
Abstract
Electrochemical biosensors hold great promise for enabling clinical analysis of biomarkers at the point-of-care. This is particularly of interest for cancer management due to the importance of early diagnostics as well as the critical need for frequent treatment monitoring. We have reviewed clinically-relevant electrochemical biosensors that have been developed over the past five years for the analysis of prostate specific antigen (PSA), a model protein target for prostate cancer management. We have critically evaluated the key performance metrics of these biosensors for clinical translation: limit-of-detection, linear range, and recovery rate in bodily fluids. These PSA electrochemical biosensors can be broadly categorized as sandwich assays, direct detection assays, and indirect detection assays. Among these, indirect detection assays deliver the lowest limit-of-detection. We have identified the development of multiplexed assays for detecting a panel of cancer biomarkers that includes a combination of protein and nucleic acids targets as a key priority for future development.
Export citation and abstract BibTeX RIS

This is an open access article distributed under the terms of the Creative Commons Attribution 4.0 License (CC BY, http://creativecommons.org/licenses/by/4.0/), which permits unrestricted reuse of the work in any medium, provided the original work is properly cited.
Early diagnosis and surveillance of cancer allow for improved prognosis, increased survival rates, and a better quality of life for patients. This has fueled research in the discovery of early-stage cancer biomarkers, as well as in the development of new and improved technologies for ultra-sensitive biomarker analysis.1 Biomarkers are measurable biologically-relevant indicators, which are expressed in either increased or suppressed levels due to the presence of a disease.2 The fluctuation of biomolecular markers such as proteins and nucleic acids from normal physiological levels found in healthy humans is used for the diagnosis, prognosis and monitoring of diseases such as cancer, and has formed the foundation for developing clinical diagnostic tests.3
Prostate cancer (PCa) is one of the most life-threatening diseases for men over the age of 50 with the lethality being ascribed to the lack of symptoms that are expressed in early stages.4 Consequently, tremendous efforts have gone into the discovery of biomarkers associated with PCa, and technology for their use in clinical applications. Prostatic acid phosphatase (PAP) was one of the first biomarkers used for the diagnosis and staging of (PCa), but due to the difficulty in accurately quantifying the localized production of the isoenzyme to the prostate, investigators spent years trying to find a more reliable diagnostic reagent.5 In 1979 Wang and colleagues isolated and purified a 33-kD glycoprotein that was distinct from PAP, but was also highly specific to prostate cells, termed prostate specific antigen (PSA).6 A subsequent study found that PSA could be detected in human serum, proving that in addition to being cell-type specific, it is released into circulation enabling minimally-invasive analysis.7 Shortly after, it was concluded that the concentration of serum PSA coincided with prostatic tumor progression and could be used for diagnosis, staging and monitoring the disease after treatment.8 PSA was also identified in healthy males; however, a level above the threshold concentration of 4 ng ml−1 has been recognized as an indication of PCa.9 Although PSA is one of the most validated biomarkers for clinical decision making in regards to PCa, it is not currently used for PCa screening due to the controversies raised in the literature regarding the specificity of PSA. Increased levels of PSA is also associated with benign prostatic hyperplasia (BPH), leading to false positive PCa diagnosis and unnecessary invasive biopsies, demonstrating the lack of specificity required for a screening biomarker.10 Despite this, PSA blood tests are commonly used in monitoring cancer progression in patients receiving treatment and for early diagnosis in combination with a digital rectal exam (DRE). Given that frequent PSA analysis is performed to monitor the efficacy of treatments in cancer patients (in the form of PSA velocity or doubling time measurement),11 there is a need for point-of-care tests with facile sample collection, easy operation, and rapid sample to answer times, similar to the home glucose test, for disease monitoring.12
Tremendous research efforts have been focused on the POC detection of proteins, and particularly PSA, using biosensors.13 Electrochemical biosensors have been central in the development of handheld bioanalytical systems as they combine sensitivity, specificity, and multiplexing with scalable and cost-effective manufacturing.14 Due to their promise in creating market translatable technologies, we focus this review on electrochemical biosensors for the detection of PSA. PSA detection using optical methods—fluorescence,15 colorimetric,16 surface plasmon resonance,17 and surface enhanced Raman spectroscopy18—have been discussed elsewhere. Several articles have been published on the electrochemical detection of PSA in the last five years (Table I) with multiple reviews focusing on a particular electrode material,19,20 biorecognition element,21 or signal transduction mechanism for PSA detection. Here, we focus on categorizing electrochemical PSA detection platforms based on their operating mechanisms, and solely include assays that have been validated with clinical samples or using complex biological matrices over the past five years. This review is meant to highlight the advantages and disadvantages of various biosensing mechanisms used for PSA detection to guide readers interested in protein detection towards choosing the right assay for their particular application case.
Table I. Summary of the recent work on electrochemical detection of PSA categorized based on the detection mechanism: (a) sandwich assay, (b) direct detection assay, (c) indirect detection assay.
| Assay components | Method of detection | Linear range | LOD | Recovery rate | References |
|---|---|---|---|---|---|
| A) Sandwich Assay | |||||
| Ab1, Ab2, HRP, H2O2 | SWV + CA | 0.001–10 ng ml−1 | 0.84 pg ml−1 | 22 | |
| Ab1, Fc labeled aptamer | DPV | 0.05–100 ng ml−1 | 0.017 ng ml−1 | 96.8%–101.4% | 23 |
| Ab1, Ab2, HRP, H2O2 | DPV | 3–15 ng ml−1, 15–100 ng ml−1 | 0.093 ng ml−1 | 103% | 24 |
| Ab1, Ab2, HRP, H2O2, MB as redox mediator | SWV | 1–18 ng ml−1 | 0.001 ng ml−1 | Serum—96.3%–98.8% | 25 |
| Urine—98.9%–99.6% | |||||
| Plasma—96– 97.8% | |||||
| Ab1, Ab2, HRP | DPV | 2 pg ml−1−200 ng ml−1 | 2 pg ml−1 | RSD 3%–10% | 26 |
| Ab1, Ab2 on gold coated magnetic NP | CV | 0.085–30 ng ml−1 | 0.085 ng ml−1 | >95% | 27 |
| Nanobodies, HRP | DPV | 0.1–100 ng ml−1 | 0.08 ng ml−1 | 90% to 101% | 28 |
| Ab1, Ab2 on BSA-Cu Nanoclusters | SWV | 0.5 pg ml−1–100 ng ml−1 | 145.69 fg ml−1 | RSE −5.0% to 5.5% | 29 |
| Ab1, Ab2-HRP-thionine | DPV | 0.1–5 ng ml−1, 5–100 ng ml−1 | 0.02 ng ml−1 | 101% and 100.6% | 30 |
| Ab1, Ab2, HP5@AuNPs@g-C3N4 hybrid nanomaterial | DPV | 0.0005–10.00 ng ml−1 | 0.12 pg ml−1 | 97.62%–99.35% | 31 |
| beta-CD/biotin-Ab1, Ab2-HRP | SWV | 10 pg–25 ng ml−1 | 6.7 pg ml−1 | 93, 96% | 32 |
| Ab1, Ab2- Pd@CuO | CA | 10−5 ng ml−1 −102 ng ml−1 | 0.002 pg ml−1 | 99.2%–101.2% | 33 |
| Ab1, Ab2- PtCu@rGO/g-C3N4/ | CA | 50 fg ml−1 to 40 ng ml−1 | 16.6 fg ml−1 | 99.7%–101% | 34 |
| Magnetic based mouse IgG-Ab1, AP tagged anti-mouse Fc IgG | CA | 1–80 ng ml−1 | 2 ng ml−1 | 35 | |
| Ab1, Ab2/HRP- polypyrrole (Ppy) Nps | CA | 0.001–40 ng ml−1 | 0.7 pg ml−1 | 36 | |
| Ab2, SiO2@polydopamine nanocarrier -MB, hydrogel surface | SWV | 10 fg mL−1–100 ng ml−1 | 1.25 fg ml−1 | 98.5%−106.5% | 37 |
| Cu2+@Ag-Au labeled Ab2 on GS-SnO2-Au@Pt | SWV | 0.01–100 ng ml−1 | 3.84 pg ml−1 | 99.6%−103% | 38 |
| B) Direct capture assay | |||||
| Aptamer, (rGO)/thionine (THI) nano composites | DPV | 0.05–200 ng ml−1 | 10 pg ml−1 | 39 | |
| Antibody on beta-CD molecules | DPV | 1 pg ml−1–1 ng ml−1 | 0.3 pg ml−1 | 98.3, 101.7, 102.1 | 40 |
| Aptamer, silica thin film-coated gold electrodes | DPV | 1–300 ng ml−1 | 280 pg ml−1 | 107.2 ± 4.55% | 41 |
| Antibody-COOH-AgPtPd/NH2-rGO composite | DPV | 0.000004−300 ng ml−1 | 4 fg mL−1 | 99.4% to 98.9% | 42 |
| Antibody, K3Fe(CN)6 as redox reporter in electrolyte | SWV + EIS | 0.055 fg mL−1−25 ng ml−1 and 1–36 ng ml−1 | 0.06 pg ml−1 and 2 ng ml−1 | 91% to 106% | 43 |
| Antibody on nanocomposite | DPV | 3 pg ml−1–60 ng ml−1 | 2 pg ml−1 | 91% to 97.5% | 44 |
| Antibody on rGO | DPV | 0.006–30 ng ml−1 | 0.003 ng ml−1 | 97% to 110% | 45 |
| Fc-PAMAM dendrimers, Ab | DPV | 0.01–100 ng ml−1 | 0.001 ng ml−1 | 97.89%–102.6% | 46 |
| E, Br−Py, AuNP-Hep), Nafion, Ab | SWV | 0.1–50 ng ml−1 | 0.08 ng ml−1 | 96.3% to 100.0% | 47 |
| Aptamer | DPV + EIS | 0.005–20 ng ml−1 | 1 pg ml−1 | 48 | |
| Aptamer +Antibody | EIS + DPV | EIS | EIS | 49 | |
| 0.1–10 ng ml−1 | 0.14 ng ml−1 | ||||
| 0.4–10 ng ml−1 | 0.42 ng ml−1 | ||||
| DPV | DPV | ||||
| 0.1–10 ng ml−1 | 0.14 ng ml−1 | ||||
| 0.1–10 ng ml−1 | 0.14 ng ml−1 | ||||
| Aptamer | EIS | 0.5–1000 ng ml−1 | 1 ng ml−1 | 50 | |
| Aptamer, poly aniline/AuNP | DPV | 0.1 pg–100 ng ml−1 | 0.085 pg ml−1 | 99.43%–106.1% | 51 |
| Antibody, HNTs@PPy-Pd/GCE | CA | 0.0001 to 25 ng ml−1 | 0.03 pg ml−1 | 100.8% to 103.2% | 52 |
| Antibody, AuNP functionalized CuO2@CeO2 | CA | 0.1 pg ml−1−100 ng ml−1 | 0.03 pg ml−1 | 98.3% to 100.2% | 53 |
| Antibody, Pd@rGO | CA | 0.01 ng ml−1–12.5 ng ml−1 | 10 pg ml−1 | 99.8% to 112% | 54 |
| Antibody - high molecular weight silk peptide @rGO | DPV | 0.1–5 ng ml−1, 5–80 ng ml−1 | 53 pg ml−1 | 55 | |
| PtCu hollow nanoframes, Antibody, H2O2 | DPV | 0.01–100 ng ml−1 | 0.003 ng ml−1 | 98.3%–102.3% | 56 |
| Aptamer, binding of MB to ssDNA vs dsDNA | DPV | 0.85–12.5 ng ml−1 | 0.75 ng ml−1 | 99.2% | 57 |
| Aptamer, binding of MB to ssDNA vs dsDNA | DPV | 0.125–200 ng ml−1 | 50 pg ml−1 | 58 | |
| MB labeled aptamer | DVP | 0.125–128 ng ml−1 | 50 pg ml−1 | 59 | |
| Aptamer, streptavidin, biotin | DPV | 0.025–205 ng ml−1 | 8 pg ml−1 | 95.0 to 100.3% | 60 |
| Aptamer, MB binding to ssDNA vs dsDNA | DVP | 1 pg ml−1 −100 ng ml−1 | 0.064 pg ml−1 | 99, 104% | 61 |
| Aptamer-modified Ag/CdO, Fe3O4/graphene oxide nanosheets (GO/Fe3O4 NSs) | DPV | 50 pg ml−1–50 ng ml−1 | 28 pg ml−1 | 97.3 to 103.4% | 62 |
| Antibody-modified nafion/rGO/aldehyde methyl pyridine composite | DPV | 0.005–90 ng ml−1 | 1.6 pg ml−1 | 95.8%−107.9% | 63 |
| C) Indirect detection assay | |||||
| MB labeled DNA/Antibody probe + Fc labeled DNA | ACV | 0.05–100 ng ml−1 | 16 pg ml−1 | 64 | |
| Hairpin assembly, Ag/Pt-poly MB | SWV | 10 fg ml−1–100 ng ml−1 | 2.3 fg mL−1 | 98.8%–103% | 65 |
| Ab1/PSA/AuNP, labeled aptamer, CuNPs | DPSV | 0.05−500 fg ml−1 | 0.020 fg ml−1 | RD 7.35-4.17 | 66 |
| Aptasensor -exonuclease-aided target recycling, AgNPs | LSV | 1 pg ml−1−160 ng ml−1 | 0.11 pg ml−1 | RSD 1.73%–6.08% | 67 |
| Aptamer, mercaptophenylboronic acid (MPBA), AgNPs | LSV | 0.5–200 pg ml−1 | 0.2 pg ml−1 | RSD 4.2–5.3 | 68 |
| DNA, PLLA NP, Antibodies | DPV | 0.05–100 ng ml−1—VEGF | 50 pg ml−1—VEGF | 69 | |
| 1–100 ng ml−1—PSA | 1 ng ml−1—PSA | ||||
| Thiolated peptides, AuNPs, Ag | DPASV | 0.1–100 ng ml−1 | 27 pg ml−1 | 91%–151% | 70 |
| Peptide, polydopamine-Au-HRP | SWV | 1 fg mL−1–100 ng ml−1 | 0.11 fg mL−1 | 71 | |
| Peptide cleavage, Fc and beta-CD | DPV | 0.001 to 30 ng ml−1 | 0.78 pg ml−1 | 101.2% and 106.3% | 72 |
Electrochemical Readout Methods for PSA Detection
Electrochemical methods rely on charge transfer and electrochemical reactions occurring at the electrode/electrolyte interface for signal generation.73 A typical electrochemical sensor consists of a working (sensing), counter, and reference electrode.74 The desired electrochemical reactions take place at the surface of the working electrode while the counter electrode creates a conduction path within the electrolyte, leading to a current flow between the working and counter electrodes. Typically, working and counter electrodes are made of conductive and chemically stable materials such as gold, platinum, carbon, and silicon.75 Reference electrode is employed to control the applied potential on the working electrode. The most frequently used reference electrodes are saturated calomel electrode (SCE) and Ag/AgCl electrode, due to their steady and distinct reference potentials.76,77 The most common electrochemical techniques employed in PSA sensing for point-of-care platforms include chronoamperometry (CA),77,78 cyclic voltammetry (CV),79,80 linear sweep voltammetry (LSV),67 differential pulse voltammetry (DPV),81 square-wave voltammetry (SWV),82 and electrochemical impedance spectroscopy (EIS).80,48 In this section, we briefly describe these methods.
In CA, a potential step is applied to the sensing electrode and the current response of the system is measured as a function of time (Fig. 1a).73 Typically, the initial potential (Ei) is chosen so that redox reactions are minimized, whereas, the final potential (Ef) results to fast surface reaction kinetics and the generation of a diffusion-limited current.83 It is difficult to sense the existence of multiple target analytes using CA, which has led to the wide-spread use of voltammetric techniques in biosensing. In LSV (Fig. 1b), the applied voltage is linearly scanned from Ei to Ef with a constant scan rate (voltage ramp), measuring the generated electrochemical current. When the working electrode potential approaches the reduction potential (Er), the cathodic current increases. As the applied potential increases, the concentration of the electroactive species on the working electrode surface decays. Due to the generation of a concentration gradient, the flux of the species towards the electrode increases and thus the current grows. As the scanning continues and the potential passes Er, the flux reaches its maximum point and then drops due to exhaustion of the analytes at the electrode surface, resulting in a peaked i-v curve.
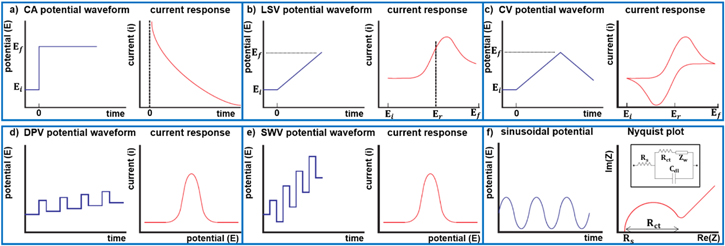
Figure 1. Electrochemical methods: (a) CA step potential and current response as a function of time; (b) LSV potential ramp and resulting i-v curve; (c) CV potential sweep and resulting CV curve; (d) DPV potential waveform and current response; (e) SWV potential waveform and current response; (f) Applied sinusoidal potential, corresponding Nyquist plot, and EIS equivalent electrical circuit (inset).
Download figure:
Standard image High-resolution imageIf the direction of the voltage sweep is reversed (Fig. 1c), the reduced species become re-oxidized at the electrode surface generating an anodic current.84 CV has an extensive application in electrochemical sensing. Valuable data regarding redox potentials and reaction rates of the electroactive species and their interactions can be collected from CV curves.73 However compared with other voltammertric techniques, they suffer from lower analytical sensitivity due to the presence of a non-Faradaic background current.
DPV and SWV are two classes of voltammetric techniques that increase analytical sensitivity by removing the background capacitive signal.84 DPV applies a sequence of voltage pulses of fixed amplitude that are overlapped on a potential ramp (Fig. 1d).84 The current measurement is performed at two specific points on each pulse, the first just before application of the pulse and the second at the end of the pulse. The reason for choosing these sampling points is to allow for the non-Faradaic current component to decay. For each pulse, the first current measurement is subtracted from the second and plotted against the base potential. The obtained peak current is directly proportional to the concentration of the electroactive analytes on the surface of the working electrode. SWV is another form of pulse voltammetry in which a symmetrical square-wave potential with a defined amplitude is overlaid on a staircase potential with a fixed step height, where the leading pulse of the square wave concurs with the staircase step (Fig. 1e). The net current is calculated by subtracting the reverse current (anodic) from the forward current (cathodic) and the current peak is centered on the redox voltage. Similar to DPV, the obtained peak current is directly proportional to the quantity of the electroactive species. SWV possesses many advantages including high sensitivity, elimination of background currents, and rapid response time.85
EIS is a highly sensitive label-free technique which can be used to detect a single biomolecule on the surface of a sensing electrode.86,87 It is based on the application of a small sinusoidal voltage (Fig. 1f) to the working electrode and measuring the complex impedance at the electrode/electrolyte interface over an appropriate frequency range.88 The obtained impedance spectrum is then fitted with an equivalent electrical circuit model composed of resistors and capacitors, providing an insight of the electrode/electrolyte interface. The corresponding circuit model of a three-electrode electrochemical cell is shown in Fig. 1f-inset, where Rs is the resistance of the electrolyte; Rct represents the charge transfer resistance caused by redox reaction of electroactive species with the working electrode; Zw is known as Warburg impedance caused by the diffusion process of reactants and Cdl represents the double-layer capacitance at the working electrode/electrolyte interface.89,90 The measured equivalent impedance is described using the Nyquist plot (Fig. 1f), where the real part of the obtained impedance is plotted on the X-axis and the imaginary part is plotted on the Y-axis. At high frequencies, the plot is illustrated by a semi-circle with Rct representing its diameter. At low frequencies, where ion diffusion dominates, the plot is basically a straight line with a slope of 45°.91 Changes in the impedance spectra are caused by the specific interactions of target proteins with bioreceptors on the surface of the working electrodes.92 EIS-based biosensors are well-known for their high sensitivity and specificity, rapid sample-to-response time, and their capability for integration with microfluidic devices. However, the drawback of using such a sensitive technique includes the interference from non-specific adsorption onto the electrode surface.90,93
Detection Mechanisms
We categorize electrochemical biosensors for PSA detection based on their approaches for translating biorecognition events into electrochemical signals. These are schematically illustrated in Fig. 2 and described below.
- 1.Sandwich assays: In these assays, the target analyte is sandwiched between two biorecognition elements, typically antibodies for capture and subsequent signal transduction. The biorecognition event is translated into an electrochemical signal by labeling one of the biorecognition elements with enzymes or nanoparticles (NPs) as described later in this review.
- 2.Direct detection assays: These assays typically use an immobilized biorecognition element for analyte capture and generate a change in electrochemical signal based on a single binding event with PSA.
- 3.Indirect detection assays: These assays rely on the capture of PSA by biorecognition elements such as aptamers or DNA/protein complexes. Upon target capture, dynamic biorecognition systems undergo structural changes that can be programmed to release or generate an additional detectable element that results in an electrochemical signal change.
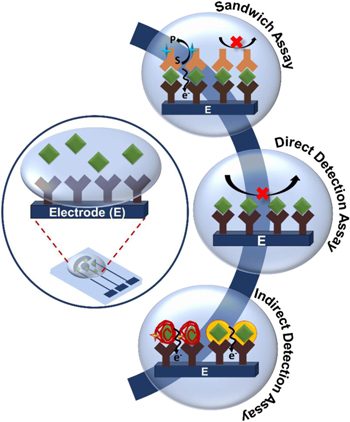
Figure 2. Schematic representation of an electrochemical chip and the detection mechanisms involved in PSA detection: Sandwich Assay, Direct Detection Assay and Indirect Detection Assay.
Download figure:
Standard image High-resolution imageSandwich assays
Enzyme linked immunosorbent assay (ELISA) is a commonly used sandwich assay for protein analysis and is typically programmed to transduce an optical signal change.94 Electrochemical analogs of ELISA use biorecognition and target labeling steps similar to those used in ELISA; but employ voltammetry or amperometry to generate an electrochemical signal. This method relies on sandwiching PSA between two highly specific biorecognition elements such as antibodies. Typically, one antibody (Ab1) is designated for the capture of the target while a second antibody (Ab2) is used for signal generation or amplification. The first interaction usually occurs between an immobilized Ab1 and the PSA molecule, followed by binding of Ab2 to the Ab1-PSA complex. Ab2 can be labeled with either an enzyme or a redox molecule to achieve an electrochemical signal upon successful capture of the target protein. Antibodies are used in the majority of sandwich assays as biorecognition elements but there are alternatives such as aptamers and nanobodies, which are also discussed in this section.
In electrochemical sandwich assays, the redox properties of the enzyme tagged Ab2 is commonly used for signal transduction. In this approach, Ab2 is directly tagged with enzymes such as horse radish peroxidases (HRP), alkaline phosphatase (ALP), glucoamylase, β-galactosidase or glucose oxidase, which catalyze the reduction of their respective substrates in the presence or absence of a mediator.83 HRP is a widely used enzyme in electrochemical sandwich assays that catalyzes the reduction of H2O2 in the presence of Hydroquinone (HQ).83 Chen et al. developed commercially available screen-printed electrode-based microfluidic devices (CASPE-MFDs) to demonstrate the detection and quantification of PSA in human serum samples.22 In this assay, magnetic beads conjugated with Ab1 were immobilized on the gold screen printed electrodes. Following PSA capture on the magnetic beads, HRP/Ab2 was introduced to the electrode using microfluidics. Magnetic beads were used to increase the surface density of Ab1 on the electrode surface and to enhance the capture of target PSA. The redox current was generated by the enzymatic and electrochemical reaction between HRP and HQ respectively using both CA and SWV. Using these devices, a linear range of 0.001–10 ng ml−1 and a limit-of-detection (LOD) of 0.84 pg ml−1 were obtained for detecting PSA in serum. HRP is the most commonly used enzyme in electrochemical sandwich assays due to its stability and fast reaction kinetics. However, it produces a background signal in the absence of the analyte due to the electrochemical activity of its H2O2 substrate. Additionally, HRP relies on the use of mediators for signal generation. ALP has been used to overcome the background and mediator limitations of HRP. For example, Zani et al. demonstrated a magnetic bead-based assay that uses mouse IgG-Ab1 for PSA capture and ALP tagged anti-mouse FcC IgG for signal transduction.35 ALP catalyzes the conversion of α-naphthyl phosphate to α-naphthol. Oxidation of α-naphthol at the electrode surfaces generates an amperometric signal (Fig. 3a). This sensor demonstrated a linear range of 1–80 ng ml−1, and a LOD of 2 ng ml−1 in human serum samples. While enzyme-based sandwich assays are highly specific to PSA, they have limitations due to the intrinsic instability of enzymes.95 These assays also require multiple aggressive washing steps to alleviate non-specific binding that may lead to false negative or false positive results.
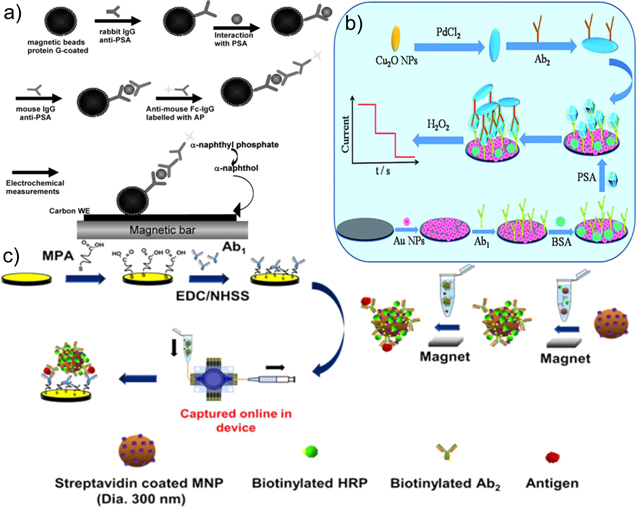
Figure 3. Sandwich assays: (a) Magnetic bead based assay for PSA detection using ALP (Reprinted from (Zani et al.; 2010)35 with permission from Electroanalysis); (b) Pd@CuO NP based detection of PSA (Reprinted from (Chu et al.; 2016)33 with permission from Royal Chemistry Society); (c) Multiplexed detection of PSA using magnetic NPs (Reprinted from (Tang et al.; 2016)26 with permission from American Chemical Society).
Download figure:
Standard image High-resolution imageTo further enhance assay sensitivity and LOD, NPs can be used as labels. NPs may serve as enzyme alternatives for directly catalyzing redox reactions, or they can function as scaffolds for increasing the loading density of enzymes or antibodies. Incorporation of NPs offers ease of labeling, tunable physical and chemical properties, and large surface-to-volume ratio. The increase in available surface area enables enhanced loading of enzymes for better signal amplification and allows for increasing the quantity of Ab1 for increasing capture efficiency.96,36 Nanomaterials such as NPs,31,27 nanoclusters,29 nanosheets,38 or cargo releasing nanocarriers37,97 have been used in electrochemical sandwich assays. Chu et al. chose biocompatible Pd@CuO NPs for enhanced catalysis towards the reduction of H2O2 (Fig. 3b).33 They constructed a sandwich assay based on the Ab1-PSA-Ab2-Pd@CuO architecture, with a linear range of 10−5–102 ng ml−1, and a LOD of 0.002 pg ml−1. This sensor showed a recovery rate of 99.2%–101.2% in human serum for PSA, which is a measure of signal preservation associated with a known amount of PSA quantified using the sensor in a biological solution that contains material that may interfere with signal generation. Feng et al. constructed a sandwich assay with Ab1-PSA-Ab2- PtCu@rGO/g-C3N4.34 In this study, bimetallic Pt-Cu NPs were integrated with 2D rGO-gC3N4 to improve electrical charge transfer and activity for the reduction of H2O2. This assay demonstrated a linear range of 50 fg ml−1−40 ng ml−1, a LOD of 16.6 fg ml−1, and a recovery rate of 99.7%–101.0% for PSA detection in human serum. Tang et al. demonstrated that integrating magnetic NPs (MNPs) labeled with Ab2 and HRP into a high-throughput microfluidic array aided in sensitive, rapid and parallel detection of PSA and three other prostate cancer protein biomarkers (Fig. 3c).26 The device consisted of a thirty two-electrode sensor-array, with eight electrodes dedicated to detecting each analyte. Antibodies for PSA, prostate specific membrane antigen (PSMA), interleukin-6 (IL-6) and platelet factor-4 (PF-4) were immobilized onto each of the eight-electrode sensing patches. The capture of each protein target was performed off-chip and in serum using MNPs labeled with four different antibodies. Following analyte capture by the respective antibodies, MNPs were loaded into the sensor by a syringe and captured by a ring-shaped magnet. A HQ/H2O2 solution was then loaded for signal generation using DPV (Fig. 3c). A LOD of 0.05–2 pg ml−1 and a dynamic range of sub pg ml−1 - above 1 ng ml−1 were achieved. The clinical relevance of the device was demonstrated by analyzing seven patient samples and comparing device performance against ELISA. The results were well-correlated with ELISA demonstrating a relative standard deviation (RSD) of ±3%–10%. The use of NPs in sandwich assays provides a promising avenue for enhancing analytical performance of biosensors. In spite of this, the development of NPs that combine chemical stability, biocompatibility and highly efficient catalysis remains a challenge.
In addition to antibodies, aptamers and nanobodies have been used for the recognition of PSA in electrochemical detection platforms. The use of these biorecognition elements is envisioned to solve the challenges related to the low stability, high non-specific adsorption, and chemical functionalization of antibodies.98 Nanobodies are clones of the various domains of an antibody; however, they do not include the fragment crystallizable (FC) regions, making them smaller and more resistant to non-specific interactions compared to antibodies.98 They can be raised against PSA in vivo and used for capture and detection antibodies.28 Using this adaptation to the universal sandwich assay, Liu et al. were able to reach a LOD of 0.08 ng ml−1 with a linear range of 0.1–100 ng ml−1 and 90%–100% recovery in three spiked human serum samples.28 This sensor was challenged with 17 patient samples, and an excellent correlation was found between this assay and a commercially available analyzer based on photometry (Roche Cobas).
Aptamers are single-stranded DNA or RNA molecules that have very high affinity and specificity to non-nucleic acids targets, such as proteins, small molecules, or whole cells. Because aptamers are selected in vitro and can be mass produced through chemical synthesis, they have emerged as promising antibody-alternatives in sandwich assays.99 Aptamers are first identified through Systematic Evolution of Ligands by EXponential enrichment (SELEX) that involves isolation from synthetic DNA libraries in vitro.100 Savory et al. were the first to identify an aptamer for PSA by applying a genetic algorithm to aptamer sequences that were selected through the SELEX process.101 Aptamer-based sandwich assays build on the same operating principles as those based on antibodies. However, since the size of aptamers are significantly smaller than antibodies, the detection aptamers are typically conjugated to small redox molecules—instead of enzymes—for directly generating an electrochemical signal. This modification was demonstrated by Meng and colleagues who replaced the detection antibody with a PSA-specific aptamer labeled with ferrocene (Fc).23 In the presence of the target, an Ab1/PSA/Apt2-Fc sandwich was formed, and the electrochemical response of Fc was recorded using DPV. This assay displayed a linear detection range of 0.05–100 ng ml−1 with a LOD of 0.017 ng ml−1 and a human serum recovery rate of 96.8%–101.4%. Chen et al. demonstrated the innovative use of nucleic acids within a sandwich assay by utilizing DNA tetrahedron as a scaffold for the immobilization of Ab1, and used HRP-tagged Ab2/Au for signal transduction.77 The DNA tetrahedron did not interact directly with the target; however they enhanced the assay sensitivity due to the increased nano-spacing for the antibody to efficiently capture PSA and Ab2 compared to double stranded DNA molecular linkers. A LOD of 1 pg ml−1 was obtained for a linear range up to 0.02 ng ml−1. The analysis of PSA in patient samples was successfully validated using the standard chemiluminescence methods.
Conventional sandwich assays have proven to be reliable and powerful tools for electrochemical PSA analysis over the past 25 years.102 These assays have demonstrated a LOD as low as 1.25 fg mL−1 and have resulted in dynamic ranges as wide as 10 fg mL−1–100 ng ml−1 (Table I). The customizability of these assays provides an excellent quantitative analysis platform that can be applied to a wide variety of protein biomarkers. While sandwich assays are considered one of the most successful strategies for detecting PSA, the multiple operational steps and washing procedures used in these assays hinder their reproducibility and limits their applicability for POC analysis. Hence, there is an urgent need for developing single-step and label-free assays for PSA analysis, which will be discussed in the next section.
Direct detection assays
Contrary to sandwich assays, direct detection assays require a single biorecognition molecule and binding event for detecting the target analytes. Elimination of the second binding event reduces assay complexity and reagent amounts, increases assay speed, decreases false positive signals associated with non-specific interactions of the detection antibody with the transducer, and allows for in situ analysis.103 In a typical design, the recognition elements, antibodies and aptamers, are immobilized onto the electrode surface and specific binding of PSA is measured. The complex that is formed through the binding of the biorecognition molecule and PSA creates an inert layer that alters the flux of electrons or redox species to the electrode surface.91,92 These effects can be measured through impedance-based, voltammetric or amperometric techniques, most commonly EIS, DPV or CA.
Impedance-based techniques have found great success for direct sensing of PSA since protein capture at the electrode surface directly influences the double layer capacitance and charge transfer resistance of the interface.104 EIS is typically used for direct PSA analysis, where models based on the Randles circuit are used to extract the resistive and capacitive parameters.105 PSA capture at the electrode hinders the ability of redox species to reach the electroactive surface, which increases the charge transfer resistance and decreases the double layer capacitance.106 Using this direct detection strategy with anti-PSA capture antibodies and bovine serum albumin (BSA) used as a blocking agent (Fig. 4a), a LOD of 0.06 ng ml−1 with a recovery rate in a range of 91%–106% in human serum was achieved.43 A major challenge with this technique is that non-specifically adsorbed molecules, in addition to specific PSA capture, can influence the measured signal. To circumvent this issue, surface blockers such as carbo-free, gelatin, ethanolamine hydrochloride and BSA have been used following surface functionalization with capture antibodies or inside target solutions for reducing non-specific adsorption.107
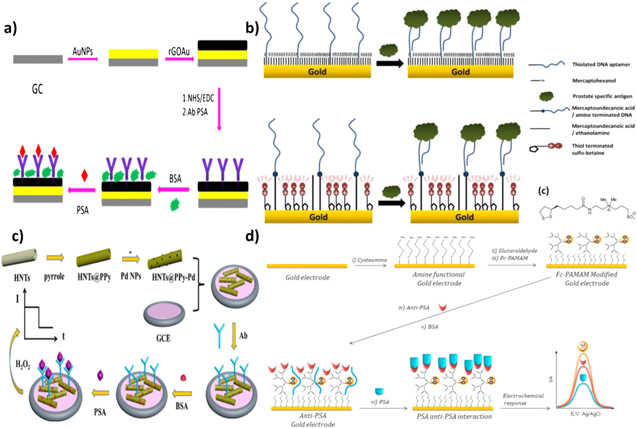
Figure 4. Direct detection assays: (a) EIS based PSA immunoassay using reduced GO decorated with AuNPs, functionalized with PSA-specific antibody, and blocked using BSA (Reprinted from (Assari et al.; 2019)43 with permission from Microchimica Acta.); (b) EIS-based PSA aptasensor Au electrodes (Reprinted from (Jolly et al.; 2015)50 with permission from Elsevier); (c) Amperometric- based PSA immunosensor on polypyrrole coated halloysite nanotubes decorated with PdNPs (Reprinted from (Li et al.; 2017)52 with permission from Springer); (d) Voltammetric-based PSA immunosensor on Au electrodes (Reprinted from (Cevik et al.; 2016)46 with permission from Elsevier).
Download figure:
Standard image High-resolution imageAptamers have also been used in similar electrochemical setups to detect PSA using EIS. Contrary to antibody recognition, upon binding to the target, the aptamers typically undergo a structural change that gives rise to a signal change. As a result of the structural changes that aptamers undergo, both decrease50 and increase49 in the Rct have been reported, depending on the mechanism that is used to detect PSA. In one mechanism, target capture results in a blocking layer that impedes the ability of redox species to interact with the electrode surface. The performance of aptamers and antibodies in direct PSA detection using such mechanism was compared on graphene quantum dot gold nanorod (GQDs-AuNR) modified screen-printed electrodes. Through the use of EIS, LODs of 0.14 ng ml−1 and 0.42 ng ml−1 (with linear ranges of 0.1–12 ng ml−1 and 0.4–12 ng ml−1) were achieved with aptamers and antibodies respectively, demonstrating an enhanced LOD with aptamers.108 The second mechanism arises from the inherent negative charge of aptamers. In this case, when PSA molecules bind to aptamers on the surface of the electrode, a decrease in the charge transfer resistance is seen. This reduction in charge transfer resistance is either related to the PSA architecture exposing more positive charges when bonded to the aptamer or due to the screening of the charges of the aptamer by the protein (Fig. 4b). In either case, this reduces the electrostatic barrier for the transport of ferro/ferricyanide anions towards the electrode surface.50 Using this mechanism, PSA concentrations lower than 1 ng ml−1 in a linear range of 0.5–1000 ng ml−1 were measured. The measurement of a decrease in Rct compared to the increase observed in the first mechanism is important for distinguishing between specific target capture and non-specific adsorption. In the first mechanism, both non-specific adsorption and selective target capture result in an increase in Rct; however in the second mechanism, target binding results in a decrease in the Rct and non-specific adsorption results in an increase in the Rct.
EIS is able to directly measure subtle changes corresponding to specific protein binding, but the practicality of this technique for clinical PSA sensing is questioned. As briefly discussed earlier, non-specific interactions of biomolecules in complex samples pose a high risk for obtaining false positive results. Additionally, EIS is sensitive to electrical noise and interference, which poses the requirement for system shielding.109 The extreme sensitivity of this method to solution and environmental conditions may be overcome by introducing multiple control experiments performed in parallel; however, these additional requirements impede on the desired simplicity of a clinical biosensor. For this reason, voltammetric and amperometric techniques have also been applied for the direct sensing of PSA. In these assays, protein capture reduces the flux of redox species to the electrode surface and decreases the electrochemical current, resulting in signal-off biosensors.110,111,54,41,40,45,42,63 These assays are operationally simple and have led to the development of sensors with a low LOD and a wide dynamic range. One example is an immunosensor fabricated by Li et al. (Fig. 4c) that consists of halloysite nanotubes with a polypyrrole shell functionalized with antibodies and deposited on a glassy carbon electrode (Ab/HNTs@PPy-Pd/GCE).52 In this assay, PSA binding decreases the signal obtained from the reduction of H2O2, leading to a LOD of 0.03 pg ml−1 and a linear dynamic range of 0.0001–25 ng ml−1. This assay demonstrated successful PSA detection in human serum with a recovery rate of 100.8%–103.2%. Another approach to direct detection of PSA using voltammetry involves incorporating redox-active moieties directly onto the electroactive surface and relying on the inhibition of efficient electron transfer caused by the formation of an insulating layer through accumulation of target protein on the surface. This configuration produces a signal-off sensor where the change in signal is dictated by steric hindrance caused by surface capture of PSA.39,55,44,47,51,56 Çevik et al. constructed a direct detection platform using Fc-cored polyamidiamine dendrimers (Fc-PAMAM) deposited on Au electrode (Fig. 4d).46 Anti-PSA antibodies were immobilized onto the electrode surface and a change in the electrochemical response of the redox core upon binding of PSA could be measured using DPV. A clinically relevant linear range of 0.01–100 ng ml−1 was reported and a detection limit of 0.001 ng ml−1 was calculated. Zhang et al. used a similar strategy of incorporating a redox active moiety onto the electrode surface, but instead of relying on the formation of an insulating layer, PSA was quantified by the dissociation of the insulating layer. An aptamer-streptavidin-DNA complex was reversibly immobilized onto a hemin-graphene/palladium NP/GCE.60 Binding of PSA to the complex prompted a dissociation event from the surface due to weak coordination chemistry used for immobilization. The result was exposed hemin at the surface and an increase in the hemin oxidation peak, detected using DPV. The linear range was found to be 0.025–205 ng ml−1 with a LOD of 8 pg ml−1. Signal recoveries ranging from 95.0%– 100.3% were found in three spiked human serum samples.
Novel catalysts are being increasingly used in direct detection assays to increase the binding-induced signal changes. AuNP functionalized CuO2@CeO2 core–shell particles have been used in such assays for harboring the capture antibodies.53 The synergistic effects of the two metal oxides with the metal and the architecture of the composite increases electron transfer efficiency and reaction efficiency towards the reduction of H2O2 for a one step Ab-PSA interaction. This immunosensor demonstrated a linear detection range of 0.1 pg ml−1–100 ng ml−1 and a LOD of 0.03 pg ml−1. A study of different concentrations of PSA in human serum demonstrated a recovery rate ranging from 98.3%–100.2%.
While steric hindrance has proven to be useful in developing assays for the direct detection of PSA, other approaches using signal reporters have been used to translate a single binding event to a measurable signal. One strategy involves incorporating redox molecules directly into the biorecognition elements, where analyte capture causes a structural change, which results in a change in the position or accumulation of the redox molecules relative to the electrode surface. Such assays have been developed using methylene blue (MB) as the redox molecule and PSA aptamer as the biorecognition element by taking advantage of the intrinsic binding properties between MB and DNA. Free MB in solution can interact specifically to exposed guanidine bases in single stranded DNA (ssDNA) or intercalate into double stranded DNA112 and can be detected using voltammetric methods.113 Both signal-on57 and signal-off61,58 aptasensors have been developed using MB/DNA interactions by immobilizing a PSA aptamer onto an electrode surface and monitoring an increase or decrease in current. Another method of employing a MB-based aptasensor is through direct conjugation of MB to one end of an immobilized PSA aptamer. The structural change of the aptamer caused by PSA binding leads to a change in electrochemical signal that can be detected by voltammetry. Sattarahmady et al. constructed a PSA sensor by immobilizing a MB modified aptamer sequence onto a nanostructured Au electrode and measured the reduction of MB using DPV.59 Aptamer folding caused by specific recognition of PSA resulted in a decreased distance between MB and Au, achieving more efficient electron transfer. This signal-on sensor demonstrated a linear range of 0.125–128 ng ml−1, a LOD of 50 pg ml−1, and successful performance in multiple human serum samples. Structure switching aptamers have also been used with metal NPs as the signal reporter rather than MB. Zhao et al. reported a PSA biosensor that relied on the structural change of a Ag/CdO NP labeled aptamer to cause a decrease in the electrochemical signal by dissociation from the electrode surface.62 Aptamer functionalized Ag/CdO NPs were assembled onto GO/Fe3O4 nanosheets, which could then magnetically adsorb onto the surface of a magnetic GCE (MGCE). A large DPV signal could be generated in the absence of PSA, but upon biorecognition, dissociation of the NPs caused by the reversible association of aptamer with the nanosheet resulted in a decrease in the generated signal. The linear range of the biosensor was found to be 50 pg ml−1–50 ng ml−1 and the LOD was calculated to be 28 pg ml−1. The recovery of PSA in four human serum samples were in the 97.3–103.4% range, demonstrating the potential for practical use. The use of the MGCE contributed to simple and automated assembly of the electroactive components onto the electrode surface while the Ag core of the NP allowed for enhanced electron transfer. The combined components of this sensor attributed to the wide, clinically relevant linear range, low LOD and ease of fabrication. The structure-switching capabilities of aptamers and their conjugation with small redox molecules enables the development of direct signal-on biosensors, which has been challenging to develop using antibodies.
Direct detection assays reduce the amount of analytes and washing steps that are required in sandwich assays and decrease the complexity of PSA analysis. Additionally, these assays can be used for monitoring analytes in situ.52,114 The shortcoming of these assays is that non-specific adsorption of materials onto the sensing surface often produces a signal that is difficult to decouple from the signal generated through target binding.91
Indirect detection assays
While there is value in the simplicity of direct detection assays and the versatility of sandwich assays, "indirect" assay designs not belonging to these categories have been developed to increase the sensitivity of biosensors, diminish the need for expensive reagents, and to enable multiplexing. As with sandwich and direct detection assays, indirect detection assays rely on the specific biorecognition of PSA using antibodies or aptamers. However, this single binding event does not result in a detectable signal. Instead they rely on additional interactions, such as DNA strand displacement,64 to either release a surrogate target, or indirectly induce a reaction that generates an electrochemical signal correlating to PSA concentration. Typically, the signal generated by these assays is transduced using voltammetric techniques.
Through the indirect detection mechanisms, it is possible to introduce functional components for signal enhancement and target recycling for signal amplification. Non-enzymatic target recycling was reported by Zhao et al. where a series of DNA strand displacement reactions initiated by PSA capture led to the capture of a dual reporter DNA immobilized metal-polymer.65 PSA aptamers were immobilized onto magnetic beads along with a partially complementary DNA strand that could be released upon biorecognition of PSA (Fig. 5a). The release of the partially complementary strand could hybridize with hairpin DNA that was immobilized onto a GCE/AuNP electrode. This hybridization event exposed a toehold region, through which a catalytic DNA segment could displace the partially complementary DNA strand while maintaining the open form of the immobilized hairpin DNA. This open form left a region of the immobilized hairpin DNA that was complementary to the reporter probe DNA unprotected and available for hybridization. The reporter probe DNA was conjugated with Au/Pt-polymethylene blue (PBM) composites that when bound to the hairpin DNA through complementary base pairing with the reporter probe generated a large current signal. The release of the partially complementary strand could further open other hairpin DNA molecules, demonstrating surrogate target recycling. SWV was used to measure the electrochemical signal and H2O2 was used to catalyze the redox reaction of the PBM reporter, further enhancing the signal. A wide linear range and an ultralow LOD were reported as 10 fg ml−1 and 2.3 fg ml−1−100 ng ml−1, respectively. This sensor proved its clinical applicability with successful recovery rates of 98.8%–103% in two human serum samples, and acceptable relative error when compared to chemiluminescence immunoassays. Enzymatic amplification has also been performed, utilizing rolling circle amplification (RCA), to generate long DNA sequences that could associate with CuNPs.66 This assay involved two recognition elements, an immobilized antibody and an aptamer/RCA primer loaded AuNP. A sandwich structure could be formed between the loaded AuNPs and antibodies in the presence of PSA, triggering the generation of RCA products. The generated products specifically interacted with CuNPs that could be subsequently dissociated and measured using differential pulse stripping voltammetry on an Au electrode. The cascade of signal amplification processes resulted in a linear range of 0.5–500 fg ml−1 and a LOD of 0.02 fg ml−1. The sample analysis performed in human serum was compared to results obtained using an electrochemiluminescence immunoassay and a relative deviation of less than 8% was reported. Miao et al. constructed an ultrasensitive signal-off sensor driven by target-triggered dissociation, measured by LSV.67 They utilized DNA origami as a molecular scaffold, PSA aptamers as recognition elements, AgNPs as signal reporters and an exonuclease as a signal amplification component (Fig. 5b). The PSA aptamer sequence was designed with an NH2 terminus for the attachment of AgNPs. Immobilization of the unlabeled aptamer to the electrode surface was done via complementary base pairing with a sequence in the DNA scaffold. Upon recognition of PSA, the conformational change in PSA aptamer resulted in its dissociation from the complementary strand. Signal transduction was done through the addition of AgNPs onto the sensor after introducing the sample. In the absence of the target, a large signal was obtained using LSV; however, in the presence of PSA, the lack of the amino-terminated aptamer resulted in a reduced signal. Prior to the addition of AgNPs, exonuclease was introduced to free captured PSA by degrading the aptamer, allowing for target recycling and enhanced sensitivity. A LOD of 0.11 pg ml−1 was achieved with a linear range of 1 pg ml−1–160 ng ml−1. The performance of the device was challenged in six undiluted human serum samples and was compared to the results obtained through an immunoradiometric assay. The RSD ranged from 1.73%–6.08% showing acceptable agreeance between the two detection methods.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Indirect detection assays: (a) Enzyme-free target recycling based assay for the detection of PSA using dual metal PMB (Reprinted from (Zhao et al.; 2018)65 with permission from Elsevier); (b)Target-triggered dissociation based assay for PSA detection with enzyme assisted amplification (Reprinted from (Miao et al.; 2018)67 with permission from Elsevier); (c) Simultaneous detection of PSA and VEGF using ssDNA modified GO and PLLA (Reprinted from (Pan et al.; 2016)69 with permission from Elsevier); (d) Enzymatic peptide cleavage based assay for the detection of PSA using host-guest interaction of Fc and β-CD (Reprinted from (Xie et al.; 2015)72 with permission from the Royal Society of Chemistry).
Download figure:
Standard image High-resolution image{kind=link}
Many assays have reported great success not only because of the use of target recycling for amplification but also because of the use of other signal generation strategies, such as metal NPs. Xia et al.68 was able to demonstrate how metal NPs can be used to greatly amplify the electrochemical signal without the use of enzymes or other target recycling tactics. Using an aptamer as a biorecognition element, PSA could be captured at the electrode surface and subsequent reactions could be performed to exploit the saccharide groups of the glycoprotein upon immobilization. The PSA-aptamer complex was decorated with 4-mercaptophenylboronic acid (MPBA) that induced in situ network formation of AgNPs. The solid-state Ag/AgCl reaction from the AgNP network could be measured using LSV and a linear range of 0.5–200 pg ml−1 was obtained, as well as a LOD of 0.2 pg ml−1. When challenged with clinical samples, acceptable RSDs were achieved, validating the clinical performance of the sensor. In this assay, signal generation is achieved by the aggregation of AgNPs onto protein saccharides; therefore, it can be easily applied to the detection of other relevant glycoproteins.
Non-metallic NPs have also been used in the indirect detection of PSA. An assay designed by Pan et al. was able to simultaneously detect PSA and vascular endothelial growth factor (VEGF), a non-specific tumor protein biomarker, by incorporating poly-L-lactide (PLLA) NPs for delivering the target capture elements and amplifying the electrochemical signal (Fig. 5c).69 The detection was based on the increase of steric hindrance and the resultant decrease in electron flow from [Fe(CN)6]3-/4- to the electrode in response to target binding. A solution of ssDNA and GO was incubated on an Au electrode to manufacture a sensing platform capable of capturing VEGF through association with the specifically chosen ssDNA sequence. After incubation of the electrode with the first target protein, PLLA NPs labeled with both anti-PSA and anti-VEGF were introduced and specifically interacted with the VEGF-ssDNA complex on the electrode surface. Decrease in electrochemical signal corresponding to this binding event was recorded using DPV. PSA could then bind to the PLLA NP-VEGF-ssDNA, further reducing the electrochemical signal. Linear ranges of 0.05–100 ng ml−1 and 1–100 ng ml−1 for VEGF and PSA were obtained, respectively. The LOD for both proteins were the lower limit of the linear ranges.
Antibodies and aptamers are common biomolecules used to detect protein targets such as PSA, but recently peptides have been used as biorecognition elements within indirect assays.115 Peptide sequences incorporated into PSA detection assays are not used to capture or immobilize the protein, but rather exploit the proteolytic activity of PSA to induce an electrochemical signal. One approach employs an enzymatically cleavable peptide sequence that upon interaction with proteolytically active PSA (paPSA) results in a cleavage of the portion of peptide responsible for generating a detectable signal by binding to metal NPs.70 Decrease in electrochemical signal can be directly correlated to the concentration of paPSA, generating a signal-off sensor. Conversely, the enzymatic cleavage can increase the signal magnitude by reducing steric hindrance on the electrode surface.71 In this design, the cleavable peptide sequence immobilized on the surface was chemically conjugated to BSA, enhancing surface blockage and decreasing electron transfer with the redox active electrolyte. Exposure of the electrode surface as a result of peptide cleavage by paPSA increases the electrochemical signal, presenting a signal-on method for paPSA detection. A different approach uses the cleaved peptide as a surrogate target molecule for the detection of paPSA. Xie and colleagues demonstrated this by taking advantage of both the enzymatic properties of PSA and the host-guest interaction between Fc and β-cycextrin (β-CD) to create a sensor capable of reaching a LOD of 0.78 pg ml−1 (Fig. 5d).72 Two recognition elements were immobilized in this work; a peptide sequence with a PSA cleavage site labeled with a Fc molecule onto an Fe3O4@Au magnetic bead and β-CD onto multi-walled CNT-PAMAM on a GCE. The peptide sequence acted as a surrogate target as it could be cleaved from the magnetic bead in the presence of paPSA and captured by the β-CD on the CNT PAMAM electrode surface. The peptide cleavage was transduced into an electrochemical signal using DPV and Fc as a redox probe. The increasing concentration of electroactive peptides on the surface corresponded directly to the concentration of PSA through a linear range of 0.001 ng ml−1–30 ng ml−1. Recovery of paPSA in human serum was between 101.2–106.3%. The detection of a specific isoform of PSA could be viewed as a limitation, however, paPSA has been demonstrated to be involved in PCa metastasis rendering it a suitable protein biomarker for determining the aggression level.116 In addition, the low cost, ease of synthesis, and stability of peptides make them excellent substitutes for antibodies in immunoassays.
The increased sophistication of indirect detection assays through incorporation of multiple biorecognition elements, NPs, redox labels or other biomolecular processes has led to PSA biosensors with increased sensitivity and selectivity and has provided a basis for multi-target analysis. Although the increased complexity of these assays results in a parallel increase in the number of washing steps and the amount of reagents; they have demonstrated ultralow and clinically-relevant LODs ranging from 1 ng ml−1 down to 0.020 fg ml−1 (Table I).
Conclusions and Future Direction
In recent years, there has been tremendous research efforts towards improving the sensitivity, specificity and LOD of PSA detection using electrochemical methods for clinical decision making. Based on the operating principles, we can categorize the electrochemical detection of PSA into the conventional sandwich assay, direct capture assay, and the indirect detection assay. The sandwich assays are the electrochemical analogs of ELISA, which is commonly used in biological research and clinical analysis. Although sandwich assays are widely researched; these rely on multiple steps and a large amount of reagents. An alternative to this is the direct approach where a single capture event leads to signal generation by using the concepts of EIS and voltammetry. Indirect detection assays are another alternative to sandwich assays, in which PSA binding results in the release of a functional target, which can be captured for electrochemical signal transduction. This approach typically comes at a cost of increased complexity, with the advantage of increased sensitivity and selectivity. Although sandwich and direct detection assays have demonstrated clinically-relevant performance, the indirect detection assays shine in demonstrating a record performance: an assay of this type successfully detects as low as 0.11 fg ml−1 of PSA with a wide linear range of 1 fg mL−1–100 ng ml−1.71 Despite ultra-sensitivity in detecting levels of PSA within the clinical realm, challenges exist when attempting to commercialize indirect biosensors with the main obstacles being reagent stability and the increased cost of reagents and materials.
Simultaneous multi-analyte analysis is becoming increasingly important in early cancer diagnosis and prognosis117 Multiplexed detection of relevant cancer biomarkers reduces the risk of false positives and increases the accuracy of diagnosis. While PSA serum levels have been shown to be directly correlated to PCa, there is some controversy over the association of an increased PSA concentration and benign factors such as BPH.118 To overcome this, there has been an increasing number of detection schemes that incorporate additional detection elements to analyze a suite of protein biomarkers that are known to be associated either generally with cancer or specifically with PCa.119 The analysis of the additional biomarkers in parallel to PSA is an important safeguard against obtaining false positive results. Several authors have published papers demonstrating multiplexed detection and the importance of analyzing multiple protein biomarkers. However, these devices are only a small portion of the total number of emerging protein biosensors. In this review we have covered a large number of PSA sensors that have emerged within the last five years, with only two being capable of multi-protein detection, demonstrating the urgent need for the development of multiplexed sensors. For PSA biosensors, VEGF is one of the most commonly paired protein biomarkers but other panels of PCa-specific biomarkers containing PSMA, IL-6 and PF-4 have been developed to increase detection accuracy and reliability. Nucleic acids are another class of biomolecules that have been used for cancer diagnostics since they are circulated at an early disease stage and amplification technologies allow for ultrasensitive analysis.120 Developing a universal technology for simultaneous detection of multiple classes of analytes is expected to enable earlier and more accurate cancer diagnosis.
Acknowledgments
The authors acknowledge the financial support provided for this work from NSERC and Ontario Ministry of Research and Innovation. L.S. is the Canada Research Chair in Miniaturized Biomedical Devices and is supported by the Canada Research Chairs Program. L.S. is the recipient of the Ontario Early Researcher Award. All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.