image High-resolution imageSo far the purely energetic picture, but given the simplifications made and the admittedly rather "bad" computational level, the ion-pair had to be qualified further. Looking at the Raman spectra they contain a superposition of the responses of the "free" anions and ion-pairs—as the concentrations were not low/high enough to allow for more or less only one type of species—and this would not have been practical for the measurements or creation of solutions. By a rather complex analysis looking mainly at subtraction spectra and the region of the C–N triple bond stretching there was still no conclusive evidence. By employing the polarized spectra and the computed ratios the origins of the peaks could be explained; with a calculated depolarization ratio of 0.08 for the suggested ion-pair and an experimental value of 0.09, for the band observed at ca. 654 cm−1, there was a consistent picture created of how the TADC anion interacts with the lithium cation in the solutions.
The same strategy as above, but employing the complementarity of IR and Raman spectra instead of polarized/depolarized Raman spectra, was applied to the next logic member in the family; the PATC anion and its lithium salt.39 The thinking was that adding another electron-withdrawing group would further decrease the ion-ion interaction strength. The Li-PATC salt itself had only occurred in the scientific literature within the synthesis of different pyrazole acids.50,51 Available in only minute amounts due to an explosive intermediate in the synthesis path, 0.3–4.0 M solutions of the salt in DMSO were made. While the PATC offered four unique sites of interaction, again the bidentate coordination to two ring nitrogen atoms was found to render the most stable ion-pair structure. While the computational methods employed differed somewhat, it was clear adding one more nitrile group withdrew quite a bit more negative charge from the ring (0.62e vs 0.32e) and thus the ring itself much more positive. Yet, however, the two nitrogen atoms of main interest were found to be more negative (−0.74e vs −0.50e)! This was not really reflected in the ion-ion interaction strength—they were close to that of Li-TADC—pointing to the role of an extensive conjugation within the anion(s). The energy data and partly a negative proof reasoning for the experimental and computed spectral comparison were both strong indicators for the sole existing type of ion-pair for Li-PATC being of the same nature as for Li-TADC.
Subsequently, a new purely computational study was set up, armed with the knowledge of the atomic charges within the TADC and PATC rings vs the nitrile groups and the preferred site of lithium cation interaction. This study40 mapped the full space of the Li+[N5C2n]− (0 ≤ n ≤ 5) system—consisting of eight different anions and the different sites and the most stable ion-pairs. Some of these salts had been made previously by laser ablation techniques,52 but this was anyhow considered an in silico design study. Making use of the significant advances of computational power at the time, the computations now not only covered may more anions and ion-pairs, the computations also went further in the method applied, using the very accurate (and computationally expensive) Gaussian-3 (G3) method.53 Furthermore, also the analyses made were more extensive, covering both the anion volumes by a Monte-Carlo based algorithm, the stability vs oxidation of the anions, as inferred from the HOMO energies, and the aromaticity of the anions (Fig. 2). The latter two were used in more comprehensive way to qualify the stabilities of the different anions by four different criteria; aromaticity, ring charge, chemical hardness—using Koopmans' theorem, and HOMO energies. We note in passing that the [N5]− anion was found to have an aromaticity index very close to the theoretical limit and that all [N5C2n]− anions were found to clearly be aromatic.
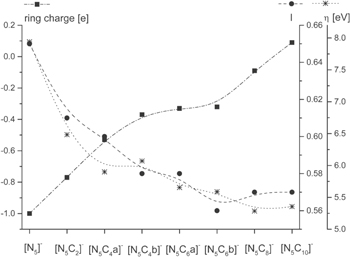
Figure 2. Ring charge, aromaticity index and chemical hardness of the Hückel anions. Reproduced from Ref. 40 with permission from the PCCP Owner Societies.
Download figure:
Standard image High-resolution imageWhile the previous computational studies38,39 were made using methods not really allowing for any strong conclusions to be drawn based only on the ion-ion interaction energies, the G3 method allows for this to be done. Again the bidentate coordination ion-pair dominates when possible—and a major decrease in the interaction strength is obtained when the anion structure lacks this site and instead bidentate coordination to two nitrile groups is preferred. For the end members of the series, the [N5]− and the [N5C10]− anions, these two sites are the only possible bidentate sites and the ion-ion interaction energy decreases from ca. −580 kJ mol−1 to ca. −460 kJ mol−1. Indeed, as compared to other WCAs used in battery electrolytes computed as references, all [N5C2n]− anions were found to be less coordinating, with the exception for BOB which only was beaten by the [N5C6b]−, [N5C8]− and [N5C10]− anions. With these results as a basis, recommendations for future synthesis efforts were made—putting focus on avoiding the possibility of bidentate ring nitrogen coordination, which is exactly what e.g. the TDI (4,5-dicyano-2-(trifluoromethyl)imidazole) anion does.54
Quite a different approach to in silico design of heterocyclic anions and their lithium salts was put forward in Ref. 41. Therein the anion family [CF3SON4C2n]− (0 ≤ n ≤ 4) was systematically explored (Fig. 3), building on the notion that except for the TADC anions, all anions suggested in Ref. 40 are difficult to synthesize. The strong electron withdrawing character of the CF3S(=O)– moiety promises low ion-ion interaction and the inherent asymmetry should also make these salts less prone to crystallization. The HF computational level was adequate for a first round of screening, which again showed the bidentate coordination to two ring nitrogen atoms to be preferred by the lithium cation, and second to that a monodentate coordination to the sulfonyl oxygen atom. As previously, the ion-ion interactions decreased globally by the number of –CC≡N substituents and to a large extent lower than for the standard WCAs. A novel aspect covered in the very short paper was the suggested synthesis path for one of the anions (n = 2) by simple precursors and conditions. While resorting to have some fluorine in the structure, the use of standard precursors could hold promise for easy synthesis and characterization to validate the in silico design predictions.
Figure 3. The Hückel anions studied in Ref. 41.
Download figure:
Standard image High-resolution imageFollowing the same path of in silico design, a much broader, and larger family of anions/lithium salts were next in line: imidazoles and benzimidazoles.42 In 2009–2010 these were attacked by a large screening effort looking at what the ring substituents a cyano group, a trifluoromethyl, or a pentafluoroethyl, would do to both the ion-ion interaction and the stability vs oxidation (Fig. 4). Mixed types of substitutions were also covered, which made the total computational effort rather large despite using reasonable DFT levels (B3LYP and VSXC functionals). While aiming, as before, for suggesting new anion structures, this study had the distinct advantage that the LiTDI salt already had been made and tested as a battery electrolyte component,54 which could be used to "correct" the likely underestimated stabilities of oxidation for a few other anions. The finding that even minor alterations, such as the exact positioning of a substituent, could alter the ion-ion interactions significantly is very valuable, especially as these anions, due to their larger size and more substituents, would require increased synthesis efforts for each anion targeted.
Figure 4. The Hückel anion families studied in Ref. 42 exemplified by two members: TDI and PTB.
Download figure:
Standard image High-resolution imageQuite a different approach was taken in the next computational study on Hückel anions43—targeting a single anion and lithium salt—the by now increasingly popular LiTDI, but using a range of methods to accurately account for the solvation, using both implicit (C-PCM) and explicit solvent molecules (acetonitrile and water). The latter study was called for due to the nature of LiTDI forming a stable hydrate. The results were comprehensively compared to Raman spectra collected for different concentrations of LiTDI, solvents including PEO as a SPE system. Again, as in Refs. 54 and 39, the rather simple ion-pair models accounted largely accurately for both the correct site of interaction, a bidentate coordination of a ring nitrogen atom and a fluorine from the –CF3 group, as well as the resulting spectral changes—qualitatively. All solvated models, however, showed the lithium cation interaction with the –CF3 group to loose significance and more or less a monodentate Li-TDI coordination to result—and this came with a far better correspondence to all the experimental Raman spectra. The study subsequently went to quite some effort to also quantitatively assess the use of both implicit and explicit solvation models. Strengths and weaknesses were noted for each and that combining them only made for more complex computations, but no better interpretation or quality. The resulting recommendation was to primarily apply implicit solvation models, unless a certain ion-solvent interaction is to be studied.
Building on the expertise gained in modeling the solvation details and spectroscopic assessments, all fluorine-free electrolytes were created by employing e.g. the LiTADC salt in PEGDME or PC in a PAN matrix.44 This study was very applied; the characterization and testing was towards ion conductivity and impedance, phase and thermal stability, stability vs oxidation and aluminum corrosion, and practical cell tests in Li/LiFePO4 cells—including capacities and coulombic efficiencies. While many measures were promising, the final recommendation was that alternative salt and solvent combinations were needed to be explored from a performance point of view.
Almost like a side-note, quite a few of the Hückel family anions from Ref. 40 were used in a general computational benchmarking study of ESWs for WCAs.45 One crucial advantage used here was that these Hückel anions have rather few atoms and electrons in their structures and thus a comparatively low computational cost. Yet, the full CCSD(T)/CBS level of computation was beyond the limits of the resources available for these anions—but they could be used to create a wider set of WCAs in an approximate approach coined ΔCBS.
Finally, the unique features of the TDI anion, rigid and semiplanar, was contrasted with the omnipresent TFSI anion, flexible and elongated, in a study to outline why the speciation turns out quite differently for very similar systems.46 The observation is that for high concentrations the LiTDI salt can create two different ionic species in systems where the LiTFSI salt would create only one.55 This feature was probed by a combined DFT and MD study of the two salts in two different glymes, G3 and G4, at different concentrations up to those possible to form solvated ionic liquids (1:1 ratio salt:solvent). The ion-ion coordination details here depend on the force-field employed in the MD simulations, but it is clear that the monodentate and rigid ligand nature of the TDI anion dictates the formation of polyanion species formation, in stark contrast to the multidentate and flexible TFSI anion. Also the "second site" of the TDI anion, seems to assist in the formation of polyanions—something only occurring at the highest concentrations (Fig. 5). The "side-effect" of this is the creation of "free" lithium cations—and hence the disproportionation of these systems is not only explained, but also a promising way to design for fast lithium ion transport.
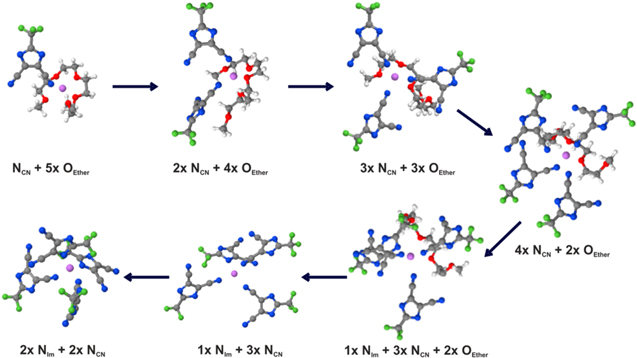
Figure 5. The suggested mechanism of polyanion species creation in Ref. 46. Reprinted with permission from P. Jankowski, M. Dranka, W. Wieczorek, and P. Johansson, J. Phys. Chem. Lett., 8, 3678 (2017). Copyright (2017) American Chemical Society.
Download figure:
Standard image High-resolution imageIn all, the early modeling approach to a few selected Hückel anions was used to look at very small details, but these studies and results have subsequently been enlarged, generalized and found their importance in the design of new anion structures. This goes along with initiating also rather practical studies of novel battery electrolytes including the most recent ones where the charge carrying species can be modified only by the appropriate combination of salt, solvent and concentration. We foresee that more computational studies on Hückel anions can continue to open paths for better electrolytes—be it for Li-ion batteries or other applications e.g. next generation batteries, supercapacitors, etc.
Implementation of the New Salts for Industrial Purposes and International Scientific Cooperation
In the middle of the 1990's, in response to the industrial demand, one of us (MA) proposed the synthesis of a new type of salts based on Hückel anions, which comprises an aromatic skeleton. This concept was then strongly supported by the molecular modeling (see above) performed early on at Chalmers and by the synthesis efforts at Warsaw University of Technology (WUT). Among possible Hückel anions azoles would be advantageous as they only contain carbon and nitrogen atoms. Among heteroatoms that can be members of an aromatic ring, nitrogen atoms interact only weakly with lithium cations. The first salt obtained that was designed in such a way was the LiTADC, used in SPEs.47,56 Subsequently, by making use of the previously developed synthesis route for cyclization of imidazolide57 anions, the LiTDI salt (lithium 4,5-dicyano-2-(trifluoromethyl)imidazolate) was made at large scale.58 In addition to this, other variants were made: LiPDI (lithium 4,5-dicyano-2-(pentafluoroethyl)imidazolate) and LiHDI (lithium 4,5-dicyano-2-(n-heptafluoropropyl)imidazolate)58,59–61 (Fig. 6), using the same synthesis route (Fig. 7). The synthesis route used at the time was one-step and the whole synthesis was one-pot. Synthesis route had no high requirements towards dryness of atmosphere or precursors, and no rigorous requirement of purity for the latter, in contrast to other, commercially available, lithium salts. Unlike LiPF6, the new salts were stable in the presence of moisture. It is even possible to create water solutions of LiTDI, LiPDI and LiHDI. As they are able to complex water molecule at 1:2 ratio, the salts can even act as moisture scavengers in organic, aprotic electrolytes.
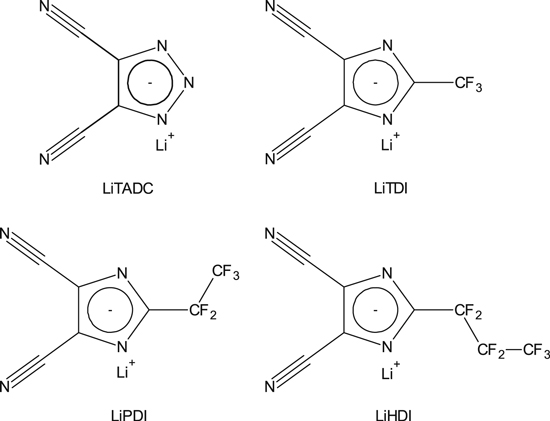
Figure 6. Synthesized lithium salts based on Prof. Armand's concept and in cooperation with WUT.
Download figure:
Standard image High-resolution image
Figure 7. LiTDI synthesis using the route developed at WUT.
Download figure:
Standard image High-resolution imageAll the above affects the salt manufacturing cost, which has a direct effect on the cell manufacturing cost. That the salts are stable in water may also affect the manufacturing costs: lower requirements for storage, handling, purity of other components and conditions for the cell manufacturing. It may also influence the production line costs by decreasing use of pure and dry inert gas for automated production or circulation and drying of dry air for manual assembly. To these manufacturing cost savings can also be added: moisture scavenging costs and energy for drying cell components before assembly.
The thermal stabilities of these salts are much improved vs LiPF6; they do not decompose before >250 °C, while LiPF6 undergoes decomposition already at ca. 70 °C. That is why fewer (or none) stabilizing additives are needed, and at the same time there is no need for HF or moisture scavengers. This is beneficial for the calendar life of the battery, as LiTDI is rigorously stable with the electrolyte, in contrast with LiPF6.
Optimization of Electrolyte Properties from the Viewpoint of Battery Application
Focusing on the LiTDI salt, the optimization of its electrolytes started with benchmarking using typical battery solvents and solvent mixtures; starting with ethylene carbonate with dimethyl carbonate (EC/DMC) 1:1 w/w ratio. 1 mol kg−1 electrolytes based on LiTDI or LiPDI showed lower ionic conductivities (ca. 6.5 mS cm−1) than those based on LiPF6 (10 mS cm−1). However, they exhibited higher lithium cation transference numbers—above 0.4, while for LiPF6 based electrolytes those are typically 0.2–0.3. These results were confirmed using PFG-NMR spectroscopy. The TDI and PDI diffusion coefficients were ca. 30% lower than that of PF6, while the lithium cation diffusion was similar (lower by ca. 10%). This was also an indirect proof of weaker cation-anion interactions, as predicted by the molecular modeling.
Preliminary tests showed compatibility of the new electrolytes with typical LIB electrodes with stable cycling using cells of graphite anodes and manganese oxide spinel cathodes. Electrolytes based on LiTDI and LiPDI were found to be more stable than those based on LiPF6 under the same conditions. While the latter exhibited the highest initial cell capacity (113 mAh g−1), it had a much faster loss of capacity and the initial cell capacities with the new electrolytes were very close: 110 mAh g−1. Also, the rate capability was more or less the same up to 1C, and with negligible differences for 2C. The LiTDI and LiPDI electrolytes were also electrochemically stable against platinum and aluminum electrodes from 0 to 4.8 V vs Li+/Li (as measured by means of CV).62
These first promising results suggested subsequent studies aiming at further optimization of electrolytes based on LiTDI. Initially, ca. 20 solvent mixtures were selected based on their low melting points and high ionic conductivities for other salts, with a focus on the lithium cation conductivity. EC was used as a base as it is the only highly polar organic carbonate used in LIB cells that is relatively cheap and compatible with graphite. EC was mixed with linear carbonates DMC, diethyl carbonate (DEC) and ethyl-methyl carbonate (EMC). Other co-solvents/additives were also used, such as dimethoxyethane (DME), fluoroethylene carbonate (FEC) and vinylene carbonate (VC).
For EC:DMC, EC:DEC, EC:DEC:DMC, EC:DEC:DME and EC:DMC:DME mixtures the two latter contain 4% DME additive, the highest content not causing serious growth of the SEI resistance (Table I). All electrolyte compositions were chosen to keep EC content high, which allows for keeping high ionic conductivity, but at the same time to keep an as low melting point as possible to fulfill requirements of sub-ambient operation. For EC:DMC the composition choice was easy, as mixture at 1:2 w/w ratio is eutectic, even though it does not exhibit a very low melting point (ca. −10 °C). Unfortunately, EC:DEC, EC:EMC and EC:DEC:DMC all have their eutectic compositions below 5% EC content.63 Based on the literature, non-eutectic mixtures were selected: EC:DEC 1:2 w/w ratio and EC:DEC:DMC at 1:1:1 w/w/w ratio. This ensures that the EC content is relatively high—33 wt%—while the melting point is low—below 0 °C. Subsequent addition of the lithium salt to the solvent mixture decreased the melting point to the level required by the industry.
Table I. Electrochemical parameters of the selected electrolyte compositions developed during EuroLiion project and those of commercial electrolytes for comparison. Conductivities measured at 20 °C.
| Electrolyte | c/mol kg−1 | σ/mS cm−1 | t+/− | σLi+/mS cm−1 |
|---|---|---|---|---|
| LiTDI in EC:2EMC | 0.3 | 4.2 | 0.54 | 2.3 |
| LiTDI in EC:2EMC | 0.7 | 4.9 | 0.52 | 2.5 |
| LiTDI in EC:DMC:EMC | 0.4 | 4.7 | 0.46 | 2.1 |
| LiTDI in EC:DMC:EMC | 0.7 | 5.5 | 0.43 | 2.4 |
| LiTDI in EC:DEC:EMC | 0.5 | 4.4 | 0.44 | 1.9 |
| LiTDI in EC:DEC:EMC | 0.8 | 5.9 | 0.50 | 2.9 |
| LiTDI in EC:DEC:DMC | 0.7 | 5.0 | 0.50 | 2.5 |
| LiPF6 in EC:DEC:DMC | 1 | 10.1 | 0.24 | 2.5 |
| LiTDI in EC:2DMC | 0.3 | 5.1 | 0.62 | 3.2 |
| LiTDI in EC:2DMC | 0.6 | 5.7 | 0.55 | 3.2 |
| LiPF6 in EC:2DMC | 1 | 10.1 | 0.35 | 3.5 |
The LiTDI-based electrolytes were investigated in a broad salt concentration range and a constant increment (≤0.1 mol kg−1), resulting in relatively high ionic conductivities already at low concentrations. The ionic conductivity maximum for the EC:DMC 1:2 w/w electrolytes is 5.7 mS cm−1 and is reached at 0.63 mol kg−1. However, 5.1 mS cm−1 is reached at a concentration as low as 0.31 mol kg−1. This result is advantageous compared to other salts, such as LiPF6, LiBF4 or LiTFSI, which require 1 mol kg−1 to reach their ionic conductivity maximum and drop quickly with the concentration.4,64 Similarly, high ionic conductivities at low concentrations of LiTDI salt were reached for other solvent mixtures, such as EC:DEC. Altogether, this means, that the lithium salt cost using LiTDI can be saved by down to one-third. This makes a considerable difference, as for LIB cell component cost calculations the salt share of the electrolyte manufacturing costs is 50%–75%. The exact share depends mostly on solvents used and additives chosen (no HF or moisture scavengers are required in case of LiTDI use). Depending on the cell size, the electrolyte share of the total cell component cost is ca. 5%–15% and the salt itself 3%–12%.
From the initial optimization study, it was concluded that addition of DME did not considerably increase the ionic conductivity of the EC:DMC system. DME addition worked better for the EC:DEC system, because unlike DMC, DEC has a much higher viscosity than DME.
The lithium cation transference numbers of LiTDI-based electrolytes are higher than for the LiPF6-based electrolytes. The lithium cation conductivity showed that despite lower total ionic conductivities over the whole concentration range, the electrolytes exhibit very similar lithium cation-only conductivities. The same is true for other solvent mixtures, for instance EC:DEC and EC:DEC:DMC.65
For high power applications (e.g. automotive/traction) 0.3 mol kg−1 electrolytes cannot be used, but the ∼0.6 mol kg−1 electrolytes work and they still have advantage of lower manufacturing cost. It also does not preclude the use of lower salt concentrations in cells for other applications, such as high energy density.
Another optimization was aimed at low temperature operation, focusing on the mixtures containing EMC that decreases the melting point.66 Five solvent mixtures with EMC contents of 33%–66% provided ionic conductivities of 2 mS cm−1 at −20 °C, and the best exhibited 3.5–4.5 mS cm−1 at 20 °C. Again, it was possible to obtain electrolytes based on LiTDI having the same lithium cation conductivity as those based on LiPF6.
Parameters of selected compositions yielded during optimization investigations and two commercially available electrolytes for comparison are gathered in Table I.
Examples of Successful Cycling of LiTDI in Half and Full Cells
Over the last 10 years since LiTDI salt was introduced to the scientific field, it has been the object of multiple tests of its properties, but also of its compatibility with other cell components. As aluminum corrosion tests were made simultaneously with the introduction of the salt, the remaining most important tests for practical purposes were those made vs active electrode materials. Various groups made experiments vs both anodes and cathodes. In the last 5 years, also full cells with LiTDI-based electrolyte have been cycled and their performance reported.
As for the anodic materials (Table II), graphite was cycled first, as it is still the most common material used in LIB cells. Pure 1 mol kg−1 LiTDI in EC:DEC (3:7 v/v) had worse rate capability performance than LiPF6 with standard additive (VC) in the same solvent mixture. However, the performance was leveled when the LiTDI-based electrolyte used the same additive—it exhibited then the same rate capability—up to 10C.67 In other tests with EC:DMC (1:1 w/w) or EC:GBL:MP solvent mixture (ethylene carbonate: γ-butyrolactone : methyl propionate; 1:1:1 w/w/w), both with FEC as additive, LiTDI had at least the same rate capability and comparable capacity retention over longer cycling (tests up to 10C).68
Table II. Anode materials tested in half-cells vs LiTDI in various solvent mixtures.
| Anode active material | Solvent mixtures |
|---|---|
| Graphite | EC:DEC with and without FEC/VC,67 |
| EC:DMC with and without FEC/VC,68 | |
| EC:GBL:MP with FEC68 | |
| LTO | EC:DEC67 |
| Si/C composite | EC:DMC,69 |
| EC:DMC with and without FEC/VC70 | |
| Silicon (pure, nano, etc.) | EC:DMC with and without FEC/VC,71,72 |
| EC:DMC:10%FEC:2%VC73 | |
| Sb2O3/C composite | EC:DMC74 |
Another anodic active material used in LIB cells on the industrial scale, LTO (spinel lithium titanate, Li4Ti5O12) has also been tested for compatibility with LiTDI-based electrolytes. Up to 10C, rate capability of 1 mol kg−1 LiTDI-EC:DEC (3:7 v/v) electrolyte both with and without additives (2% of VC or 2% of FEC) was the same as that of LiPF6 in the same solvent mixture with 2% VC additive up to 10C. It kept over 80% of nominal capacity up to 12C. Above 10C—up to 40C, LiTDI without additives started to perform worse, but with additives worked at least the same as LiPF6.67
Other very promising anode materials, tested widely for over 10 years, are silicon and graphite-silicon composites. Silicon-carbon composites with high silicon content (50%) were the object of LiTDI electrolyte compatibility tests using different composite precursors (composites produced using MPCVD). In half-cells the 0.63 mol kg−1 LiTDI-EC:DMC (1:2 w/w) electrolyte without additives showed over 800 mAh g−1 capacity and had a retention of 80% after 300 cycles,69 and compared to LiPF6, showed the electrolytes with additives (5% VC, 1% VC + 5% FEC or 2% VC + 10% FEC) to perform much better than LiPF6-based electrolytes with additives (2% VC + 10% FEC) or without. The best initial capacities were 437 mAh g−1 for LiPF6 with additives vs 807 mAh g−1 for LiTDI without additives and 630 mAh g−1 with additives. Capacity retention after 150 cycles was 197 mAh g−1 for LiPF6 with additives, 874 mAh g−1 for LiTDI without additives, and 882 mAh g−1 for LiTDI with additives. Capacity retention after 500 cycles for LiTDI with additives was 878 mAh g−1.70
Nanosilicon was also tested extensively as anodic active material in a few publications.71–73 LiPF6 causes silicon to react with always present in LiPF6 traces of HF, blocking the silicon anode with reaction products. Thus, the first study tested if LiTDI is compatible with silicon, which was confirmed after an in-depth analysis of the SEI composition,71 and stable cycling at 1200 mAh g−1 for at least 300 cycles with 0.6 mol kg−1 LiTDI in EC:DMC (1:2 w/w) with 10% FEC and 2% VC additives.72,73
A 0.75 mol kg−1 LiTDI in EC:DMC (1:1 w/w) electrolyte was also tested with a nanostructured composite of antimony oxide and carbon showing good compatibility and a capacity over 400 mAh g−1.74
Many common cathode materials were tested (Table III)—lithium cobalt oxide (LiCoO2, LCO), lithium manganese oxide (LiMn2O4, LMO), carbon coated lithium iron phosphate (LiFePO4, LFP) and mixed nickel manganese cobalt oxide (LiNi1/3Mn1/3Co1/3O2, NMC111 and LiNi0.5Mn0.3Co0.2O2, NMC532). In NCM532, the LiTDI in EC:DEC (1:1 v/v) electrolyte proved to be as good as LiPF6 in EC:DEC (1:1 v/v) under standard conditions.75 However, at higher temperatures (60 °C) the LiPF6-based electrolyte ceases to work and quickly deteriorates, while the LiTDI-based electrolyte is stable, even gaining some capacity. NMC111 vs LiTDI in EC:GBL:MP (1:1:1 w/w/w) showed cycling stability and capacity retention comparable to a LiPF6-based electrolyte.68 In rate capability tests LiPF6- and LiTDI-based electrolytes (EC:DEC 3:7 v/v) performed similarly with NMC111 and LMO (with a slightly lower capacity for the LiTDI-based electrolyte above 10C). During cycling with LFP cathode active material LiTDI rate capability was inferior to that of LiPF6 above 1C, but with LCO LiTDI performed better above 0.3C.76 Also, a 0.6 mol kg−1 LiTDI electrolyte performs much better than 1 mol kg−1, both during long cycling and during rate capability tests. Practical tests also have shown that lower concentration and total ionic conductivity of LiTDI electrolytes is not the barrier to compete with LiPF6. This has been attributed to the much higher lithium cation transference number in LiTDI-based electrolytes, increasing the lithium cation conductivity to the same level as the LiPF6-based electrolytes.
Table III. Cathode materials tested in half-cells vs LiTDI in various solvent mixtures.
| Cathode active material | Solvent mixtures |
|---|---|
| NMC532 | EC:DEC75 |
| NMC111 | EC:GBL:MP,68 |
| EC:DEC76 | |
| LFP | EC:DEC76 |
| LCO | EC:DEC76 |
| LMO | EC:DEC76 |
| Li2FeSiO4 | EC:DEC77 |
| Sulfur | DOL:DME78 |
Also Li2FeSiO4 was tested for compatibility vs LiTDI in EC:DEC (1:1 v/v).77
Full cells LTO∣LFP and LTO∣NMC assembled using a 1 mol kg−1 LiTDI-EC:DEC (3:7 v/v) electrolyte (Table IV), were very stable and long running. Comparisons were made to cells using a LiPF6 in EC:DEC (3:7 v/v) + 2 wt% VC electrolyte and after 450 cycles the LTO∣NMC cells had practically the same (low) capacity fading (within 0.5%) and the same high coulombic efficiency. For the LTO∣LFP cells 900 cycles gave close to 100% coulombic efficiency and similar capacity retentions: 87.6% and 85.4% for the LiPF6− and LiTDI-based electrolytes, respectively.67
Table IV. Electrode materials tested in full cells vs LiTDI in various solvent mixtures.
| Full cell electrode active materials | Solvent mixtures |
|---|---|
| graphite∣NMC | EC:GBL:MP68 |
| LTO∣LFP | EC:DEC67 |
| LTO∣NMC | EC:DEC67 |
Using LiTDI-EC:GBL:MP (1:1:1 w/w/w) in graphite∣NMC cells showed that LiTDI can compete with LiPF6 for long and fast cycling with standard electrodes. It has also been shown that LiTDI-based electrolytes are stable when cycled for 20–30 cycles at 1C at low (−25 °C) and high temperatures (60 °C) in contrast to LiPF6-based electrolytes.68
Finally, LiTDI has been also tested as a stabilizing additive for LiPF6-based electrolytes (Table V). LiTDI addition effectively decreased the deterioration rate of the electrolyte and cell by scavenging trace moisture and HF. Also, LiTDI have been able to increase LiPF6-based electrolyte stability when cycled in both half- and full cells above 50 °C. The stability at high temperatures is not as good as for a LiTDI-based electrolyte, but outperforms standard LiPF6-based electrolytes, even with FEC or VC additives.79–81 Addition of LiTDI also extends cell life. In NMC half-cells even 1% addition of LiTDI increases cycle life; 80% of the initial capacity remain after 1000 cycles as compared to after 200 cycles for LiPF6-based electrolytes without LiTDI.79 Apart from better performance at high temperatures and longer cycle life, a LiTDI additive also decreases the SEI layer resistance and voltage hysteresis growth rate (over 1000 cycles).
Influence of Electrolyte Structure on the Conductivity Mechanism in Both Liquid and Solid Systems
In the section describing the molecular modeling, the importance of the unique structure of Hückel type salts for the ion transport mechanism in electrolytes (both liquid and polymeric ones) was highlighted. The extensive studies on the dissociation of the salts with Hückel anions in anhydrous aprotic solvents led to the identification of typical structural motifs present in liquid and polymer electrolytes.54,55 The simplified representation of aggregation process is shown in Fig. 8. The first step is formation of ion-pairs (Fig. 8b). As seen by modeling the imidazole and triazole anions preferably use the ring nitrogen atoms to interact with the lithium cation, while it is the nitrile groups for tetracyanopyrrolide. Increasing salt concentration leads to the formation of dimers, which may rearrange into chains (Fig. 8c). The further aggregation process is different for lithium and sodium salts and also depends on the type of solvent. In lithium based systems containing simple solvents with one donor group, such as tetrahydrofuran (THF) or organic carbonates, increased salt content results in precipitation of highly aggregated solvates, with all anions representing the same type of coordination. The mechanism changes in solvents with chelating properties, such as glymes, with a tendency to disproportionate. Crystalline solvates of LiTDI with shorter glymes (G1, G3) have in their structures two types of cationic species: coordinated exclusively by anions and coordinated by solvent molecules and anions. If the length of the chain is sufficient to fully coordinate cations, as for tetraglyme (G4), the disproportionation goes further and leads to the formation of a solvate with one part of cations complexed solely by solvent molecules and other by anions alone (Fig. 8d). Such a complex, e.g. the solid electrolyte [Li2(G4)22+][Li4TDI62−], is characterized by high conductivity in solid state (1.8 × 10−5 S·cm−1, 30 °C) and an exceptionally high lithium transference number (tLi+ = 0.82) at 30 °C.55
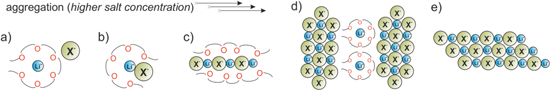
Figure 8. Aggregation modes in concentrated and solid-state electrolyte systems, (a) model of "solvate" ionic liquids, good t+, (b) ionic contact pairs, moderate t+, (c) chains or solvated aggregates, low t+, (d) disproportionated system with solvated cations and aggregated polyanions, very high t+, (e) higher aggregates, very low t+.
Download figure:
Standard image High-resolution imageIt should be stressed that there is a major difference with respect to the systems composed of lithium non-heterocyclic salt systems based on imidate anions such as TFSI. For concentrated LiTFSI solutions with glymes of appropriate chain lengths it is possible to obtain solvated ionic liquids as shown by the team of prof. Watanabe.82,83 These liquid electrolytes of high conductivity possess in their structure lithium cations fully solvated by glyme molecules and uncoordinated free anions. For LiTDI-glyme systems it is possible to go further with concentrations of salt to obtain not liquid but solid-state electrolytes having benefits of fully solvated lithium cations. The other advantages of disproportionation mechanism occurring in solid heterocyclic systems is an immobilization of anions resulting from the formation of an aggregated anionic sub-network in form of aggregated polyanions, which increases the lithium cation transference number.
A similar concept incorporating the formation of aggregated polyanions and fully solvated lithium ions to improve cation transport properties can be induced for ternary systems based on ionic liquids (ILs) with TDI anions.
The addition of the LiTDI-glyme solvate to the ternary systems based on ILs with TDI anions improve their lithium transport properties.84,85 Electrolytes based on mixtures of LiTDI and ILs: XMIm+TDI−(XMIm+ = 1-alkyl-3-methylimidazolium cation, where X stands for alkyl, E for ethyl, P for propyl, B for butyl group) were only partially successful, because of low miscibility, instability, and finally low transference numbers (tLi+ = 0.04). In particular, poor lithium conductivities were associated with the creation of a 3 dimensional structure at high salt contents. A crystalline complex where lithium cations and imidazolium anions create chain-shaped [Li(TDI)2]nn− polyanions, surrounded by XMIm+ cations, is a very good conductor of imidazolium anions, but not of lithium cations. The desirable charge carriers are trapped resulting in low ionic conductivities, tLi+ and high local viscosity. Increasing the salt content only leads to higher aggregation, where almost all lithium cations are permanently immobilized. Ternary systems composed of LiTDI-G4 solvates and IL avoid this disadvantage. Raman studies of such electrolytes reveal, that an addition of glyme modifies the electrolyte's structure, but does not simply lead to the formation of free anions or ion-pairs (Fig. 9). Instead, for the highest (1:1) LiTDI:G4 ratio the spectrum resembles the spectrum of a molten LiTDI-G4 complex, while the spectra of lower LiTDI:G4 ratios points to formation of different types of solvates, ion-pairs and free ions. The presence of both fully solvated cations or dications and aggregated lithium centered polyanions are responsible for the increased lithium transference numbers observed for the systems with higher salt contents.
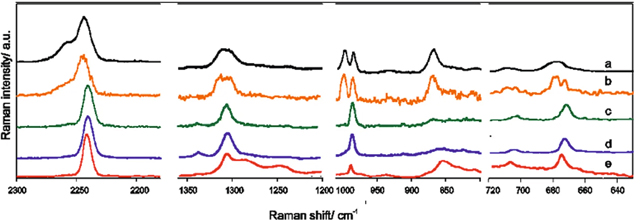
Figure 9. Raman spectra of samples: (a) molten LiTDI-G4; (b) 0.1[LiTDI·4.5(G4)]:0.9BMImTDI (subtraction spectrum); (c) 0.1[LiTDI·2.0(G4)]:0.9BMImTDI (subtraction spectrum); (d) 0.1[LiTDI·1.0(G4)]:0.9BMImTDI (subtraction spectrum); (e) BMImTDI. Reprinted from E. Karpierz, L. Niedzicki, T. Trzeciak, M. Zawadzki, M. Dranka, J. Zachara, G. Z. Żukowska, A. Bitner-Michalska, W. Wieczorek, Scientific Reports, 6, 35587 (2016). Reference 84 CC BY 4.0.
Download figure:
Standard image High-resolution imageThe disproportionation also takes place in semi-crystalline PEO-based SPEs86,87 with O/Li (Na) ratios of 15–20. Increasing the salt content, O/M ≤12, results in formation of crystalline PEO-salt complexes. Based on previous studies correlating structural and spectroscopic data, the lithium and sodium cations seem surrounded by TDI anions and EO units are likely to maintain a coordination number of 6 and 8, respectively. In LiTDI-PEO systems there are two distinct polymer-salt complexes, having anions representing different types of salt coordination; TDI anions interacting with Li+ by a ring nitrogen atom and a fluorine atom and the remaining four coordination sites being oxygen atoms of PEO, or each lithium cation being linked to a ring nitrogen atom, a fluorine atom, and also a nitrile nitrogen of an another TDI anion, leaving only three coordination sites available for PEO oxygen atoms. The former thus is a contact ion-pair (CIP), and the latter an ionic chain.
The same type of coordination was found in the NaTDI-PEO system,88–90 but additionally, for the most diluted electrolytes, a third type of complex with anions decoupled from the cations is formed (SSIP). Melting temperatures of the phases having the TDI in ion-pairs are either lower (40 °C) or comparable (70 °C) to the melting temperature of the crystalline phase of PEO (65 °C), while the aggregated chain-type complexes melt above 100 °C. As a result, the thermal dependence of the conductivity of the SPEs exhibits several jumps (Fig. 10).

Figure 10. Influence of the phase composition on the conductivity of the samples with O/Li equal 20 (red) and 6 (purple). Reprinted with permission from P. Jankowski, G. Z. Żukowska, M. Dranka, M. J. Marczewski, A. Ostrowski, J. Korczak, L. Niedzicki, A. Zalewska, W. Wieczorek, J. Phys. Chem. C, 120, 23358 (2016). Copyright 2016 American Chemical Society. Reference 87.
Download figure:
Standard image High-resolution imageIn the more concentrated system, the chain-type phase partially dissolves during heating, above the melting point of the CIP phase, but the systems remain partially crystalline. On the other hand, melting of the crystalline phase of PEO in the more dilute systems leads to an increased salt dissociation in the amorphous phase. For the concentrated system, melting of the CIP phase results in a release of ion-pairs and a decreased percentage of mobile charge carriers in the amorphous phase. A particularly interesting effect was found for O/Li = 6, with three jumps in the conductivity. The distinct increase in conductivity at 60 °C is due to the melting of the PEO phase, which, however, was not present at room temperature, but likely a consequence of disproportionation above 40 °C, after the melting of the CIP phase, and leads to the formation of both crystalline PEO and chain-type phases.
The melting of SSIP-PEO-NaTDI, or a eutectic mixture thereof, takes place at relatively low temperature, ca. 30 °C–40 °C, and the melting of CIP-PEO-NaTDI or PEO is preceded by partial dissolution in the amorphous phase. As a result, only one distinct jump in the conductivity between 50 °C and 70 °C is observed, and this corresponds to overlapping melting of crystalline phases.89
To summarize it has been shown that previously described molecular modeling studies not only help to design and synthesize novel families of Hückel type salts, but also tend to correspond very well to the experimental structures and spectroscopic findings, shedding light on ionic transport mechanisms.
Application of Hückel Salts in Post Lithium-ion Batteries
Sodium-ion battery electrolytes
Currently, the dominant battery technology for portable devices and electric vehicles is LIBs. However, the sources of lithium are not sufficient to meet all needs, and the price of lithium is increasing.91 This makes it necessary to search for new technologies that can replace or supplement LIBs—next generation batteries (NGBs). A natural alternative to replace lithium seems to be sodium due to its availability and low price.
The development of sodium-ion batteries (SIBs) began simultaneously as for LIBs in the early 70 s and 80 s, but the success of the LIB chemistry made research on SIBs less of interest. Comparison of the sodium ion size (1.02 Å) to the lithium ion size 0.76 Å, as well as its lower polarity creates significant differences in transport properties. The above differences also have consequences for coordination, crystal structures and lattice constants. These differences will have an impact on the charge transfer at the electrode-electrolyte interface. It has been proven that the desolvation energy is about 25%–30% less for sodium than for lithium92,93 and this should have a positive effect on the kinetics.
A very important perspective of the work on the Hückel anions was the development of their sodium salts (and SIB electrolytes) (Fig. 11)94:
- sodium 4,5-dicyano-2-trifluoromethylimidazolate (NaTDI),
- sodium 4,5-dicyano-2-pentafluoroethylimidazolate(NaPDI),
- sodium pentacyanopropenide (NaPCPI),
- sodium 2,3,4,5-tetracyanopyrrolate (NaTCP),
- sodium 2,4,5-tricyanoimidazolate (NaTIM).

Figure 11. Structures of anions (other than TDI and PDI) for sodium ion electrolytes. Reference 94 CC BY 4.0.
Download figure:
Standard image High-resolution imageThese salts showed promising properties from the point of view of application i.e. excellent thermal stability and high electrolyte conductivities: 0.75 mol kg−1 NaTDI (EC/DMC): 11.9 (mS cm−1); 0.75 mol kg−1 NaPDI (EC/DMC): 6.0 (mS cm−1), and a very wide ESW >4.5 V vs Na).
Together with electrodes made by tape casting and MPCVD cells were made, SbxOy/C ∣∣ Cathode, and the anode materials showed very good capacity (425 mAh g−1)74 and stability over 100 cycles using a LiTDI electrolyte and the cathode materials showed good capacity ∼100 mAh g−1 using NaTDI and NaPDI electrolytes (Fig. 12).95
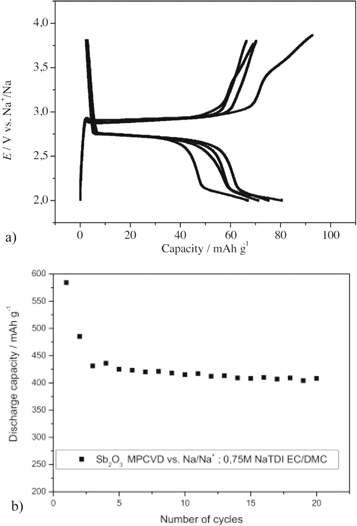
Figure 12. Examples of compatibility of sodium electrolytes. (a) Cathodic. Voltage vs capacity graph of NaxCoO2 ∣ 0.75 mol kg−1 NaTDI-EC:DMC (1:1) ∣ Na half-cell at C/10 rate,95 Reprinted from Electrochimica Acta, vol. 222, A. Bitner-Michalska, A. Krztoń-Maziopa, G. Żukowska, T. Trzeciak, W. Wieczorek, and M. Marcinek, "Liquid electrolytes containing new tailored salts for sodium-ion batteries," p. 108, Copyright (2016) with permission from Elsevier. (b) Anodic Discharge capacity of the SbxOy/C thin-film nanocomposite anode and 0.75 mol kg−1 NaTDI EC/DMC during long-term cycling at room temperature at C/10 rate.74 Reprinted from Electrochimica Acta, vol. 210, A. Bitner-Michalska, K. Michalczewski, J. Zdunek, A. Ostrowski, G. Zukowska, T. Trzeciak, E. Zero, J. Syzdek, and M. Marcinek, "Microwave Plasma Chemical Vapor Deposition of SbxOy/C negative electrodes and their compatibility with lithium and sodium Hückel salts-based, tailored electrolytes," p. 395, Copyright (2016) with permission from Elsevier.
Download figure:
Standard image High-resolution imageThe sodium salts were also applied in SPEs using hot-pressing to eliminate the use of solvents from the production, minimizing cost and improve environmentally friendliness. SPEs of NaTCP and NaTIM in PEO, O/Na = 10–50 showed maximum conductivity at n = 16, and for NaPCPI, the maximum conductivity was obtained for n = 20 at 90 °C (Fig. 13). Overall, NaTCP is the most promising salt for SPEs rendering very good mechanical properties and high ionic conductivity, for 16:1 it is >1 mS cm−1 at 70 °C.94,96
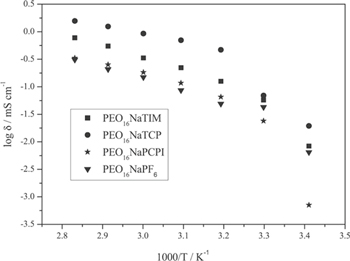
Figure 13. Comparison of conductivity of PEO based sodium electrolytes.94 CC BY 4.0.
Download figure:
Standard image High-resolution imageRecent progress, practical issues, and future perspective of Na-ion systems clearly indicates they should be considered as a promising alternative. However, the similar chemistries of lithium and sodium should not be interpreted as identical.97
Multivalent battery electrolytes
Another group of battery technologies emerging as serious candidates for NGBs are those based on multivalent cations like calcium, magnesium and aluminum.98–100 At present the most advanced of these are magnesium based batteries. Magnesium ions are divalent, the metal anode has a high capacity per volume and furthermore, and very important from an economy point of view, is commonly found in the Earth's crust and relatively cheap.101–103 The possibility of electrochemical deposition of metallic magnesium without dendrite formation has been demonstrated and in addition, magnesium is less reactive than lithium, and thus magnesium metal batteries will be safer.
Efficient batteries with metallic magnesium and aprotic electrolyte have been already demonstrated in early 2000, but issues remain104 for the mechanism of formation, structure and properties of the passivation layers on magnesium metal, the transport mechanism and the structure of magnesium ions complexes in the electrolyte, the electrochemical stability of the electrolytes, the process at the electrolyte—cathode interface as well as intercalation and the mechanism of magnesium ion transport in cathode materials.102,103
Two different magnesium salts based on Hückel anions have been tested, mainly for proof of concepts. As conventional magnesium salts, such as halides, perchlorates or imides seem to be incompatible with the magnesium metal surface, the use of Hückel anions may offer new possibilities.
The first electrolyte is based on (ƞ5-cyclopentadienyl) magnesium, also known as magnesocene (MgCp2, Mg(C5H5)2) dissolved in the tetrahydrofuran (THF).105 The cyclopentadienyl anion (C5H5−) possesses six π electrons as well as planar structure and form sandwich type complexes with metal cations (Fig. 14). The 0.5 mol kg−1 MgCp2/THF electrolyte made possible reversible magnesium plating and stripping with low over-potentials. The main drawback is the low oxidation stability of the electrolyte; depending on the electrode material ca. 1.4–1.6 V vs Mg/Mg2+, which of course limits its potential application to only low voltage cells like with Mo6S8 Chevrel phases.
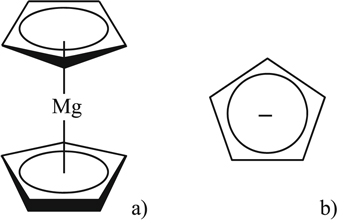
Figure 14. Scheme of (a) (ƞ5-cyclopentadienyl)magnesium (magnesocene, MgCp2, Mg(C5H5)2); (b) cyclopentadienyl anion (C5H5−).
Download figure:
Standard image High-resolution imageThe second electrolyte system is based on MgTDI2 dissolved in dimethyl formamide (DMF),106 which shows good electrochemical stability and highly reversible reduction and oxidation reactions suggesting intercalation and deintercalation of magnesium into natural graphite.
Lithium-sulfur battery electrolytes
Another NGB technology where Hückel anion based salts find application are lithium-sulfur (Li-S) batteries. In 2009 that there was a significant breakthrough for Li-S batteries, with a high theoretical capacity of 1672 mAh g−1, due to the use of porous carbon materials.107–111 Commercialization of Li-S battery cells is, however, laden with several problems. One of the biggest issues is the capacity fading, which can be directly linked to continuous reactions between soluble polysulfides and Li metal (polysulfide shuttling mechanism), which leads to the formation of an insulating layer on the anode.107–109 This pushes researchers to find ways to reduce the diffusion of the polysulfides between electrodes, mainly new electrolytes or by using specially designed membranes. Today the most popular electrolytes are based on LiTFSI dissolved in dioxolane and dimethoxyethane (DOL:DME) 1:1 v/v mixture.
The first implementation of electrolytes with Hückel anion based salts in Li-S batteries was reported by Dominko et al.,112 aiming to explain the reaction mechanism during operation. The electrolyte salt was changed from LiTFSI to LiTDI for mainly two reasons: i) removing a sulfur-containing species (TFSI) from the electrolyte, and ii) curiosity on advantages of LiTDI vs LiTFSI. The electrochemical cycling showed the LiTDI-based electrolyte to be similar to the LiTFSI-based electrolytes, and thus the same reaction mechanism.
LiTDI in Li-S battery electrolytes was more deeply investigated in two other reports.113,78 It was shown that interactions with polysulfides may be controlled by the salt anion113 and diffusion coefficients of each of the components of several Li−S electrolytes using different anions (FSI, OTf, TFSI, and TDI) were correlated with cell performance. The various ion−ion and ion−solvent interactions influence the anion mobility and thereby the performance, rendering better cell performance for the TDI-based electrolyte, as the TDI anion has lower mobility and weaker interactions with the lithium polysulfides.
In another work, LiTDI was used as a supporting electrolyte salt to effectively decrease the polysulfide solubility and thereby minimize the polysulfide shuttle mechanism.78 AIMD simulations revealed that the TDI anion coordination affects the polysulfide disproportionation to form Li4S8 dimers and reduces solubility. Furthermore, with the incorporation of 0.1% Li2S8 and 0.2 mol kg−1 LiNO3 additives, excellent performance was obtained using the LiTDI-based electrolyte; 300 cycles with a residual discharge capacity of 600 mAh g−1.
The industrialization and commercial development of Hückel anions based salts
In 2010, Arkema started a collaboration with Prof. Michel Armand on what was at the time a promising new fluorinated lithium salt: LiTDI. Arkema is a specialty chemical and advanced materials company. The development of a new lithium salt like LiTDI fitted in the diversification strategy of the fluorochemicals business unit. The purpose of this collaboration was to develop an industrial process and to investigate the advantages of LiTDI. Starting from lab scale synthesis, Arkema developed a new process that eliminates the impurities responsible for electrochemical performance degradation. This new process has been successfully upscaled in a 400 L reactor to make several kg of LiTDI per batch to start the commercial development.
Moreover, thanks to internal tests and customers' feedback, several LiTDI benefits have been demonstrated, especially the use of LiTDI is used as an additive. First, LiTDI improves upon the SEI stability obtained with traditional carbonates or SEI additives (FEC, VC and others). LiTDI also decreases the irreversible capacity and the SEI resistance. Table VI shows the resistance for a pouch cell (200 mAh, NMC622/Graphite) using a conventional electrolyte: 1 mol kg−1 LiPF6 in EC:EMC 3:7 v/v + 2 wt% FEC, and for a LiTDI-based electrolyte: 0.95 mol kg−1 LiPF6 + 0.05 mol kg−1 LiTDI in EC:EMC 3:7 v/v + 2 wt% FEC.
Table VI. Impedance of pouch cells (NMC622/Graphite) with different electrolytes after SEI formation by two cycles of 100% DOD at C/10.
| Electrolyte | Conventional | LiTDI-based |
|---|---|---|
| Cell impedance (Ω) | 0.357 | 0.076 |
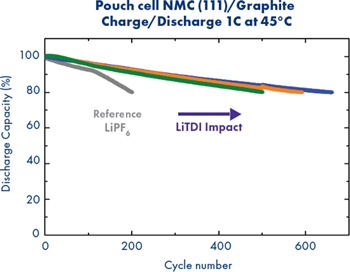
The addition of LiTDI decreases the overall cell resistance by playing an unknown role in the SEI formation: inorganic charge or co-polymerization. Moreover the presence of LiTDI in the SEI improves the lifetime, especially at high temperatures. In collaboration with Hydro-Québec, lifetime studies have been performed at pouch cell level (NMC111/Graphite) with different electrolytes with or without LiTDI (Fig. 15).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 15. Discharge capacity of pouch cell cycled with 1C cycling rate at 45 °C for different electrolytes: 1 mol kg−1 LiPF6 in EC/DEC 3/7 v/v +2 wt% of FEC (Grey), 0.95 mol kg−1 LiPF6 + 0.05 mol kg−1 LiTDI in EC/DEC 3/7 v/v + 2 wt% of FEC (blue), 0.9 mol kg−1 LiPF6 + 0.1 mol kg−1 LiTDI in EC/DEC 3/7 v/v + 2 wt% of FEC (orange) and 0.8 mol kg−1 LiPF6 + 0.2 mol kg−1 LiTDI in EC/DEC 3/7 v/v + 2 wt% of FEC (Green).
Download figure:
Standard image High-resolution image{kind=link}
The presence of an optimum LiTDI concentration markedly improves the lifetime at higher temperatures. LiTDI stabilizes the SEI on graphite, but also limits the degradation of LiPF6. Indeed, thermal tests on LiPF6-based electrolytes show a better stability in presence of LiTDI. Thanks to its cyano groups, LiTDI traps water and/or HF and as a result prevents LiPF6 degradation.
For Arkema, LiTDI is clearly a promising additive to improve the performance of LIBs, especially for those aimed at the electric vehicles market, by enabling faster charge and longer lasting batteries. The next step for Arkema will be the up-scaling of LiTDI production to several metric tons in order to start the commercialization.
Conclusions
The above paper describes the successful development of the new family of salts based on Hückel type anions. Starting from the pioneering idea of Prof. Armand through the molecular design and optimization of salt synthesis followed by structural characterization and studies of new electrolytes in variety of half and full cell systems the story ends with the commercialization of the first and better known member of the family—LiTDI. The product itself can be successfully used as main component of the LIB electrolyte or as a useful additive to the electrolyte formulation. The use of the Hückel type salts acting as water and HF scavengers results not only in the improvement of the performance of cells based on currently available technologies but also in the increase of safety and possibly considerable reduction in the battery production costs.114 As has been shown above Hückel type salts can be a very useful component of electrolytes for novel LIBs (e.g. silicon anode technology)70 as well as for post LIBs such as SIBs, Li-S and multivalent batteries. Moreover, thanks to the unique structure of the concentrated LiTDI electrolytes both in liquid and solid (polymeric) systems, new opportunities with utilization of this salt in water- or glymes-based electrolytes55 are wide open and are being currently explored. Finally, the commercialization step, which is no doubt the highlight of the story, results from multiple co-operations between scientists with variety of backgrounds representing both academia and industry as well as spanning several different countries.
Acknowledgments
The authors wish to emphasize that the distinct part of the presented work was carried out in the frame of multilateral collaboration within the Alistore-ERI network as well as within the European Community projects EuroLiion and SirBatt involving Alistore-ERI members.