
Abstract
Intense literature and research efforts have focussed on the exploration of complex hydrides for energy storage applications over the past decades. A focus was dedicated to the determination of their thermodynamic and hydrogen storage properties, due to their high gravimetric and volumetric hydrogen storage capacities, but their application has been limited because of harsh working conditions for reversible hydrogen release and uptake. The present review aims at appraising the recent advances on different complex hydride systems, coming from the proficient collaborative activities in the past years from the research groups led by the experts of the Task 40 'Energy Storage and Conversion Based on Hydrogen' of the Hydrogen Technology Collaboration Programme of the International Energy Agency. An overview of materials design, synthesis, tailoring and modelling approaches, hydrogen release and uptake mechanisms and thermodynamic aspects are reviewed to define new trends and suggest new possible applications for these highly tuneable materials.
Export citation and abstract BibTeX RIS
1. Introduction
The shift towards 'green' and sustainable energy production, storage, and use requires efficient and widespread development of renewable energy technologies to tackle climate change and implement a sustainable energy and ecological transition. Many technological energy solutions have been investigated and developed in the past decades, one of those includes hydrogen technology and hydrogen storage materials to boost energy storage and distribution of intermittent renewables.
Hydrogen, as a promising energy carrier, forms covalent bonds to non-metallic elements in the periodic table, some metalloids, such as boron and silicon, most of the late d-metals, but also light elements characterised as 'weak' metals, such as beryllium and aluminium [1]. Complex hydrides include many classes of materials suitable for solid-state hydrogen storage with high gravimetric and volumetric hydrogen capacities, that allow hydrogen storage in compact and light-weight tanks [2]. This review focuses on complex hydride systems based on a select elements, i.e. nitrogen, boron, aluminium, and some transition metals (e.g. manganese, iron, cobalt and nickel). Many reviews have considered these systems recently, and the vivid research activities underline the interest in looking into structural, thermal and reactivity properties of these compounds, towards the developments of new materials. For this reason, the present review aims at collecting possible paper of interest and resuming in a concise way the relevant research of the last 20 years, focusing on hydrogen storage properties in complex hydrides, and suggesting new trends and possible future research directions beside hydrogen storage.
Non-metals and metalloid elements tend to form molecular compounds with hydrogen and the lighter ones are gaseous under ambient conditions [1]. Most of these gaseous compounds are toxic, reactive, or otherwise harmful and the only exceptions appear to be water (H2O) and methane (CH4). Nitrogen can react with hydrogen and form ammonia (NH3), which is a thermodynamically stable molecule (i.e. free energy of formation,  ). Boron has a rich chemistry with hydrogen and forms a diversity of boranes, but all are thermodynamically unstable (ΔGo > 0). The electron deficient molecule, monoborane (BH3), dimerises readily to form diborane (B2H6) even at low partial pressures. Diborane has the characteristic three-centre-two-electron bonds also observed in solid polymeric alane (AlH3), which is also thermodynamically unstable. However, alane and borane react similarly with an ionic metal hydride, e.g. lithium hydride LiH, via an additional reaction to form thermodynamically stable tetrahydridoalanate and tetrahydridoborate, i.e. LiAlH4 and LiBH4, respectively. In contrast, ammonia reacts with a similar ionic metal hydride, e.g. LiH, via an elimination reaction to form lithium amide (LiNH2) and hydrogen. Thus, for the complex hydrides the hydrido ion (H−) acts as a ligand and forms homoleptic complexes, tetrahydridoaluminate ion (AlH4
−) or tetrahydridoborate ion (
). Boron has a rich chemistry with hydrogen and forms a diversity of boranes, but all are thermodynamically unstable (ΔGo > 0). The electron deficient molecule, monoborane (BH3), dimerises readily to form diborane (B2H6) even at low partial pressures. Diborane has the characteristic three-centre-two-electron bonds also observed in solid polymeric alane (AlH3), which is also thermodynamically unstable. However, alane and borane react similarly with an ionic metal hydride, e.g. lithium hydride LiH, via an additional reaction to form thermodynamically stable tetrahydridoalanate and tetrahydridoborate, i.e. LiAlH4 and LiBH4, respectively. In contrast, ammonia reacts with a similar ionic metal hydride, e.g. LiH, via an elimination reaction to form lithium amide (LiNH2) and hydrogen. Thus, for the complex hydrides the hydrido ion (H−) acts as a ligand and forms homoleptic complexes, tetrahydridoaluminate ion (AlH4
−) or tetrahydridoborate ion ( ), which are often briefly denoted alanate and borohydride, respectively, as well as nitrogen-based complex ions, amide (
), which are often briefly denoted alanate and borohydride, respectively, as well as nitrogen-based complex ions, amide ( ) and imide (NH2−). Ammine coordination complexes can be formed as well by neutral coordination of the complex anion. Alkali or alkaline earth metals can act as counter ions (cations) and stabilise these complex ions to form stable ionic solids [3].
) and imide (NH2−). Ammine coordination complexes can be formed as well by neutral coordination of the complex anion. Alkali or alkaline earth metals can act as counter ions (cations) and stabilise these complex ions to form stable ionic solids [3].
The late transition metals, e.g. Mn, Fe, Co, and Ni, generally have a low affinity to hydrogen, but they do react with hydrogen when alloyed or mixed with a metal with a higher affinity, such as magnesium. Distinct to the metallic hydrides, they only exist with fixed stoichiometry, e.g. Mg2FeH6, Mg2CoH5, and Mg2NiH4 [4–7]. The structural analysis of these solids reveal that they also consist of a homoleptic coordination complex with the hydrido anion (H–) as a ligand, forming octahedral [FeH6]4−, square-pyramidal [CoH5]4−, and tetrahedral [NiH4]4− groups [8, 9].
Complex hydrides have relatively high hydrogen content, see table 1, which have provided significant interest in their properties and possible uses. Magnesium iron hydride is remarkable with an extreme volumetric hydrogen density of ρV ∼ 150 g H2 l−1, which is over twice that of liquid hydrogen, i.e. ρV = 71 g H2 l−1, and magnesium nickel hydride is one complex hydride, out of very few, to store hydrogen reversibly at moderate conditions [10–12].
Table 1. Properties of selected complex metal hydrides. M: molecular weight, ρ: density, ρm: gravimetric hydrogen density, ρv: volumetric hydrogen density, ΔHdec: decomposition enthalpy, Tdec: reported decomposition temperature.
| M (g mol−1) | ρ (g ml−1) | ρm (wt% H2) | ρV (g H2 l−1) | ΔHdec (kJ mol−1) | Tdec a (°C) | References | |
|---|---|---|---|---|---|---|---|
| LiBH4 | 21.78 | 0.66 | 18.4 | 122 | 69 | ∼400 | [13] |
| NaBH4 | 37.83 | 1.07 | 10.8 | 116 | 108 | ∼500 | [14] |
| Mg(BH4)2 | 53.99 | 0.79 | 14.8 | 117 | 120 | ∼350 | [15, 16] |
| LiAlH4 | 37.95 | 0.92 | 10.6 | 97 | 10 | ∼150 | [17, 18] |
| Li3AlH6 | 53.85 | 1.02 | 11.2 | 114 | 25 | ∼200 | [19–21] |
| NaAlH4 | 54.00 | 1.28 | 7.3 | 93 | 33 | ∼230 | [22, 23] |
| Na3AlH6 | 102.00 | 1.45 | 5.9 | 86 | 49 | ∼275 | [24] |
| LiNH2 | 22.96 | 1.17 | 8.8 | 103 | 66b | ∼250 | [25, 26] |
| Mg2FeH6 | 110.50 | 2.74 | 5.5 | 150 | 98 | ∼400 | [4, 10, 27–30] |
| Mg2CoH5 | 112.58 | 2.89 | 4.5 | 126 | 86 | ∼360 | [6, 31–36] |
| Mg2NiH4 | 111.34 | 2.70 | 3.6 | 98 | 64 | ∼325 | [7, 37–40] |
aDecomposition temperatures strongly depend on the physical conditions for the measurement and the published data scatters significantly. bDecomposition of LiNH2 to NH3 and H2.
The decomposition of the family of complex hydrides, MAHx , can in general be illustrated by the intermediate formation of a neutral hydride by the central atom in the complex, AHy and an ionic hydride by the counter metal cation, MHz , in the solid [3]. As mentioned above, the neutral counter part to the complex hydride (AHy ) is usually thermodynamically unstable, in particular at elevated temperatures, and will immediately decompose. In case the neutral counter part is a gas, it may be released from the solid hydrogen storage material during decomposition, e.g. ammonia appears to be an intermediate for the amide-imide system and it may be released from the solid state [41]. Similarly, lithium borohydride may release diborane when decomposed in vacuum or reacted with an additive such as silicon dioxide [42]. Thus, the family of complex hydrides has significantly more complex mechanisms for release and uptake of hydrogen as compared to metallic and ionic hydrides, and most of them remain not fully understood.
Basic research activities have demonstrated that the dehydrogenated states of a complex hydride usually has relatively high stability and hydrogen uptake only occurs at extreme conditions [43]. Further investigations are needed to fully describe and model those systems and to explore possible real applications at the prototype scale [44, 45]. Therefore, materials containing complex ions were identified as possible hydrogen storage materials until the late nineties, where titanium was found to catalyse the release and uptake of hydrogen in NaAlH4 [46]. Possible strategies of tailoring properties of complex hydrides include anion or cation substitutions, nanoconfinement, or the design of proper mixtures, either forming eutectics or so called reactive hydride composites (RHCs).
The present review examines the latest advancements (mainly in recent times, but also including relevant investigation over the last 20 years) in complex hydride systems (section 2), to define their synthesis, structural properties, thermodynamics, and characteristics of their reactivity to fully model systems as a function of composition towards a rational design of promising new materials for real applications. The main classes discussed include alanates (section 2.1), borohydrides (section 2.2), boranes (section 2.3), ammines (section 2.4), amides and imides (section 2.5), transition metals complex hydrides (section 2.6), and RHCs (section 2.7).
Trends in properties and new applications are discussed in section 3, especially underlining the intense outputs of research groups and collaborative projects of the International Energy Agency Hydrogen Technology Collaboration Program Task 40 experts.
2. Systems
2.1. Alanates
Aluminium is the most abundant metal in the crust of the earth. Therefore, hydrides based on aluminium are attractive for future large-scale applications such as energy storage. Alane (AlH3) is among the most promising candidate materials for hydrogen storage, owing to its high hydrogen capacities, ρm = 10.1 wt% H2 and ρV = 149 g H2 l−1, and it has been proposed both as a possible fuel for rockets and for mobile propulsion [47, 48]. Alane readily forms metal alanates by addition reactions with more stable hydrides. In the mid-nineties, the discovery of reversible hydrogen release and uptake for titanium-catalysed sodium tetrahydridoaluminate (NaAlH4) provided a paradigm shift for the research field covering solid-state hydrogen storage [48, 49].
Another research direction is the decomposition of alanates in the presence of aluminium sulphides, e.g. the system 6KAlH4–Al2S3, which release 71% of the theoretical hydrogen content below 300 °C, i.e. at 65 °C lower than for pure KAlH4, via several unknown compounds [50]. The NaAlH4–Al2S3 system releases 4.9 wt% of H2 starting at T < 100 °C without the need for a catalyst, via complex decomposition processes that involve multiple new sulphur-containing hydride compounds. The system shows partial H2 reversibility, without the need for a catalyst, with a reversible capacity of ∼1.6 wt% H2 over 15 cycles in the temperature range of 200 °C–300 °C. This absorption capacity is limited by the need for high H2 pressures (>280 bar) to drive the absorption process at the high temperatures required for reasonable absorption kinetics. The large number of new phases discovered in this system suggests that destabilization of complex hydrides with metal sulphides is a novel research avenue for hydrogen storage materials [51].
Nanoconfinement represents another promising strategy for promoting (de)hydrogenation kinetics and reversibility that has been the subject of intense recent investigations. Nanoconfinement of NaAlH4 in carbon scaffolds significantly reduces the hydrogen release temperatures due to a promoting effect of the scaffold, which is independent of maximum pore size (Dmax), and a nano-size effect observed for pores in the range 7 ⩽ Dmax ⩽ 39 nm [52]. Carbon dioxide activation of carbon aerogels increases both pore volume and surface area, and increasing CO2-activation tends to facilitate the infiltration process [53]. Synergetic effects between nanoconfinement and a catalytic effect from a TiCl3 functionalized carbon aerogel scaffold have been achieved, with onset of hydrogen release at Tonset = 33 °C and maximum release at 125 °C [54, 55]. It has also been shown that NaAlH4 nanoconfined in carbon scaffolds can be a potential anode material for batteries [56]. The decomposition of NaAlH4 at 450 °C forms Al and Na, where the Na can be removed under vacuum leaving a porous aluminium scaffold [57]. The specific surface area of the obtained scaffold was determined to be 7.9 ± 0.1 and 6.0 ± 0.5 m2 g−1 by the Brunauer–Emmet–Teller method and from small-angle x-ray scattering measurements, respectively. This aluminium scaffold has been used to nano-confine various complex metal hydrides, allowing for a decrease in decomposition temperature between 150 °C and 250 °C from that of the bulk [58]. NaAlH4 has also been melt infiltrated into these Al scaffolds, resulting in a reduced decomposition temperature, in fact hydrogen release is observed from 100 °C [59].
In addition to NaAlH4, nanoconfinement has also proven a successful strategy for other aluminum-based hydrides. One example is LiAlH4, which has often been overlooked, despite its high gravimetric capacity, due to its intrinsic metastability. The bulk material releases hydrogen in three steps, the first of which (the conversion to Li3AlH6) is weakly exothermic by −10 kJ mol−1 H2. However, recent work combining synthesis, computational modeling, and advanced characterization to demonstrate that metastable LiAlH4 can be thermodynamically stabilized by nanoconfinement inside the pores of nitrogen-functionalized CMK-3 carbons [60], making the hydrogen uptake and release reversible. The theoretical investigation uncovered two critical factors to achieve full reversibility of the nanoconfined system. First, by computing cluster structures and energies of related Li–Al–H compounds, the authors discovered that the undesired Li3AlH6 phase hindering full reversibility is significantly destabilized upon nanosizing. This is consistent with the lack of any indication of Li3AlH6 in the experimental data. Second, electronic structure calculations and ab-initio molecular dynamics simulations revealed that the nitrogen sites change the density of states of CMK-3 in the vicinity of the Fermi level, effectively acting as solvation sites for lithium ions and stabilizing the fully hydrogenated LiAlH4 phase. This enables LiAlH4 regeneration under 1000 bar H2, which was previously assumed to be infeasible. On the other hand, it has been achieved at 100 bar via the formation of solvent adducts [61–63].
Another recent study demonstrated reversible dehydrogenation of metastable alane (AlH3) through a similar nanoconfinement approach [64]. Analogous to the first stage of LiAlH4 decomposition, hydrogen release from AlH3 is exothermic. As a result, extraordinary pressures (of the kbar-range) are required for full rehydrogenation [65]. The authors discovered this rehydrogenation becomes feasible by stabilizing molecular AlH3 within the nanopores of a bipyridine-functionalized covalent triazine framework (AlH3@CTF-bipy). Density functional theory was used to compute geometric and electronic structures, energies, and spectroscopic features of standalone versus confined AlH3 nanoclusters. The calculations illuminated a surprising and non-intuitive mechanism: dissociative AlH3 binding to bipyridine sites accompanied by single-electron transfer forms AlH2 groups on nanopore surfaces, that serve as nucleation sites for AlH3 cluster formation. This proposed mechanism was fully validated by nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) experiments. Through first-principles thermodynamic models, the authors predicted that these nucleated AlH3 clusters are much more stable than bulk AlH3, consistent with rehydrogenation experiments of AlH3@CTF-bipy at pressures more than tenfold lower than the bulk (700 bar vs >7000 bar).
In general, the hydrogen storage performance of complex hydrides is not only controlled by intrinsic properties, but it can also be severely affected by potential contaminants introduced during synthesis and preparation. Along these lines, White et al [66] explored the effect of native surface oxidation on the kinetics of hydrogen release fom Ti-catalysed NaAlH4 [66]. They used near-ambient-pressure in-situ x-ray photoelectron spectroscopy (XPS) to track the dynamical evolution of various intermediates in the surface oxide region. Providing unbiased interpretation of spectroscopy results for interfaces is notoriously challenging because bulk-like species are rarely formed, and it is difficult to find reference spectra for phase-pure compounds. Accordingly, the authors combined the experiments with direct simulation of XPS signatures of key chemical species identified in complex structure models. They determined that surface oxides can impede diffusion in the later stages of the reaction, but can actually aid formation of chemical intermediates. First-principles simulations further revealed that hydrogen release is facilitated by activating the hydridic-protic H···H interaction through the appearance of surface oxygen. As such, the authors concluded that, whereas bulk oxide layers can slow reversible (de)hydrogenation of NaAlH4, atomically thin surface oxide or hydroxide layers could actually be beneficial. This work highlights the need to revisit 'well-known' properties, while emphasizing the importance of controlling the dynamical behaviour of their surfaces to achieve kinetic improvements.
2.2. Borohydrides
The past few decades have revealed a multitude of novel metal borohydrides, M(BH4)n, and this class of materials is further expanded by a variety of bi-metallic and some tri-metallic compounds [3, 67]. Thus, metal borohydrides have extremely rich compositional chemistry and fascinating structural flexibility [68]. The structural features provide a range of physical properties within diverse fields, e.g. hydrogen storage, solid-state batteries, and optical and magnetic properties, demonstrating several possible applications for this class of materials [3, 49, 69–71], as described in section 3. The evaluation of the thermodynamics of pure borohydrides has been implemented in the past to evaluate the possible interaction among different compounds in binary and higher mixtures [43–45, 72–74]. Experimental and theoretical approaches can be combined during thermodynamic assessments by the calculation of phase diagrams (CALPHAD) method as evidenced in the development of complex hydride databases [75]. Chemical manipulation of the materials has further provided several classes of derivatives, e.g. anion and cation substituted compounds and introduction of neutral molecules, which open up new routes for rational materials design and thus tailoring of desired properties [76]. Mixtures of borohydrides to form reactive hydride composited have been reported and tested in some lab-scale prototype tank for hydrogen storage [77–79].
2.2.1. Synthesis
Sodium borohydride (NaBH4) is a versatile reducing agent used in a number of industrial processes. The commercial production is based on a synthetic route developed by Brown and Schlesinger in the 1950s [80]. Mechano-chemistry is the typical synthesis method of novel metal borohydrides, which can be formed through a metathesis reaction between LiBH4 or NaBH4 and a metal halide, MXn . The approach often yields the monometallic borohydride and the lithium or sodium halide salt [81–88]:
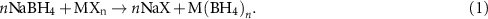
The stability of the metal halide salt is believed to be the driving force for forming the monometallic borohydride. Contrarily, the heavier alkali metal borohydrides, MBH4 (M = K, Rb, Cs), often provide bimetallic borohydrides [3, 68]. The metal halide is an unwanted by-product and the mechano-chemical synthesis may lead to a reaction between the metal halide and the metal borohydride, which forms either a solid solution or a mixed metal borohydride-halide compound [3, 68, 89–93]. Hence, additional purification steps are required to obtain a single-phase metal borohydride sample. High purity grade metal borohydrides for research purposes are available from mainstream chemical suppliers at the lab-scale. Applicative-oriented projects, however, have demonstrated their scale up to reach the kilogram-level. Some research laboratories, such as Aarhus University (Denmark) and KatChem (Czech Republic), successfully up scaled the production of novel borohydrides, by either ball milling and/or wet chemistry methods [94].
Pristine metal borohydrides are successfully prepared by solvent based methods, which may also provide specific structural polymorphs depending on the reaction conditions [95]. Specifically, the reaction between an ionic or polar-covalent metal hydride and a borane (BH3) donating neutral complex, e.g. dimethyl sulphide borane, (CH3)2SċBH3, in an anhydrous solvent follows a nucleophilic addition/hydride-transfer mechanism to form the M(BH4)n . Subsequent purification and/or de-solvation of the metal borohydride may be necessary, providing a high purity product and some degree of polymorphic control [95].
Solid-gas or solid-liquid reactions are efficient in reacting metal borohydrides with a variety of neutral molecules, e.g. NH3, THF, DMS etc, to produce a compound where the metal is fully coordinated by the neutral molecules, which act as a ligand, via a lone pair [95–97]. Thus, there is limited or no control of the composition of the reaction product, but this may be modified by subsequent heat treatment or by mechano-chemical treatment with appropriate ratios of the pristine metal borohydride [96, 98–100].
Interestingly, mechano-chemistry has also devised a new approach to access pure, crystalline compositions of metal borohydrides that cannot be prepared by any other synthesis method, e.g. most di- and tri-metallic borohydrides [70, 101–108].
2.2.2. Structural properties
The structural diversity of metal borohydrides is closely related to the  group acting as a flexible ligand. The complex ligand can coordinate via a corner (B–H, κ1), an edge (B–H2, κ2), or a face (B–H3, κ3) to a metal, or act as a counter anion in the anionic compound. The latter usually occurs if the metal is fully coordinated by stronger ligands, e.g. NH3 [96, 109, 110]. Cations with high charge density, i.e. charge (q) over volume (V), q/V, and relatively high Pauling electronegativity, χP, are polarising and they have a tendency for directionality in the bonding and formation of (polar-) covalent bonds. Zirconium borohydride is a solid consisting of isolated, neutral, molecular Zr(BH4)4 complexes and thus has a low sublimation temperature, Tsub = 29 °C, due to the lack of an interconnecting network [111]. Titanium borohydride also forms a molecular borohydride, Ti(BH4)3, but it is much more reactive, Tdec ∼ 0 °C, which is attributed to the electron configuration, d1. Transition metals with d-electron configurations, d0, d5, or d10 form metal borohydrides stable at ambient conditions [112]. Transition metal borohydrides, TM(BH4)2, TM = Cr2+ (d4), Fe2+ (d6), and Co2+ (d7) are only stable in solution (T < −30 °C), but can be stabilised by neutral molecules, such as ammonia, which coordinate to the metal and prevent redox reactions with the borohydride complex [113].
group acting as a flexible ligand. The complex ligand can coordinate via a corner (B–H, κ1), an edge (B–H2, κ2), or a face (B–H3, κ3) to a metal, or act as a counter anion in the anionic compound. The latter usually occurs if the metal is fully coordinated by stronger ligands, e.g. NH3 [96, 109, 110]. Cations with high charge density, i.e. charge (q) over volume (V), q/V, and relatively high Pauling electronegativity, χP, are polarising and they have a tendency for directionality in the bonding and formation of (polar-) covalent bonds. Zirconium borohydride is a solid consisting of isolated, neutral, molecular Zr(BH4)4 complexes and thus has a low sublimation temperature, Tsub = 29 °C, due to the lack of an interconnecting network [111]. Titanium borohydride also forms a molecular borohydride, Ti(BH4)3, but it is much more reactive, Tdec ∼ 0 °C, which is attributed to the electron configuration, d1. Transition metals with d-electron configurations, d0, d5, or d10 form metal borohydrides stable at ambient conditions [112]. Transition metal borohydrides, TM(BH4)2, TM = Cr2+ (d4), Fe2+ (d6), and Co2+ (d7) are only stable in solution (T < −30 °C), but can be stabilised by neutral molecules, such as ammonia, which coordinate to the metal and prevent redox reactions with the borohydride complex [113].
Alkaline earth and d-block metals with moderate charge density and Pauling electronegativity, e.g. divalent metals, M2+, mainly form framework structures. These compounds typically contain borohydride complexes bridging two or three metals, which provide a high degree of structural flexibility. This is highlighted by the many polymorphs and an amorphous phase observed for magnesium borohydride, Mg(BH4)2. Next to its five known and two yet unsolved polymorphic crystal structures, the amorphous phase of Mg(BH4)2 has been experimentally revisited recently in a detailed study [15, 114, 115]. The amorphous phase can be formed in three different ways [116–118]: (a) reactive ball milling of MgB2 in H2 atmosphere, (b) mechano-chemically induced polymorphic transition, and (c) by a pressure collapse. A recent study utilised the product of synthesis (b) and investigated it by total scattering and pair distribution function (PDF) analysis, which is possible as hydrogen (H) has an oxidation state of −1, and, as such, it is characterized by an electron density. The local ordering, up to 5.1 Å, resembles the ordering in γ-Mg(BH4)2 with Mg–BH4–Mg building blocks. Up to 12.3 Å, the PDF suggests that the interpenetrating channels known from the γ-phase are somewhat present, although with a loss of its functional porosity, whereas above 12.3 Å, a featureless PDF pattern was found, confirming the amorphous nature of this phase [114, 115, 117].
Metals with even lower charge density and Pauling electronegativity have increasingly ionic interactions in the solid state, i.e. increasing degree of charge transfer from the metal to the  complex [119]. This is illustrated by the alkaline earth metal borohydrides, where Be(BH4)2 forms a polymeric, partly covalent structure, while Ba(BH4)2 is more ionic. The dominating ionic bonding becomes clearer for the heavier alkali metal borohydrides, MBH4, M = Na, K, Rb, and Cs, which form rock salt (NaCl) type solids. The most hydrogen dense solid material, ammonium borohydride, NH4BH4, is isostructural to this series, revealing pronounced ionic bonding in this compound [120]. This contrasts the bonding in the more stable ammonia borane, NH3BH3, which is a Lewis acid-base pair adduct with a covalent N–B bond.
complex [119]. This is illustrated by the alkaline earth metal borohydrides, where Be(BH4)2 forms a polymeric, partly covalent structure, while Ba(BH4)2 is more ionic. The dominating ionic bonding becomes clearer for the heavier alkali metal borohydrides, MBH4, M = Na, K, Rb, and Cs, which form rock salt (NaCl) type solids. The most hydrogen dense solid material, ammonium borohydride, NH4BH4, is isostructural to this series, revealing pronounced ionic bonding in this compound [120]. This contrasts the bonding in the more stable ammonia borane, NH3BH3, which is a Lewis acid-base pair adduct with a covalent N–B bond.
Among the rare-earth metal (RE) borohydrides, the majority form trivalent RE(BH4)3, but due to a stable +2 oxidation state of RE2+ = Sm, Eu, Yb, they usually form divalent RE(BH4)2 directly or during heating by reduction from RE3+ to RE2+ [88, 91, 95, 97, 121]. The crystal structures of the divalent RE(BH4)2 are isostructural to the alkaline earth metal borohydrides with similar ionic radii, i.e. RE(BH4)2 (RE2+ = Sm, Eu) are isostructural to Sr(BH4)2 and Yb(BH4)2 is isostructural with the different polymorphs of Ca(BH4)2 [88, 91, 95, 97, 121]. The trivalent RE(BH4)3 can crystallize in three different structures, α-, β-, and r-RE(BH4)3, with space group symmetry Pa-3, Fm-3m, and R-3c, respectively. They are all related to the rhenium oxide (ReO3) structure type, where β-RE(BH4)3 is the ideal ReO3 structure, while α- and r-RE(BH4)3 are obtained by tilting of the [RE(BH4)6] octahedra, as shown in figure 1(a). La(BH4)3 is the only compound which only crystallizes in the r-RE(BH4)3 structure, while the intermediate sized RE3+ = Ce, Pr are the only compounds that can crystallize in both the α-, β-, and r-RE(BH4)3 polymorph [97, 122–124]. The smaller RE3+ = Nd–Lu crystallize in either the α- or β-RE(BH4)3 polymorph, where the synthesis conditions allow for some polymorphic control [97, 125]. The volume of the RE(BH4)3 correlates linearly with the volume of the RE-ions, and the volume increases in the order r- < α- < β-RE(BH4)3, as shown in figure 1(b).
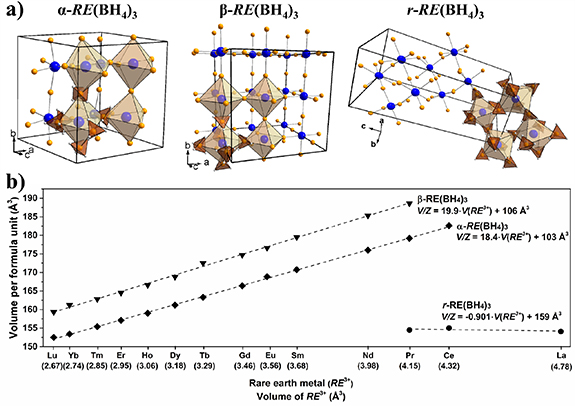
Figure 1. Trends in the crystal structures of the trivalent rare-earth borohydrides. (a) Crystal structures of α-RE(BH4)3 (Pa-3), β-RE(BH4)3 (Fm-3m) and r-RE(BH4)3 (R-3c). Colour scheme: RE3+ (blue), B (orange), H (grey). (b) Volume per formula unit (V/Z) of RE(BH4)3 as a function of the volume of the RE-ion (V(RE3+)) at room temperature. V/Z data is obtained from [86, 97, 124, 125] and the ionic radii are from [126].
Download figure:
Standard image High-resolution imageThus, the bonding of mono-metallic borohydrides spans from pronounced covalent to ionic bonding and can lead to unexpected structural topologies including interpenetrated frameworks or some degree of porosity. On the other hand, the bimetallic compounds, e.g. containing an alkali metal and an alkaline earth metal or a d- or f-block metals, have a tendency for formation of discrete composite complexes. This structural feature is due to the often-significant difference in Pauling electronegativity of the two metals. The less electronegative alkali metal tends to act as a counter ion in the solid state and the more electronegative metal tend to coordinate with a dominantly covalent interaction to  . The alkali metal scandium borohydrides, MSc(BH4)4, M = Li, Na, K, Rb, Cs, are an illustrative example all containing distinct [Sc(BH4)4]− complexes [127–129]. It is worth noting that scandium borohydride, Sc(BH4)3, has not been isolated in a pure form. There are also bimetallic borohydrides with polynuclear complexes, such as LiZn2(BH4)5, containing [Zn2(BH4)5]− as an interpenetrated framework, or LiCe(BH4)3Cl, containing tetranuclear anionic clusters [Ce4Cl4(BH4)12]4− with a distorted cubane Ce4Cl4 core which are charge-balanced by Li+ cations [130].
. The alkali metal scandium borohydrides, MSc(BH4)4, M = Li, Na, K, Rb, Cs, are an illustrative example all containing distinct [Sc(BH4)4]− complexes [127–129]. It is worth noting that scandium borohydride, Sc(BH4)3, has not been isolated in a pure form. There are also bimetallic borohydrides with polynuclear complexes, such as LiZn2(BH4)5, containing [Zn2(BH4)5]− as an interpenetrated framework, or LiCe(BH4)3Cl, containing tetranuclear anionic clusters [Ce4Cl4(BH4)12]4− with a distorted cubane Ce4Cl4 core which are charge-balanced by Li+ cations [130].
Ammonium borohydride, NH4BH4, is interesting from a dynamical and hydrogen storage perspective, due to the extreme hydrogen densities of ρm = 24.5 wt% H2 and ρV = 157 g H2 l−1, and to the significant amount of dihydrogen interactions arising in the compound between  and
and  . NH4BH4 is unstable at ambient conditions with a 'half-life' of ca. 6 h [131, 132]. However, by reacting this material with Ca(BH4)2, a composite with intermediate stability was formed, with Tdec ∼ 100 °C [106, 133]. This inspired the design and synthesis of a large variety of ammonium-metal borohydrides, (NH4)x
M(BH4)y
, with M = Li, Mg, Al, Sc, Sr, Y, Mn, La, Gd, all with high ρm of 9.2–24.5 wt% H2 [108, 134–136]. No reaction was observed with NaBH4, while a solid-solution was formed with KBH4 [108]. This exposed an intriguing structural variety, ranging from structures built from isolated tetrahedral, five-fold or octahedral anionic [M(BH4)n
] complexes, to structures built from one-dimensional chain-like frameworks, two-dimensional layers to three-dimensional framework structures, which are summarized in figure 2. In all cases, the
. NH4BH4 is unstable at ambient conditions with a 'half-life' of ca. 6 h [131, 132]. However, by reacting this material with Ca(BH4)2, a composite with intermediate stability was formed, with Tdec ∼ 100 °C [106, 133]. This inspired the design and synthesis of a large variety of ammonium-metal borohydrides, (NH4)x
M(BH4)y
, with M = Li, Mg, Al, Sc, Sr, Y, Mn, La, Gd, all with high ρm of 9.2–24.5 wt% H2 [108, 134–136]. No reaction was observed with NaBH4, while a solid-solution was formed with KBH4 [108]. This exposed an intriguing structural variety, ranging from structures built from isolated tetrahedral, five-fold or octahedral anionic [M(BH4)n
] complexes, to structures built from one-dimensional chain-like frameworks, two-dimensional layers to three-dimensional framework structures, which are summarized in figure 2. In all cases, the  was considered as a counter ion, similar to the alkali metals in bimetallic borohydrides. Dihydrogen interactions between complex
was considered as a counter ion, similar to the alkali metals in bimetallic borohydrides. Dihydrogen interactions between complex  and
and  ions contribute to the structural diversity and flexibility, but due to the similar size of r(
ions contribute to the structural diversity and flexibility, but due to the similar size of r( ) = 1.48 Å, r(K+) = 1.38 Å, and r(Rb+) = 1.52 Å, several of the NH4
+ compounds show resemblance to the K- and Rb-analogues [108]. However, a new composition and structure type was reported for (NH4)3La2(BH4)9, which forms a new two-dimensional layered structure. Interestingly, it was found that structures with three-dimensional framework display significantly longer shortest-dihydrogen bonds (>2.18 Å), while structures with lower dimensionality all displayed shorter bonds with the shortest dihydrogen bonds in the range 1.59–1.82 Å [108].
) = 1.48 Å, r(K+) = 1.38 Å, and r(Rb+) = 1.52 Å, several of the NH4
+ compounds show resemblance to the K- and Rb-analogues [108]. However, a new composition and structure type was reported for (NH4)3La2(BH4)9, which forms a new two-dimensional layered structure. Interestingly, it was found that structures with three-dimensional framework display significantly longer shortest-dihydrogen bonds (>2.18 Å), while structures with lower dimensionality all displayed shorter bonds with the shortest dihydrogen bonds in the range 1.59–1.82 Å [108].

Figure 2. Crystal structures of selected ammonium metal borohydrides showing isolated complex anions (a)–(c) and one-dimensional (d), two-dimensional (e) and three-dimensional (f) structures. (a) NH4Y(BH4)4, (b) (NH4)2Gd(BH4)5, (c) (NH4)3Gd(BH4)6, (d) NH4Li(BH4)2, (e) (NH4)3La2(BH4)9 and (f) NH4Ca(BH4)3.
Download figure:
Standard image High-resolution imageQuasielastic neutron scattering (QENS) can be used to determine the dynamic processes, such as jump rotation or diffusion. Borohydrides are a near ideal probe, when the boron-11 isotope is used, as hydrogen motions can be probed due to its strong incoherent cross section. All motions are noted as a visible broadening next to the elastic line. QENS has been employed in many different borohydrides over the years and a sensible overview has been published in [137]. QENS has been employed on the γ- and amorphous phase of Mg(BH4)2, and on Pr(BH4)3 [115, 124]. Mg(BH4)2 shows a definite broadening of the elastic line after the sample has turned amorphous, implying that this broadening is a result of the high rotational mobility of the  group [115]. Pr(BH4)3 was also investigated, due to its unusual structural variety, with a stepwise negative thermal expansion [123]. QENS data displayed a definite thermal response during the heating procedure and the mean square displacement value also showed a dependency on the cooling rate, meaning that the high temperature structure was well preserved during cooling [124]. The structural and vibrational properties of NH4BH4 have recently been investigated with inelastic and QENS, which revealed the onset of fast NH4
+ dynamics at ∼−223 °C (∼50 K), while the dynamics are almost frozen below this temperature [138, 139]. Upon further heating, the
group [115]. Pr(BH4)3 was also investigated, due to its unusual structural variety, with a stepwise negative thermal expansion [123]. QENS data displayed a definite thermal response during the heating procedure and the mean square displacement value also showed a dependency on the cooling rate, meaning that the high temperature structure was well preserved during cooling [124]. The structural and vibrational properties of NH4BH4 have recently been investigated with inelastic and QENS, which revealed the onset of fast NH4
+ dynamics at ∼−223 °C (∼50 K), while the dynamics are almost frozen below this temperature [138, 139]. Upon further heating, the  dynamics has an onset at ∼−148 °C (∼125 K). The reorientational motions were investigated with QENS, revealing a fast and complex reorientation of
dynamics has an onset at ∼−148 °C (∼125 K). The reorientational motions were investigated with QENS, revealing a fast and complex reorientation of  , while
, while  can be described as a cubic arrangement with 50% occupancy on the H positions [138], the latter in agreement with the proposed crystal structure determined by x-ray diffraction [132]. Density functional theory (DFT) calculations revealed a non-directional nature of the dihydrogen bonds, and only a weak tendency for long-range ordering [139].
can be described as a cubic arrangement with 50% occupancy on the H positions [138], the latter in agreement with the proposed crystal structure determined by x-ray diffraction [132]. Density functional theory (DFT) calculations revealed a non-directional nature of the dihydrogen bonds, and only a weak tendency for long-range ordering [139].
2.2.3. Derivatives of metal borohydrides with neutral molecules
Introduction of neutral molecules into metal borohydrides is a valuable approach to further increase their compositional and structural diversity and to tailor their chemical and physical properties. The selected molecules in most cases contain an electron pair donating atom, such as N, O, or S, which tends to coordinate to the metal in the structure via a covalent bond. Thus, a range of solvated metal borohydrides have been observed through the preparation of metal borohydrides or during subsequent reaction with a solvent [95]. The complete series of rare-earth metal borohydrides (REBs) with dimethyl sulphide (S(CH3)2) has been investigated, where RE3+ = La form multiple different solvates, La(BH4)3ċxS(CH3)2, but none of the structures have yet been solved [97, 122]. For the heavier RE3+ = Ce, Pr, two polymorphs have been reported, α- and β-RE(BH4)3ċS(CH3)2, where α-RE(BH4)3ċS(CH3)2 is isostructural with the majority of the rare-earths RE3+ = Y, Ce, Pr, Nd, Gd, Tb, Dy, Ho, Er, Tm and Lu [87, 95, 97, 140]. Interestingly, the rare-earths that are sensitive to reduction, RE3+ = Sm, Yb, form different solvates, RE(BH4)3ċxS(CH3)2, and appear to be a complex mixture of different compounds, where during thermolysis, the solvent is released together with the reduction from RE3+ to RE2+ [95, 97].
These neutral molecules often contain hydrogen atoms bonded to a more electronegative atoms, which can form dihydrogen bonds with the hydrogen in the borohydride group, e.g. B–Hδ
−⋅⋅⋅+δ
H–N, in the solid state. This is illustrated by complexes with ammonia borane, e.g. Mg(BH4)2ċ2NH3BH3, which possess strong dihydrogen bonds (length < 2.0 Å) and the layered structure of α-Sr(BH4)2ċNH3BH3 consisting of alternating layers of Sr(BH4)2 and NH3BH3, partly stabilised by dihydrogen bonds [141, 142]. Finally, the Al(BH4)3ċNH3BH3 consists of heteroleptic complexes (Al(BH4)3ċNH3BH3), where the aluminium cation coordinates to three  groups and one NH3BH3 molecule [143]. The complexes are ordered in a three-dimensional crystal structure due to weak dihydrogen bonds between the
groups and one NH3BH3 molecule [143]. The complexes are ordered in a three-dimensional crystal structure due to weak dihydrogen bonds between the  and the –NH3, which are often bifurcated on the latter side and thus reaching HċċċH distances of more than 2.6 Å [143].
and the –NH3, which are often bifurcated on the latter side and thus reaching HċċċH distances of more than 2.6 Å [143].
2.2.4. Anion substitutions
Different types of anion substitution in metal borohydrides have been described, which can be well illustrated by the behaviour of the halides [3]. The small fluoride ion, F− (1.33 Å), resembles the hydride ion, H− (1.40 Å), and fluorides and hydrides containing the same metal cation are often found to be isostructural. Therefore, fluoride substitution can occur in the borohydride anion forming  complexes [144, 145]. This was obtained for various MBH4–MBF4 systems, M = Na or K, e.g. by formation of NaBH2.1F1.9, observed from 200 °C to 215 °C by synchrotron radiation x-ray powder diffraction (SR-PXD) [145, 146]. Theoretical calculations of thermodynamic properties of orthorhombic o-LiB(H,F)4 reveal a tendency of fluorine to prefer boron atoms which already is bonded to F, rather than being statistically distributed over all the available
complexes [144, 145]. This was obtained for various MBH4–MBF4 systems, M = Na or K, e.g. by formation of NaBH2.1F1.9, observed from 200 °C to 215 °C by synchrotron radiation x-ray powder diffraction (SR-PXD) [145, 146]. Theoretical calculations of thermodynamic properties of orthorhombic o-LiB(H,F)4 reveal a tendency of fluorine to prefer boron atoms which already is bonded to F, rather than being statistically distributed over all the available  complexes [144, 147]. This is in agreement with the experimentally determined low thermal stability and release of boron trifluoride and diborane gases and the formation of significant amounts of M2B12H12 [145, 146, 148]. Thus, fluorine appears to have too strong of a destabilising effect.
complexes [144, 147]. This is in agreement with the experimentally determined low thermal stability and release of boron trifluoride and diborane gases and the formation of significant amounts of M2B12H12 [145, 146, 148]. Thus, fluorine appears to have too strong of a destabilising effect.
In contrast, the heavier halides, i.e. Cl−, Br−, or I−, chemically resemble the borohydride complex,  , and they may substitute this anion in the solid state. An easy anion substitution occurs if the metal borohydride and the metal halide are isostructural. This effect can be observed by moderate heat treatment of mechano-chemically mixed samples and it has been observed for LiBH4–LiBr, LiBH4–LiI systems, because β-LiBr, β-LiI, and h-LiBH4 are isostructural, as well as in NaBH4–NaCl mixtures, given that NaBH4 and NaCl are also isostructural [149–152]. These phenomena occur because volume contraction is facilitated by mechano-chemical treatment, i.e. dissolution of the compound with the smaller volume per formula unit (V/Z) into the compound with the larger one. On the other hand, moderate heat treatment lead to thermal expansion, which may facilitate formation of the solid solution with the larger V/Z. Though, upon prolonged heating, solid solutions tend to merge into just one. On the contrary, if the metal borohydride and the metal halide are not isostructural, anion substitution can occur in both compounds, and it may lead to two terminal solid solutions.
, and they may substitute this anion in the solid state. An easy anion substitution occurs if the metal borohydride and the metal halide are isostructural. This effect can be observed by moderate heat treatment of mechano-chemically mixed samples and it has been observed for LiBH4–LiBr, LiBH4–LiI systems, because β-LiBr, β-LiI, and h-LiBH4 are isostructural, as well as in NaBH4–NaCl mixtures, given that NaBH4 and NaCl are also isostructural [149–152]. These phenomena occur because volume contraction is facilitated by mechano-chemical treatment, i.e. dissolution of the compound with the smaller volume per formula unit (V/Z) into the compound with the larger one. On the other hand, moderate heat treatment lead to thermal expansion, which may facilitate formation of the solid solution with the larger V/Z. Though, upon prolonged heating, solid solutions tend to merge into just one. On the contrary, if the metal borohydride and the metal halide are not isostructural, anion substitution can occur in both compounds, and it may lead to two terminal solid solutions.
In the past few years, interest in intrinsic entropy effects in the solid state has been raised, illustrated by the fact that some polymorphs, e.g. h-LiBH4, β-Mg(BH4)2, and β-Ca(BH4)2 readily dissolve metal halides in the solid state [130, 153]. This effect was assigned to dynamics and an orientational disorder, which often occur in high temperature polymorphs. Therefore, anion substitution often stabilises this structure to lower temperatures.
Since 2009, halide substitution has been adopted to enhance Li+ conductivity in LiBH4 [154], stabilizing the high temperature polymorph at room temperature. In the last years, even though the Li(BH4)1−α (Br)α hexagonal solid solution has been well studied as a solid-state-electrolyte [155], few experimental data exist for hydrogen storage in the LiBH4–LiBr system [150, 156], and only recently a thermodynamic assessment has been reported [157]. The LiBH4–LiBr phase diagram was recently explored experimentally, by means of in-situ and ex-situ powder x-ray diffraction (PXD), differential scanning calorimetry (DSC), and thermodynamically assessed using the CALPHAD method, coupled with ab-initio calculations. The melting behaviour has been determined and a peritectic reaction at 380 °C and 0.60 LiBr molar fraction have been reported [157]. By combining DSC and PXD analyses, an enthalpy of mixing (ΔHMix = −1.0 ± 0.2 kJ mol−1) for the formation of the h-Li(BH4)0.6(Br)0.4 solid solution has been obtained, which was used for the CALPHAD assessment. The resulting phase diagram is reported in figure 3 [157], where it can be observed that the stability of the hexagonal Li(BH4)1−α (Br)α solid solution at room temperature is in the 0.30 ⩽ α ⩽ 0.55 range. LiBr can stabilize the hexagonal structure of LiBH4 at room temperature, whereas LiCl is soluble in LiBH4 only at high temperatures in the hexagonal phase, whereas nearly no solubility has been observed in the orthorhombic phase, as shown in figure 3 [158]. Being isostructural, LiBr and LiCl show a full solubility in the cubic phase [159].
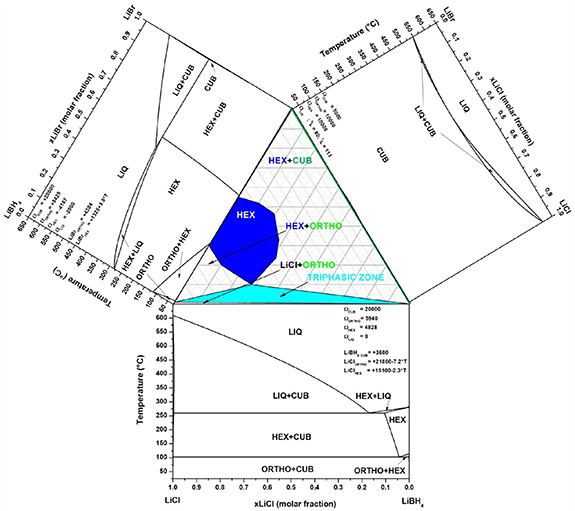
Figure 3. Isothermal section at room temperature of the LiBH4–LiBr–LiCl system, together with assessed pseudo-binary phase diagrams for LiBH4–LiBr, LiBH4–LiCl and LiBr–LiCl systems. CALPHAD assessed interaction parameters and lattice stabilities are reported as J mol−1 of compound [74, 155, 157, 159–161].
Download figure:
Standard image High-resolution imageA ternary hexagonal solid solution, h-Li(BH4)1−α−β
(Br)α
(Cl)β
, containing both chloride and bromide can be stabilized at room temperature (figure 3) [155]. The h-Li(BH4)1−α−β
(Br)α
(Cl)β
lattice parameters has been defined by PXD analysis and Rietveld refinement. It has been reported that both the a and the c lattice parameters, as well as the volume, of the h-Li(BH4)1−α−β
(Br)α
(Cl)β
phase, decrease with the increase of halides content. The contraction of lattice parameters is almost linear with increasing Cl− concentration. When Br− replaces the borohydride anions, the decrease of lattice parameters and volume is less pronounced with respect to Cl−. This behaviour is expected after halogenation, due to the smaller ionic radii of Cl− (r(Cl−) = 1.81 Å) and Br− (r(Br−) = 1.96 Å) [126] compared to that of  (r(
(r( ) = 2.03 Å) [68]. Finally, the effect of the composition and the structure dimensions of the h-Li(BH4)1−α−β
(Br)α
(Cl)β
solid solution on the Li-ion conductivity has been investigated, showing that the highest value of room temperature Li-ion conductivity in the ternary solid solution is reached for the h-Li(BH4)0.7(Br)0.2(Cl)0.1 sample (1.3 × 10−5 S cm−1 at 30 °C) [155]. It is worth noting that the incorporation of chlorine in the hexagonal phase leads to a lower weight of the solid solution, i.e. increasing the energy density in terms of electrolyte materials [155].
) = 2.03 Å) [68]. Finally, the effect of the composition and the structure dimensions of the h-Li(BH4)1−α−β
(Br)α
(Cl)β
solid solution on the Li-ion conductivity has been investigated, showing that the highest value of room temperature Li-ion conductivity in the ternary solid solution is reached for the h-Li(BH4)0.7(Br)0.2(Cl)0.1 sample (1.3 × 10−5 S cm−1 at 30 °C) [155]. It is worth noting that the incorporation of chlorine in the hexagonal phase leads to a lower weight of the solid solution, i.e. increasing the energy density in terms of electrolyte materials [155].
In contrast to metal borohydride-halide solid solutions with fully anion disordered structures, a few compounds have fully ordered structures, such as KZn(BH4)Cl2 [162] and lithium REB halides, LiRE(BH4)3X, (RE = La, Ce, Pr, Gd, Sm; X = Cl, Br, I [86, 89, 90, 92, 121, 163].
Fluoride substitutions have been obtained in RHCs, such as (CaH2/CaF2)–MgB2, Mg–LiBH4–FeF3, LiH–LiF–MgB2, and Ca(BH4)2–MgF2 system, which often have been cycled a few times with hydrogen release and uptake, but they suffer from decreasing capacity due to the high stability of metal fluorides, MFx , and lose of boron due to formation of gaseous boron trifluoride, BF3 [164–171].
2.2.5. Cation substitutions
Tailoring approaches to improve thermodynamics and kinetics of hydrogen release and uptake in borohydrides have been evidenced when mixed cations solid solutions or eutectic mixtures have been investigated [112, 172, 173]. Available phase diagrams in cation mixed pseudo binary systems are presented in figure 4, together with information on solid and liquid phases. As a matter of fact, after literature and experimental analysis on LiBH4–NaBH4 [160], LiBH4–KBH4 [174], and NaBH4–KBH4 [175], pseudo-binary systems were fully assessed, describing solid and liquid phases involved in each phase diagram [2, 161].
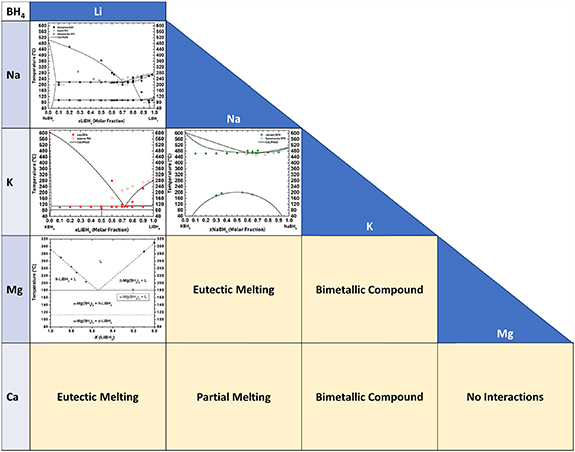
Figure 4. Available phase diagrams and information in pseudo-binary mixed cations systems. Data taken from [2, 160, 161, 173, 176–179].
Download figure:
Standard image High-resolution imageEutectic melting is clearly observed in the lithium borohydride-potassium borohydride (LiBH4–KBH4) phase diagram, by a rather deep melting point at Tmp = 105 °C [174]. This feature was utilised to nanoconfine the LiBH4–KBH4 system via melt infiltration into a CMK-3 type carbon, which successfully absorbed 2.5–3 wt% H2 in five consecutive absorption-desorption cycles at T = 400 °C–500 °C [180]. A stoichiometric bimetallic compound, LiK(BH4)2 can be prepared mechanochemically from the pure borohydrides, because the volume per formula unit (V/Z) of this compound is smaller than that of the reactants [174]. Interestingly, the binary compound does not form upon cooling.
In contrast, the sodium borohydride-potassium borohydride (NaBH4–KBH4) system forms a solid solution upon thermal treatment at 200 < T < 450 °C, apparently with solubility in the full range of compositions. Upon cooling, a solid solution, Nax K1−x (BH4) easily crystallises. The difference in melting properties is due to the fact that the volume of the solid solution is larger than that of the reactants and its formation is facilitated by thermal expansion [174, 175].
When multiple cations are involved in mixtures of borohydrides, the liquid phase is usually stabilized at lower temperature with respect to pure components, either because of the formation of eutectics or thermal minima. Due to this liquid stabilisation, hydrogen evolution is obtained from the liquid at lower temperature. The formation of a stable liquid at low temperature is also beneficial for nanoconfinement approaches to form nanosized materials, with enhanced hydrogen kinetics and cyclability properties [181–184]. The favourable effect of nanosizing has recently been confirmed and evidenced in theoretical studies and calculations for the decomposition of pure Ca(BH4)2 [185].
Furthermore, when complex mixed cations in ternary, quaternary, or even quinary equimolar mixtures are investigated, the formation of low-melting borohydrides is confirmed, with a limited formation of solid solutions and stoichiometric compounds [186, 187]. Mixed cation borohydrides interact more in the liquid phase than in the solid. On the other hand, the study of double charged cations mixture, as in the case of the Mg(BH4)2 and Ca(BH4)2 system, evidences a very limited interaction in the solid state, while a low temperature liquid was not observed, but a decreased decomposition temperature through complex reaction steps has been reported [179].
2.2.6. Thermal properties and reactivity
Hydrogen release and uptake from metal borohydrides remains not fully understood and the reactions are often relatively slow and require high temperatures and pressures to bypass the high kinetic barriers. This is illustrated by general difficulties in measuring reliable thermodynamic data for metal borohydrides by the Sievert's method [188]. In some cases, more than one decomposition reaction appears to be involved, which may depend on physical conditions, such as partial pressures, temperature and/or composition of the samples. Melting reactions, formation of viscous liquids with associated bubbling, and frothing during thermolysis of metal borohydrides is also a challenge for physical measurements, such as thermogravimetric analysis or manometric gas release analysis.
An illustrative example is the decomposition of lithium borohydride, LiBH4. The effect of pressure and temperature on equilibrium phases for LiBH4 decomposition processes has been assessed by the CALPHAD method [74]. It mainly decomposes to Li2B12H12 at elevated hydrogen partial pressures, p(H2) = 50 bar during heating to 600 °C for 5 h [189]. At lower hydrogen pressures, an increasing amount of amorphous boron is observed, and Li2B12H12 may decompose to hydrogen-poor Li2B12H12−x [190]. Thus, Li2B12H12 may be avoided depending on the decomposition conditions, to facilitate a successive hydrogen uptake, since Li2B12H12 is not able to be hydrogenated back into LiBH4, even under extreme conditions (e.g. T = 400 °C and p(H2) ⩽ 970 bar) [191].
Divalent metals appear to have a higher tendency to form metal borides as compared to alkali metals. Under hydrogen atmosphere, magnesium borohydride decomposition is somewhat suppressed and melting is observed at 270 °C, followed by a first crystallisation of MgH2, then Mg, and finally MgB2 at 510 °C [192]. However, non-crystalline closo-boranes may also act as intermediate products during decomposition of Mg(BH4)2 in vacuum [189]. It should be recognized that the decomposition of Mg(BH4)2 is complex [193] and sensitive to the nature of the starting materials and reaction conditions; reviews by Saldan [194] and Zavorotynska et al [15] have tabulated many of the reported pathways. The formation of metal borides, MBx , during decomposition has been considered key to enable rehydrogenation into the respective metal borohydride, e.g. studies on MgB2–MHx (M = Li, Na, Mg, Ca) systems, revealing the formation of M(BH4)n (M = Li, Na, Ca) either by direct hydrogenation or by ball-milling under elevated hydrogen pressures [195–197]. Similarly, 75% and 60% of Mg(BH4)2 and Ca(BH4)2 was formed by direct hydrogenation at high hydrogen pressure (>700 bar) and elevated temperature of MgB2 and CaB6, respectively [198, 199]. The high-pressure hydrogenation of MgB2 also results in the macroscopic fusing of particles, which is thought to prevent further hydrogenation [200]. Lastly, AlB2 as an additive to MHx (M = Li, Na, Ca) also proved to enable the formation of the respective metal hydride and up to 83 mol% of NaBH4 was formed [201].
The decomposition pathway of Mg(BH4)2 is different in dynamic vacuum and lower temperatures where Mg(B3H8)2 was observed as the major decomposition product at 265 °C and MgB4H10 was also proposed as an intermediate product [202, 203]. Lower dehydrogenation temperatures over longer time tend to increase the amount of larger borates and a greater quantity of Mg(B3H8)2 was obtained after five weeks at 200 °C in vacuum [204]. Interestingly, Mg(B3H8)2 takes up hydrogen at 250 °C and p(H2) = 120 bar, to transform into Mg(BH4)2 after 48 h [204]. Composites of Mg(B3H8)2ċ2THF–MgH2 (1:2) are even more prone to absorb hydrogen, e.g. at p(H2) = 50 bar and T = 200 °C after only 2 h [205].
Al(BH4)3ċNH3BH3 showed particular promise as a hydrogen storage material as the hydrogen release temperature was observed at 70 °C in an endothermic event (ΔH ∼ 39 kJ mol−1), although rehydrogenation attempts at moderate conditions, i.e. 70 °C–100 °C and p(H2) = 150 bar, were unsuccessful [143]. Further derivatives of the compound were investigated, e.g. Al(BH4)3ċCH3NH2BH3, but the slight modification of the ligand accompanies large differences in the hydrogen storage properties and a preferential release of B2H6 instead of H2 was observed [206].
Bi- and trimetallic borohydrides tend to dissociate to the monometallic borohydrides upon heating to the temperature of the least stable component, and then the metal borohydrides decompose individually. In some cases, they may also form eutectic melting composites, as described above. In case the bi- and trimetallic borohydrides are created from less stable metal borohydrides, i.e. they contain the metals Al, Sc, Mn, Zn, or Cd, then the corresponding metal borohydride compositions will decompose upon dissociation, often involving release of diborane [130, 207, 208].
The stability of metal borohydrides is to some extend influenced by the Pauling electronegativity of the metal, i.e. the higher χP, the lower the decomposition temperature, Tdec [119]. However, the opposite tendency was recently observed for the series of rare earth metal borohydrides, as shown in figure 5 [97]. It was also found that the RE(BH4)2 is more stable than the RE(BH4)3, which is ascribed to the lower charge density on the RE-ion.

Figure 5. Decomposition temperature (Tdec) of metal borohydrides as a function of Pauling electronegativity. Data is obtained from [3, 15, 81, 95, 97, 107, 173, 199, 207, 209].
Download figure:
Standard image High-resolution imageAdditives have been much debated in the scientific literature regarding the identification of a catalyst for the formation/breaking of the B–H bond. To date, no clear evidence of the existence of such a catalyst exists, but some additives appear to have a positive effect on the reaction kinetics, as grain refiners or enhancing the nucleation sites formation for reaction products. In most cases, the high reactivity of metal borohydrides leads to chemical reactions also with typically inert additives, such as gold and quartz [210].
Nanoconfinement has been explored as an effective strategy to alter the thermodynamic and kinetic properties of borohydrides for hydrogen storage. In addition to affecting reaction enthalpy, changing sizes or morphology can also alter reaction pathways, by shifting the thermodynamic equilibrium for forming different compounds or phases. Other advantages include the promotion of reversible hydrogen desorption/adsorption through accelerated kinetics. In combination with synthesis and characterization, advanced simulation techniques have been used to provide mechanistic understanding of how the thermodynamic and kinetic properties are tuned by nanoconfinement. One target material for these studies has been the Mg–B–H system, which has one of the highest theoretical capacities among the borohydride systems. The effect of pressure and temperature on equilibrium phases for Mg(BH4)2 has been assessed by the CALPHAD method [16]. Jeong et al [211] evaluated the free energies and nucleation barriers for phase transformation between the α, β, and γ-phases of Mg(BH4)2 using classical nucleation theory and revealed that there is a subtle balance between the thermodynamic driving force and nucleation kinetics that controls polymorphism in Mg(BH4)2 [211]. These simulations successfully explained the experimental observations of obtaining distinct Mg(BH4)2 phases by following different synthetic routes. In addition, the simulation results provided guidance for future control of borohydride phases through synthesis within well-controlled thermal and chemical environments.
Apart from introducing physical interactions to preserve particle size, a nanoconfining host can also chemically interact with metal hydrides to directly modify dehydrogenation pathways. One example for confined borohydrides can be found in the recent study by Schneemann et al [212], in which a metal-organic framework (MOF), Zr6O4(OH)4(bpydc)6 (bpydc2− = 2,2'-bipyridine-5,5'-dicarboxylate) was used to confine Mg(BH4)2 and enhance hydrogen desorption kinetics. First-principles simulations confirmed that observed improvements for hydrogen desorption are attributable to the Mg(BH4)2 interaction with the MOF functional groups (both the ligands and metal centre). It was shown that the activation energies for B–H dissociation are greatly reduced at the Mg sites that are directly coordinated with the N and O atoms in the MOF. The chemical interactions between infiltrated Mg(BH4)2 and the MOF host were probed by comparing simulated and measured x-ray absorption spectra, demonstrating the power of an integrated theory-experiment approach to unravel the reaction mechanisms and identify thermodynamic and kinetic limitations for multistep processes. More generally, the study affirms the merit of tuning complex metal hydride dehydrogenation reaction kinetics by manipulating local chemistry at host-hydride interfaces.
Other recent studies have focused on the Mg–B–H system from the fully dehydrogenated MgB2 product to identify pathways and limitations. One such fundamental investigation focused on the structure of the MgB2 surface, which significantly impacts the nature of initial H2 interactions during phase decomposition [213]. The authors combined first principles-derived global optimization and free energy sampling methods with experimental synthesis and spectroscopic characterization to investigate the surface atomic structure. They showed that longstanding assumptions in the chemical community regarding the static nature of boron surface structures in MgB2 and related layered metal diboride compounds may be incorrect; instead, the surfaces are disordered and fluctuate dynamically under conditions relevant to hydrogenation. This dynamic and highly heterogeneous surface environment introduces intrinsic heterogeneity of surface reactivity for H2 activation and initial H binding, which can translate to uneven hydrogen uptake behaviour as the reaction progresses. Another study by Ray et al [214] directly probed the interaction of exposed MgB2 edges with H2 using ab-initio molecular dynamics accelerated by elevated temperature and pressure conditions. Building on an earlier investigation hypothesizing that such edges can uptake limited quantities of H2 at far more reasonable conditions than the bulk material [215], the authors generated edge-enriched models to show that these areas are indeed active for hydrogenation. They subsequently mapped out possible chemical reaction pathways in the pre-nucleation regime, illustrating how chemistry might proceed at the buried solid interfaces.
To increase the concentration of reactive edges, Li et al [216] synthesized nanoscale Mg–B materials in the absence of a nanoconfining host using a solvent-assisted ball milling approach [216]. Although such morphologies are difficult to maintain upon cycling, they provide insight into the independent size effects that govern nanoscale behaviour. The authors showed that partial hydrogenation could be achieved at 280 °C, which is 100 °C below the threshold for bulk MgB2 hydrogenation, with a tenfold enhancement in the H–H dissociation rate. DFT calculations were used to reveal the fundamental surface chemistry and interaction with remaining oleate solvent molecules, which were found to further mitigate oxidation.
2.3. Borates
Boranes, Bx
Hy
, are known to have a very diverse chemistry, with all known neutral compounds being flammable, sometimes even explosive, when coming in contact with air, and they are usually rather toxic. However, anionic complexes also exist, where some are highly stable and relevant for design and development of novel 'energy materials', both for solid-state batteries or hydrogen storage. Here, the review focuses on 'smaller borates', such as  , and 'larger borates' dominated by the closo-borates, such as B10H10
2− and B12H12
2− [217].
, and 'larger borates' dominated by the closo-borates, such as B10H10
2− and B12H12
2− [217].
As introduced above, Mg(B3H8)2 is of interest due to its potential to undergo a reversible dehydrogenation-rehydrogenation cycle with Mg(BH4)2 at moderate temperatures [204]:
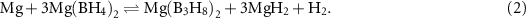
Most syntheses of Mg(B3H8)2 result in compounds containing coordinated solvent molecules, and it is only recently that a solvent-free synthesis has been reported [218], albeit with poor crystallinity and two equivalents of NaBr intermixed within the solid. Monovalent KB3H8 is more readily synthesized, solvent-free, and it is interesting to compare this compound with Mg(B3H8)2. KB3H8 exhibits a complex structural behaviour, with four polymorphs observed between −3 °C and 57 °C [219, 220]. The structural changes are related to the reorientational dynamics of the  anion, in which bridging H atoms are situated outside only 2 of the 3 sides formed by the triangle of B atoms. Similar complex polymorphism has recently also been reported for CsB3H8 [221]. The dynamics and lack of three-dimensional crystallinity are likely reasons why
anion, in which bridging H atoms are situated outside only 2 of the 3 sides formed by the triangle of B atoms. Similar complex polymorphism has recently also been reported for CsB3H8 [221]. The dynamics and lack of three-dimensional crystallinity are likely reasons why  forms during dehydrogenation of Mg(BH4)2, despite unfavourable thermodynamic predictions [222]. Similarly, another recent DFT study confirmed that the solid-state reactive environment provides a unique energy landscape that can stabilize formation of
forms during dehydrogenation of Mg(BH4)2, despite unfavourable thermodynamic predictions [222]. Similarly, another recent DFT study confirmed that the solid-state reactive environment provides a unique energy landscape that can stabilize formation of  and other species kinetically [223].
and other species kinetically [223].  has not been observed during KBH4 dehydrogenation because the decomposition occurs at temperatures above which KB3H8 is stable.
has not been observed during KBH4 dehydrogenation because the decomposition occurs at temperatures above which KB3H8 is stable.
The thermal decomposition of KB3H8 and Mg(B3H8)2 shows some similarities, but their reaction with hydrogen and metal hydrides is noticeably different, as shown in figure 6. Mg(B3H8)2 begins to lose H2 at ≈100 °C [224], while for KB3H8 it initiates at ≈150 °C–200 °C [220]. Both disproportionate into the corresponding borohydride and some combination of closo-borates B10H10
2− and B12H12
2−. According to equation (2) above, hydrogenation to the corresponding borohydride will require both H2 gas and a metal hydride. However, this does not appear to take place for KB3H8 + KH. Although a slightly different balance of  and closo-borates was observed compared to heating KB3H8 alone in an inert atmosphere, this appears to be because less dehydrogenation took place after heating at 200 °C under 460 bar H2 [220]. PXD suggested that the amount of KH had not changed significantly, but B9H9
2−, B10H10
2−, and B12H12
2− were all observed after heating under high H2 pressure. Only B12H12
2− formed when heating under an inert atmosphere. In contrast, Mg(B3H8)2 ball-milled with MgH2 is readily hydrogenated to borohydride with just 6 bar H2 pressure (figure 6) [224].
and closo-borates was observed compared to heating KB3H8 alone in an inert atmosphere, this appears to be because less dehydrogenation took place after heating at 200 °C under 460 bar H2 [220]. PXD suggested that the amount of KH had not changed significantly, but B9H9
2−, B10H10
2−, and B12H12
2− were all observed after heating under high H2 pressure. Only B12H12
2− formed when heating under an inert atmosphere. In contrast, Mg(B3H8)2 ball-milled with MgH2 is readily hydrogenated to borohydride with just 6 bar H2 pressure (figure 6) [224].

Figure 6. Percentage of  and closo-borates formed during thermal decomposition and hydrogenation of KB3H8 and Mg(B3H8)2. Data taken from [220, 224].
and closo-borates formed during thermal decomposition and hydrogenation of KB3H8 and Mg(B3H8)2. Data taken from [220, 224].
Download figure:
Standard image High-resolution imageThis is surprising given that the reverse reaction, formation of Mg(B3H8)2, is observed during the decomposition of Mg(BH4)2. However, the nature of Mg(B3H8)2 is different in the two cases: the material which is hydrogenated starts as a discrete and partially crystalline form, while that made during Mg(BH4)2 decomposition is amorphous and intimately mixed with hydride and other borate anions. This substantially changes the formation energy for  [223, 225, 226]. The reason for the stark difference between hydrogenation of KB3H8 and Mg(B3H8)2 is not clear and should be investigated in future studies.
[223, 225, 226]. The reason for the stark difference between hydrogenation of KB3H8 and Mg(B3H8)2 is not clear and should be investigated in future studies.
Lewis bases, such as ether solvents, also affect the reaction chemistry of hydridoborates. Tetrahydrofuran (THF) and dimethoxyethane (monoglyme) promote dehydrogenation of Mg(BH4)2 at lower temperature, and direct the products toward Mg(B3H8)2 and MgB10H10 [227], instead of the irreversible MgB12H12. The chain length of glymes has a strong and surprisingly non-systematic effect on the dehydrogenation products [228]. Stoichiometric additions of monoglyme, triglyme, or tetraglyme all promote dehydrogenation at low temperature (200 °C or less) while the diglyme complex appears to be stable. Increasing the quantity of monoglyme increases the quantity of  undergoing dehydrogenation, but reduces selectivity toward B10H10
2−, while increasing the quantity of tetraglyme shuts down dehydrogenation completely [228]. These results suggest that Lewis bases undergo complex interactions, most likely with both the metal ion as well as neutral boranes and potentially anionic hydridoborates. For example, THF may assist the formation of
undergoing dehydrogenation, but reduces selectivity toward B10H10
2−, while increasing the quantity of tetraglyme shuts down dehydrogenation completely [228]. These results suggest that Lewis bases undergo complex interactions, most likely with both the metal ion as well as neutral boranes and potentially anionic hydridoborates. For example, THF may assist the formation of  by 'shuttling' BH3 between borohydrides and leaving a hydride [228]:
by 'shuttling' BH3 between borohydrides and leaving a hydride [228]:
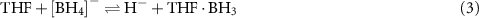
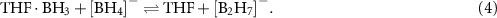
Further reaction of  with BH3 and elimination of H2 would form the observed product
with BH3 and elimination of H2 would form the observed product  . A similar mechanism involving a complex of THF with a neutral borane has been proposed in the synthesis of hydridoborates [229]. This work showed how the THF adduct of the neutral triborane B3H7 is formed from an acidic solution of KB3H8. Subsequent reaction of THF·B3H7 with Na or Li borohydride forms the corresponding alkali octahydridoborate:
. A similar mechanism involving a complex of THF with a neutral borane has been proposed in the synthesis of hydridoborates [229]. This work showed how the THF adduct of the neutral triborane B3H7 is formed from an acidic solution of KB3H8. Subsequent reaction of THF·B3H7 with Na or Li borohydride forms the corresponding alkali octahydridoborate:
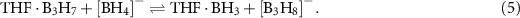
This synthetic study is consistent with the dehydrogenation studies showing that utilizing Lewis base adducts provides an appraoch for directing reactions toward favourable, i.e. reversible, borates.
Larger arachno-borates, such as  , also show great promise as energy materials. Their stability is generally in between the smaller borates and the larger closo-borates. As such, it has been shown that the
, also show great promise as energy materials. Their stability is generally in between the smaller borates and the larger closo-borates. As such, it has been shown that the  anion can degrade to B11H13OH− upon heating in an acidic environment [230]. The hydrated LiB11H14ċ(H2O)n
and NaB11H14ċ(H2O)n salts show exceptional ionic conductivity above 1 × 10−4 S cm−1 and 1 × 10−3 S cm−1 at room temperature, respectively [230].
anion can degrade to B11H13OH− upon heating in an acidic environment [230]. The hydrated LiB11H14ċ(H2O)n
and NaB11H14ċ(H2O)n salts show exceptional ionic conductivity above 1 × 10−4 S cm−1 and 1 × 10−3 S cm−1 at room temperature, respectively [230].
The closo-borates, such as B10H10 2− and B12H12 2−, only have terminal hydrogen atoms, i.e. bonded to boron with a single covalent bond (2c, 2e bond) and are therefore rather stable. Metal closo-borates are often synthesised by cation exchange in aqueous solutions, this includes calcium decahydrido-closo-decaborate, α- and β-CaB10H10, where a variety of hydrates are also described, CaB10H10ċxH2O, x = 1, 4, 5, 6, 7 [231]. Owing to their high stability, this class of material has been considered a 'hydrogen sink' for the release and uptake of hydrogen and it was expected to hamper their reversible hydrogen storage.
Hydrogen release and absorption reactions in boron-based hydrides has been debated in the past and a full description has not yet been obtained due to the complex chemistry of borates. To an extent, this is due to the possibility of several simultaneous reaction mechanisms, which depend on physical conditions, such as hydrogen partial pressures and temperature. Additional computational studies will be of great value to provide further insight into the reversibe pathways.
Initially, the composites, M2B12H12–10MH, M = Li, Na or K were hydrogenated at somewhat harsh conditions, p(H2) = 1000 bar and 500 °C for 72 h, where the corresponding metal borohydrides were obtained as the reaction products, MBH4, M = Li, Na or K [180, 215]. Later, hydrogenation studies of closo-polyborate containing composites, M2B10H10–8MH and M2B12H12–10MH, M = Li or Na, using less harsh conditions, were carried out allowing partial hydrogenation and formation of the respective metal borohydride, MBH4, at T = 300 °C, p(H2) = 500 bar over 24–48 h. It became clear that the M2B10H10–8MH composites react more readily with hydrogen gas compared to M2B12H12–10MH and that the sodium-containing composites are more reactive compared to the lithium analogues. Several intermediate compounds were observed, but not fully identified, which may suggest that the two closo-borate cages, B10H10 2− and B12H12 2−, are hydrogenated via different mechanisms.
Recently, a wide range of metal closo-borates, both mono- and divalent metals, such as Ag2B10H10, Ag2B12H12, SrB10H10, and MnB10H10, have been prepared, characterized and investigated [232, 233]. Two types of anion substitution have been shown to be possible for these materials, which is best illustrated by iodine substitution in the solid state and formation of Ag3B10H10I and Ag3B12H12I [232], while a series of partially iodinated closo-decaborates, M2[B10H10−n In ] (M = Li, Na; n = 1, 2, 10), have been observed [234]. Closo-borates can also be per-halogenated, illustrated by the series of compounds Na2B12X12, X = Cl, Br, I [235, 236]. Interestingly, a range of other substituents can also be associated to the closo-cages with high stability, illustrated by investigation of hydroxylated closo-dodecaborates M2B12(OH)12, M = Li, Na, K, and Cs. Derivatives with neutral molecules have also been discovered, mainly with protic hydrogens which coordinate via hydrogen bonding, but also others containing donor atoms which can coordinate to a metal in the structure, such as NH3, H2O etc observed for e.g. CaB10H10ċxH2O, x = 1, 4, 5, 6, 7 [231].
Interestingly, other solvated closo-borate derivatives show low temperature melting behaviour with Li2B12H12 solvated with either acetonitrile or THF melting below 150 °C [237]. The molten closo-borates can then be melt-infiltrated into porous scaffolds and then demonstrate high ionic conductivity in the molten state (∼1 × 10−4 S cm−1) [237]. The high stability also allows substitution directly in to the closo-polyborate anions, e.g. with carbon creating a distinct class of closo-carborate based materials. Currently, these new materials have high academic interest, but their complicated synthetic route, and therefore high price, hamper their widespread utilisation in all-solid-state batteries.
2.4. Ammines
Ammonia readily reacts with metal borohydrides and ammine derivatives are described for the majority of the known metal borohydrides, with ammonia contents ranging from x = 0.5–8 per metal ion. There are also few reports of ammine metal closo-borates, which appear to coordinate a greater amount of NH3, possibly stabilized by the larger anion [238–240]. Ammine metal-dodecahydro-closo-dodecaborates, Mx B12H12·nNH3 (M = Li, Na, Ca, n in the range 2–7), but also Na2B10H10·nNH3 (n = 1, 2) have been described recently [238, 241]. Table 2 provides an overview of all known ammine metal borohydrides and the corresponding structural dimensionality. The highest amount of ammonia is absorbed in Zr(BH4)4ċ8NH3 [111, 242], while low charge-density metal ions such as MBH4 (M = Na, K, Rb and Cs) do not coordinate ammonia [3]. The largest range of structures are observed for Y(BH4)3ċxNH3, with x = 1, α− 2, β− 2, α− 3, β− 3, 5, 6, 7, where α and β refer to a low- and high temperature polymorph, respectively [96, 98, 110, 243, 244]. The yttrium compounds are isostructural with several members of the ammine REBs [96].
Table 2. Ammine metal borohydrides with known composition and the dimensionality of the structures. 3D: three-dimensional frameworks; 2D: two-dimensional layers; 1D: one-dimensional chains; 0D: neutral molecular complexes; CI: complex cations and anions; L: liquid at ambient conditions; U: unknown crystal structure.
| Ammonia content, x in M(BH4)n ċxNH3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compounds | ½ | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | References |
| Mono-metallic | ||||||||||
| LiBH4ċxNH3 | 2D | 1D | La | Ua | — | — | — | — | — | [245–247] |
| Be(BH4)2ċxNH3 | — | — | — | — | CI | — | — | — | — | [248] |
| Mg(BH4)2ċxNH3 | — | 1D | 0D | 0D | — | — | CI | — | — | [249–251] |
| Ca(BH4)2ċxNH3 | — | 3D | 2D | — | 0D | — | CIa | — | — | [109, 252, 253] |
| Sr(BH4)2ċxNH3 | — | 2D | 2D | — | 0Da | — | — | — | — | [109, 253, 254] |
| Ba(BH4)2ċxNH3 | — | 3D | 2Da | — | — | — | — | — | — | [255] |
| Al(BH4)3ċxNH3 | — | 0D | — | — | — | — | CI | — | — | [256, 257] |
| Ti(BH4)3ċxNH3 | — | — | — | U | — | U | — | — | — | [258] |
| V(BH4)3ċxNH3 | — | — | — | 0D | — | — | — | — | — | [259] |
| Mn(BH4)2ċxNH3 | — | U | 0D | 0D | — | — | CI | — | — | [251] |
| Fe(BH4)2ċxNH3 | — | — | — | — | — | — | CIa | — | — | [113] |
| Co(BH4)2ċxNH3 | — | — | — | — | — | — | CIa | — | — | [113] |
| Zn(BH4)2ċxNH3 | — | — | 0D | — | U | — | — | — | — | [260] |
| Zr(BH4)4ċxNH3 | — | — | — | — | — | — | — | — | CI | [242] |
| Y(BH4)3ċxNH3 | — | 2D | 1D (α, β) | 0D (α, β) | — | CI | CI | CI | — | [96, 98, 110, 243, 244] |
| La(BH4)3ċxNH3 | — | — | — | 0D | 0D | — | CI | — | — | [96, 98, 110] |
| Ce(BH4)3ċxNH3 | — | — | — | 0D | 0D | CI | CI | — | — | [96, 98, 110] |
| Pr(BH4)3ċxNH3 | — | — | — | 0D | 0D | CI | CI | CI | — | [96] |
| Nd(BH4)3ċxNH3 | — | — | — | 0D | 0D | CI | CI | CI | — | [96] |
| Sm(BH4)2ċxNH3 | — | 2D | 2D | — | — | — | — | — | — | [96] |
| Eu(BH4)2ċxNH3 | — | 2D | 2D | — | — | — | — | — | — | [96] |
| Gd(BH4)3ċxNH3 | — | — | — | 0D | — | CI | CI | CI | — | [96, 110] |
| Tb(BH4)3ċxNH3 | — | — | — | 0D | — | CI | — | CI | — | [96] |
| Dy(BH4)3ċxNH3 | — | — | — | 0D | — | CI | CI | CI | — | [96, 110] |
| Ho(BH4)3ċxNH3 | — | — | — | 0D | — | CI | — | CI | — | [96] |
| Er(BH4)3ċxNH3 | — | — | — | 0D | — | CI | — | CI | — | [96] |
| Tm(BH4)3ċxNH3 | — | — | — | 0D | — | CI | — | CI | — | [96] |
| Yb(BH4)3ċxNH3 | — | — | — | — | — | CI | — | CI | — | [96] |
| Yb(BH4)2ċxNH3 | — | 3D | 2D | — | 0D | — | — | — | — | [96] |
| Lu(BH4)3ċxNH3 | — | — | — | — | — | — | — | CI | — | [96] |
| Bi-cationic | ||||||||||
| LiMg(BH4)3ċxNH3 | — | — | 3D | — | — | — | — | — | — | [261] |
| Li2Mg(BH4)4ċxNH3 | — | — | — | — | — | — | CI | — | — | [110, 262] |
| NH4Mg(BH4)3ċxNH3 | — | — | 3D | — | — | — | — | — | — | [108] |
| Li2Al(BH4)5ċxNH3 | — | — | — | — | — | — | CI | — | — | [263] |
| LiSc(BH4)4ċxNH3 | — | — | — | — | 1D | — | — | — | — | [259] |
| Li2Ti(BH4)5ċxNH3 | — | — | — | — | — | U | — | — | — | [258] |
| LiMn(BH4)3ċxNH3 | — | — | 3D | — | — | — | — | — | — | [108] |
| Li2Mn(BH4)4ċxNH3 | — | — | — | — | — | — | CI | — | — | [251] |
| NH4Mn(BH4)3ċxNH3 | — | — | 3D | — | — | — | — | — | — | [108] |
| Li2Fe(BH4)4ċxNH3 | — | — | — | — | — | — | CI | — | — | [113] |
| NaZn(BH4)3ċxNH3 | — | — | CI | — | — | — | — | — | — | [264] |
| NH4Y(BH4)4ċxNH3 | — | CI | — | — | — | — | — | — | — | [108] |
| (NH4)2Y(BH4)5ċxNH3 | — | CI | — | — | — | — | — | — | — | [108] |
| NH4La(BH4)4ċxNH3 | — | 1D | — | — | — | — | — | — | — | [108] |
| NH4Gd(BH4)4ċxNH3 | — | 1D | — | — | — | — | — | — | — | [108] |
| Solid solutions | ||||||||||
| Mg1−y Mny (BH4)2ċxNH3 | — | — | — | — | — | — | CI | — | — | [251] |
| Co(BH4)2−y Cly ċxNH3 | — | — | — | — | — | — | CIa | — | — | [113] |
As described above, the crystal structures of metal borohydrides often consist of three-dimensional frameworks formed by bridging  groups, resulting in connected tetrahedral [M(BH4)4] or octahedral [M(BH4)6] complexes [68]. Ammonia interrupts these frameworks, and the dimensionality of the structures decreases with increasing ammonia content. In all structures of ammine metal borohydrides, NH3 coordinates to the metal-ion via its lone pair, while the coordination of
groups, resulting in connected tetrahedral [M(BH4)4] or octahedral [M(BH4)6] complexes [68]. Ammonia interrupts these frameworks, and the dimensionality of the structures decreases with increasing ammonia content. In all structures of ammine metal borohydrides, NH3 coordinates to the metal-ion via its lone pair, while the coordination of  is more flexible and it may act as a bridging and terminal ligand and as a counter ion, accommodating the desired coordination of the metal ion. This is well illustrated by the series of Y(BH4)3ċxNH3 [96, 98, 110]. Y(BH4)3ċNH3 forms two-dimensional layers through bridging
is more flexible and it may act as a bridging and terminal ligand and as a counter ion, accommodating the desired coordination of the metal ion. This is well illustrated by the series of Y(BH4)3ċxNH3 [96, 98, 110]. Y(BH4)3ċNH3 forms two-dimensional layers through bridging  groups, connecting the Y(NH3)(BH4)5 octahedra. The layers are held together by dihydrogen interactions between NH3 and
groups, connecting the Y(NH3)(BH4)5 octahedra. The layers are held together by dihydrogen interactions between NH3 and  . Y(BH4)3ċ2NH3 forms one-dimensional zig-zag (α) or straight (β) chains via bridging
. Y(BH4)3ċ2NH3 forms one-dimensional zig-zag (α) or straight (β) chains via bridging  groups connecting the Y(NH3)2(BH4)4 octahedra. Both α- and β-Y(BH4)3ċ3NH3 are built from neutral Y(NH3)3(BH4)3 octahedral complexes, while Y(BH4)3ċ5NH3 is built from [(NH3)5(BH4)2]+ complex ions, and it is a rare example of borohydride groups acting as both a coordinating ligand and counter ion in the same compound. Y(BH4)3ċxNH3 (x = 6 and 7) are both built from cationic Y(NH3)x
3+ complexes, where all
groups connecting the Y(NH3)2(BH4)4 octahedra. Both α- and β-Y(BH4)3ċ3NH3 are built from neutral Y(NH3)3(BH4)3 octahedral complexes, while Y(BH4)3ċ5NH3 is built from [(NH3)5(BH4)2]+ complex ions, and it is a rare example of borohydride groups acting as both a coordinating ligand and counter ion in the same compound. Y(BH4)3ċxNH3 (x = 6 and 7) are both built from cationic Y(NH3)x
3+ complexes, where all  act as counter ions. Note that the structure of Y(BH4)3ċ5NH3 and both the structure and composition of Y(BH4)3ċ3NH3 were recently revised, where the latter was previously suggested to have the composition Y(BH4)3ċ4NH3 [96, 110, 243, 244].
act as counter ions. Note that the structure of Y(BH4)3ċ5NH3 and both the structure and composition of Y(BH4)3ċ3NH3 were recently revised, where the latter was previously suggested to have the composition Y(BH4)3ċ4NH3 [96, 110, 243, 244].
In an 'open' system, the thermal stability of ammine metal borohydrides and the composition of the released gas, i.e. H2 vs NH3, can be correlated to the M–N bond strength and to the thermal stability of the metal borohydrides, as shown in figure 7 [3, 96, 109]. Other factors have also been suggested to affect the thermal stability and gas-composition, e.g. strong dihydrogen bonds and the relative amount of NH3 to  , but these hypotheses have been disproved by the ammine metal borohydrides with a Pauling electronegativity (χP) less than 1.05.
, but these hypotheses have been disproved by the ammine metal borohydrides with a Pauling electronegativity (χP) less than 1.05.
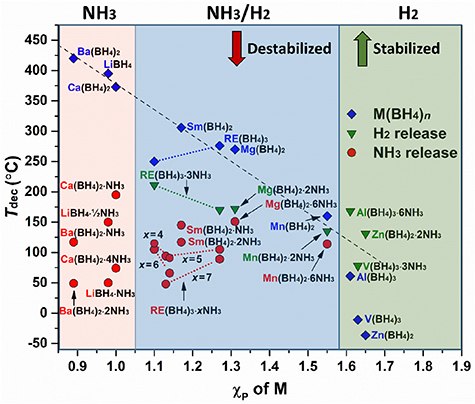
Figure 7. Decomposition temperature (differential scanning calorimetry peak temperature) as a function of the Pauling electronegativity (χP) for selected metal cations. The colored regions divide compounds releasing NH3 (red), both NH3 and H2 (blue), and H2 (green) in an 'open system'. Ammine metal borohydrides with χP < 1.58 are destabilized by ammonia, while they are stabilized for χP > 1.58. Symbols: M(BH4)n (blue squares), M(BH4)n ċxNH3 that mainly release H2 (green triangles), M(BH4)n ċxNH3 that mainly release NH3 (red circle). The black dashed line show the linear correlation between Tdec and χP for the metal borohydrides (M(BH4)n ) [96]. The dotted lines with shorter spacing refers to the series of RE(BH4)3 and RE(BH4)3ċxNH3 [96].
Download figure:
Standard image High-resolution imageAs shown before, the thermal stability of metal borohydrides are approximately inversely proportional to the χP of the metal, illustrated by the black dotted line in figure 7 [119, 265]. Thus, the composition of the released gas during thermolysis of ammine metal borohydrides in an open system can roughly be divided into the red, blue and green regions in figure 7 as regarding the Pauling electronegativity:
- (a)Red region (χP < 1.05): these compounds have a low charge-density cation (weak M–N bond) and a high thermal stability of the metal borohydride, resulting in the release of NH3.
- (b)Blue region (χP ∼ 1.05–1.58): these compounds often release NH3 when the NH3/BH4 ratio is higher than 1. When the ratio is ⩽ 1, the thermal stability of the ammine metal borohydride and the metal borohydride is often similar, i.e. the
 groups are sufficiently destabilized to react with NH3, resulting in the release of H2. However, lowering the NH3 release temperature by vacuum may favor the release of NH3 over H2 [3].
groups are sufficiently destabilized to react with NH3, resulting in the release of H2. However, lowering the NH3 release temperature by vacuum may favor the release of NH3 over H2 [3]. - (c)Green region (χP > 1.58): these compounds have a higher thermal stability than the corresponding metal borohydrides. Theoretical calculations indicate that metal borohydrides are thermodynamically destabilized by ammonia, but NH3 may provide a kinetic stabilization due to a shielding effect, which obstructs the usual decomposition mechanism [259, 266]. Consequently, these compounds mainly release H2.
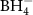
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The divalent RE(BH4)2ċxNH3 (RE = Sm, Eu, Yb) are exceptions to this simple relation with χP, as they only release NH3 during thermolysis, despite having a χP ∼ 1.1–1.2. This is related to the higher thermal stability of the metal borohydride [3, 97]. Calculations of the ionic potentials of the cations may provide a more accurate correlation, as demonstrated for the metal borohydrides [136, 267, 268].
The partial pressure of ammonia during decomposition strongly affects the decomposition temperature and the composition of the released gas. The ammonia release temperatures are significantly lowered by vacuum compared to a closed capillary, as observed for RE(BH4)3ċxNH3 (RE = La, Ce) [98]. A high partial pressure of NH3 can facilitate the release of H2, as observed for thermolysis of LiBH4ċNH3 and Ca(BH4)2ċxNH3 (x = 1, 2) in a closed vessel [252, 269]. Alternative strategies that can promote hydrogen evolution include fluorine-substitution [270], catalysis using metal nanoparticles or metal halides [271–273], or by nano-confinement [274, 275].
2.5. Amides and imides
The reaction of ammonia with alkali and alkaline earth metals or metal hydrides leads to the formation of amides such as LiNH2, NaNH2, KNH2, Mg(NH2)2, Ca(NH2)2 and Ba(NH2)2. The partial decomposition of the amides often leads to the formation of imides, for instance Li2NH, Na2NH, MgNH, CaNH and BaNH. The amides first came to the limelight in the early 2000's, when it was reported that the combination of LiNH2 and LiH releases reversibly about 6.5 wt% hydrogen at around 250 °C [276]. This is unlike the pure LiNH2 and LiH that release hydrogen at temperature above 300 °C and 550 °C, respectively. In fact, the enthalpy and entropy associated with the decomposition of LiNH2 + LiH to hydrogen is ΔH = 38.9 kJ mol−1 H2 and ΔS = 111.98 J K−1 mol−1 H2, respectively, implying that an equilibrium pressure of p(H2) = 1 bar at 74 °C is expected [277]. Hence, the use of different catalysts was explored to reduce the decomposition temperature of the mixture. This also led to several efforts to explore other metal amides and their combinations with metal hydrides for hydrogen storage applications [278–282].
Although research on applications of amides/imides for hydrogen storage materials is still progressing [283–293], most recent efforts have been focused on their application as catalysts or co-catalysts in ammonia synthesis and decomposition, as solid-state electrolytes for batteries, and novel amide/imides with multifunctional energy applications [285, 294–302]. For instance, by ball milling mixtures of lithium amide, lithium imide and lithium nitride–hydride, Makepeace et al [296] recently reported an amide-imide solid solution with wide-ranging anion compositions. Samples from majority of the lithium amide through to pure lithium imide and majority lithium nitride–hydride show a highly disordered anti-fluorite structure, with evidence for greater nitride–hydride incorporation at high temperatures. In contrast, the tetragonal lithium amide and nitride–hydride structures show very limited compositional variation.
Raman spectroscopy of lithium amide–lithium imide samples show that the amide and imide groups in stoichiometric and nonstoichiometric environments can be differentiated by the different frequency of the N–H stretch, as they show distinct bands in both environments. Thus, continuous variation in the local NHx environment(evidenced by N–H strecht) is observed as a function of the composition [303]. This rich library of stoichiometric compounds and solid solutions is a basis for expanded investigations into composition control and tailored hydrogen storage properties in M–N–H materials or mixtures. Additionally, they have been proposed for variety of applications such as ion conduction, ammonia storage, catalysts/co-catalysts for ammonia synthesis and catalytic decomposition [296]. Furthermore, it has been recently reported that novel compounds are formed in the LiBH4–LiNH2 system, and nanocomposites of them with mesoporous silica have been explored [301]. After heating or ball milling mixtures of LiBH4 and LiNH2, the presence of multiple Li(BH4)1−x (NH2)x phases is clearly observed by PXD and further corroborated with DSC and diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) analysis. By DSC, different LiBH4–LiNH2 phases can be distinguished by their specific melting temperature, revealing up to three different phases; such as Li2(BH4)(NH2), Li3(BH4)(NH2)2 and Li4(BH4)(NH2)3 in the prepared LiBH4–LiNH2 mixtures, depending on the composition of the starting materials and the synthesis temperature [304]. The different phases show different properties both for reversible hydrogen release and Li ion conduction. For instance, lower hydrogen release temperature is favoured for the phases with low LiNH2 content, while the Li-ion conductivity increases with increasing LiNH2 (up to 50 mol% LiNH2) content and then decreases above 50% LiNH2 in the mixture [301].
In terms of hydrogen storage properties of amide-imides, Gizer et al [284] recently investigated the influence of a K2Mn(NH2)4 additive on the hydrogen sorption properties of the Mg(NH2)2 + 2LiH (i.e. Li–Mg–N–H) system. It was demonstrated that the addition of K2Mn(NH2)4 (5 mol%) leads to a decrease of the peak dehydrogenation temperature from 200 to 172 °C, with a hydrogen storage capacity of 4.2 wt% maintained over 25 hydrogen soprtion cycles. In-situ synchrotron radiation-PXD analysis revealed that K2Mn(NH2)4 decomposes into Mn4N and a potassium compound, most likely amorphous or nanocrystalline KH. Therefore, the increased hydrogen sorption kinetics was ascribed to the presence of KH and Mn4N, which alter the hydrogen sorption properties of the system [284]. In a related study, the hydrogen sorption properties of the ternary amide-hydride LiAl(NH2)4–LiAlH4 system was investigated by means of mechanical milling and thermal methods. It was shown that the mixture decomposes spontaneously at ambient conditions during mechanical milling, releasing 70% of the H2 (six equiv. of H2 or 5.7 wt% H2) within 2 h of mechanical milling. The hydrogen release rate increases with increasing amount of LiAlH4, suggesting an ion migration mediated dehydrogenation. The hydrogen release reaction occurs via the formation of Li3AlH6 as an intermediate, and LiH and AlN as the final products, with a total of eight equiv. of H2 (7.5 wt%). For the physically mixed LiAl(NH2)4–LiAlH4 (1:3) which are not in intimate contact, NH3 mediated dehydrogenation is prone to occur. However, the decomposed residue, LiAl(NH)2 reacted with LiAlH4, leading to rapid hydrogen release. The basicity of the hydrides was found to be an important point in triggering amide-hydride interactions, which leads to low hydrogen release temperatures [286]. In another recent study, Zhang et al [291] examined the effects of Na addition on the reversible hydrogen uptake of Mg(NH2)2–MgH2 system. The addition of the Na leads to the formation of NaMgH3. They demonstrated that the replacement of NaMgH3 for MgH2 suppresses ammonia release and improves the hydrogen desorption property of the Mg(NH2)2–NaMgH3 system. At high temperature (190 °C), the formation of NaMg(NH2)(NH) was detected, suggesting that the addition of Na alters the reaction pathway. The authors demonstrated that the formation NaMg(NH2)(NH) is responsible for the partial reversibility (1.8 wt% of hydrogen at 10 bar and 190 °C) observed in the Mg(NH2)2–NaMgH3 system [291].
The system Li2NH and LiBH4 was recently studied. The Li2NH–LiBH4 sample (in a molar ratio of 1:1) shows very good hydrogenation kinetics, with hydrogen absorption starting at 37 °C at 50 bar, which is more than 100 °C lower than that of pristine Li2NH, and amongst the mildest reported for amide systems [285]. The Li5(BH4)3NH complex hydride, obtained by ball milling LiBH4 and Li2NH in various molar ratios, has been identified. The crystal structure has been solved in space group Pnma, and refined coupling DFT and synchrotron radiation x-ray powder diffraction data [305].
Finally, lithium amide/lithium nitride system has a high gravimetric capacity (10.3 wt%), making it an attractive material for hydrogen storage applications. However, in the bulk, the H2 release temperature exceeds 400 °C, and the compounds exhibits slow hydrogen release kinetics and poor reversibility, hindering their applications for reversible hydrogen storage. It has been shown recently that nanoconfinement using porous carbon can lower the dehydrogenation temperature of the material, which was attributed to the thermodynamically penalizing role of internal 'nanointerfaces' [306]. More recent work is tackling this mechanism in greater detail, focusing on the additional role of hydride-host interactions to understand changes in reactivity. Through a combination of ab-initio molecular dynamics and electronic structure calculations alongside experimental nanoconfinement studies in N-doped carbons, the effect of nanoporous carbon host on Li–N–H thermodynamics has been explicitly probed [307]. Authors have found that charge transfer and confinement effects exerted by carbon hosts can significantly stabilize the dehydrogenated Li3N phase over the hydrogenated phases. This points to the strong interfacial interaction between a nanoporous carbon host and Li3N as an additional tunable factor for achieving lower dehydrogenation energy in Li–N–H system.
2.6. Transition metals complex hydrides
The first known transition metal complex hydride, K2ReH9, was fully described in 1964 [308]. Despite the fact that there are now more than a hundred known types of hydrides divided in almost 50 structure types discovered over past decade [8, 9] not many of them bring too much of the attention as potential materials for hydrogen and heat storage, mainly due to the high cost of materials, high thermal stability, not straightforward reversibility, etc.
Mainly due to the relatively low cost, complex hydrides based on iron (Mg2FeH6), nickel (Mg2NiH4) and cobalt (Mg2CoH5) are being intensively studied till now, since they display attractive properties, which can be relevant for hydrogen storage applications. For instance, although its gravimetric hydrogen density, ρm = 5.5 wt% H2 is lower than that of MgH2, Mg2FeH6 exhibits the highest volumetric hydrogen density, ρV = 150 g H2 l−1, among the known metal hydrides and it has excellent hydrogen soprtion cycling properties. For instance, no irreversible capacity losses were observed over 600 cycles carried out between 450 °C and 550 °C and at pressures between 65 and 75 bar [27]. These properties make Mg2FeH6 a promising hydrogen storage material for stationary applications at high temperatures (near 450 °C) [309].
Ball milling is the most commonly used method for mixing Mg and Fe powders and subsequent hydrogenation at high temperature under hydrogen pressure produces Mg2FeH6. Recently, cold roll milling (CRM) has also been adopted to produce Mg/Fe nanocomposite powder with a layered structure [310]. In the CRM process, Mg and Fe powders are premixed with a small amount of oleylamine as a process control agent. The pre-mixed powders were milled using an in-house-made vertical roll milling machine at 50 rpm in air. The milling machine has two vertical rolls of 100 mm in diameter, a vibrational powder feeder capable of regulating the amount and speed of the powder feeding, and an inlet chute for stable spill-free powder entry between the rolls. The fine layered structure improves the hydrogenation reaction kinetics by reducing the diffusion distance of Mg and/or MgH2 through the refinement of Fe particles, which is particularly critical when the hydrogenation temperature is lower than 400 °C [311, 312]. The layer thickness of Fe particles can be controlled by varying the number of CRM passes. The Mg and Fe layers become thinner with increasing number of rolling passes, namely down to 10 nm in the case of Fe, which is enough to support the fast formation of Mg2FeH6. If CRM process is done in air, the preferential oxidation of Mg is unavoidable and the higher the number of CRM passes, the higher the MgO content in the Mg/Fe composite powder. Repeated CRM effectively decreased the Fe layer thickness up to a sufficient value for the fast formation of Mg2FeH6, but too much CRM passes decreased the hydrogen storage capacity due to inevitable oxidation of Mg in air. The thickness of the Fe layer plays an important role in the initial stage, whereas the degree of oxidation and the distribution of oxides have more influence on the formation of Mg2FeH6 in the long term by consuming Mg. Both microstructure refinement and minimal oxidation are the prerequisites for efficient Mg2FeH6 synthesis, with the former condition being achievable by optimizing the number of millings, and the latter by performing CRM under an inert atmosphere.
Recently the synthesis of Mg2FeH6 by ball milling and subsequent sintering was conducted with the use of both plain and highly alloyed austenitic stainless steel (AISI 316L) instead of pure iron [313, 314]. The idea was to see if the alloying elements interfere and influence the synthesis process, as well as if they change the thermodynamic properties of the obtained compound. Using steel (or even steel scrap or scrap in general [315, 316]) might be a way of reducing the price of the material when it is supposed to be used in high quantities as a heat or hydrogen storage medium. It was found that, in the case of plain steel, the properties of the obtained material do not change and the carbon present in the steel does not interact with the hydride nor form solid solutions. It was confirmed, however, that the mechanical properties of the steel (high hardness) may influence the milling process itself, thus contributing to the lowering of the reaction yield. In case of manufacturing from AISI 316L steel, the main finding was that the single phase, austenitic structure of the steel during ball milling undergoes the mechanically induced martensitic transformation and the martensite readily reacts with the magnesium hydride. The final product is likely composed of quaternary Mg–Fe–Cr hydride, possessing different equilibrium pressure as compared to pure Mg2FeH6 and Mg2NiH4.
The synthesis of Mg-based transition-metal complex hydrides containing Fe, Co, or Ni can also be achieved in one processing step by ball milling elemental powder mixtures in a reactive hydrogen atmosphere [102, 317]. The reactive mechanochemical synthesis has the advantage of enhancing hydrogen sorption kinetics, due to the formation of fresh surfaces and reduced particle sizes, allowing hydride formation after only a few hours of milling. It should be noted that, while fresh surfaces are consumed with hydrogen cycling and are critical during the milling process, reduced particle sizes are retained upon cycling [318], playing a beneficial role in maintaining good hydrogen cycling kinetics. Reactive milling has been also successfully used for the synthesis of Mg-based complex hydrides with more than one TM, resulting in the formation of quaternary hydrides such as Mg2(FeH6)0.5(CoH5)0.5 [319, 320].
Recently, the properties of Mg2Fex
Co(1−x)Dy
complex deuterides with different Fe–Co and D contents (0 ⩽ x ⩽ 1 and 5 ⩽ y ⩽ 6) were reported and the results were compared with ternary Mg2CoD5 and Mg2FeD6 compounds [34]. Deuterium was used to allow the structural study with powder neutron diffraction. The crystal structure characterization of the milling products shows that, depending on the relative content of Fe and Co (Fex
Co1−x
), the following hydrides are formed: Mg2(FeD6)0.09(CoD5)0.91, Mg2(FeD6)0.3(CoD5)0.7, Mg2(FeD6)0.4(CoD5)0.6, Mg2(FeD6)0.5(CoD5)0.5, Mg2(FeD6)0.7(CoD5)0.3 and Mg2(FeD6)0.9(CoD5)0.1. In all cases, Fe and Co randomly occupy the same crystallographic site, creating solid solutions containing both FeD6
4− and CoD5
4− complex anions. For x = 1, Mg2(FeD6)0.09(CoD5)0.91 was found to be isostructural either with Mg2CoD5 (tetragonal P4/nmm), whereas for x ⩾ 5, the quaternary hydrides have the same cubic Fm
 m structure as Mg2FeD6 and Mg2CoD5 at high temperatures. For x = 0.3, the presence of a two-phase region, where the tetragonal and cubic phases coexist, was found and supported by the assessment of the stability of the hydrides from experimental and literature data.
m structure as Mg2FeD6 and Mg2CoD5 at high temperatures. For x = 0.3, the presence of a two-phase region, where the tetragonal and cubic phases coexist, was found and supported by the assessment of the stability of the hydrides from experimental and literature data.
All Mg2Fex Co(1−x)Dy quaternary complex hydrides were shown to have a similar hydrogen desorption process and to be relatively stable, but their desorption temperature (maximum desorption temperature Tdec ≅ 277 °C) was below that of MgH2. The activation energy Eades (87–89 kJ mol−1) and enthalpy ΔHdes (about 70 kJ mol−1 H2) of desorption were reported to be independent from the relative amount of complex anions. Desorption of hydrogen results in the formation of Mg and (FeCo) solid solutions, which are found unevenly distributed. Nonetheless, the reversible hydrogenation to form the quaternary hydrides is observed at 30 bar of H2 and 400 °C for Mg2(FeH6)0.5(CoH5)0.5.
Complex transition-metal hydrides containing hydride complexes with hydrogen coordination number exceeding six are quite rare, due to the presence of high energy barriers for their formation process [321]. In addition, if the temperature is raised to promote the formation reaction, the target hydrides will decompose, and, conversely, the reaction will have difficulty in proceeding below the decomposition temperature. One way of overcoming this situation is to apply high hydrogen pressures, of the order of several GPa. As a result, the target hydrides are thermodynamically stabilized, and the temperature can be raised without decomposition to a region where the decomposition reaction would proceed under ambient pressure. Several new hydrides, including Li5MoH11 with the nine-fold H coordination hydride complexes MoH9 3− have been synthesized recently [322–329] using a high-temperature and high-pressure technique, as developed by Fukai et al [330].
2.7. RHCs
Many borohydrides possess very high gravimetric energy densities (i.e. LiBH4-18.5 wt%, MgBH4-14.8 wt%), which makes them interesting for hydrogen storage applications. However, their use in practical applications is hindered while they are thermodynamically stable and poorly reversible. Combining borohydrides, with other hydrides, the reaction enthalpy of the system is decreased, compared to the decomposition enthalpy of the single components. These systems are called 'RHCs', referring to mixtures of different metal hydrides reacting with each other during dehydrogenation, rather than decomposing individually. Similar approach was already shown by Reilly and Wiswall in 1967, where MgH2 and MgCu2 reacts with each other to form Mg2Cu upon hydrogen release [331]. Major disadvantage of this system is the significant capacity reduction due to addition of MgCu2, which does not contribute to the total hydrogen capacity. First example of RHCs were reported by Chen et al in 2002 (LiNH2 + LiH system). Following this work, Bark [196, 332] and Vajo [333] reported destabilization of LiBH4 by combining it with MgH2. Desorption of this system is a two step reaction process. When the backpressure of 5 bar of H2 is supplied (above the equilibrium pressure of LiBH4 alone), firstly MgH2 decomposes and later on, magnesium reacts with with LiBH4 to form MgB2. Absorption reaction can be carried out in single step for the MgB2–LiH composite at moderate pressure-temperature conditions (250 °C, 50 bar) [334]. So far, extensive research is conducted on the RHCs to understand the reaction mechanisms, cycling properties and suitable additives which may help to lower the operation temperatures further [335–342]. In a recent study, Zhao et al developed a novel process, where MgH2 is ball milled in conjunction with aerosol spraying of LiBH4 dissolved in THF [343]. Hydrogen release could be carried out in three steps: (a) H2 release from the decomposition of nano–LiBH4 and then decomposition product Li2B12H12 reacts with nano–MgH2 to release H2, (b) H2 release from the decomposition of nano–Mg(BH4)2, and (c) H2 release from the decomposition of nano–MgH2. These desorption reactions lead to 4.1 wt% H2 release below 265 °C, which is one of the highest quantities for this system up to date. However, H2 capacity of the composite degrades gradually over hydrogenation and dehydrogenation cycles at 265 °C, mainly due to gradual increases in crystallite and particle sizes. From one side, designing the systems at nano-scale gives the opportunity to establish RHCs in solid state with improved kinetics and lower operating temperatures. From the other side, stability of these nano-composites at the operating temperatures of RHCs is of great importance, since it is known that two main reactants, LiH and MgB2 segregate continuosly upon cycling [344]. Therefore, the use of a scaffolding material is an option which may help to improve the cycling stability while keeping the reactants confined. Le et al introduced an inert polymer, poly(4-methyl-1-pentene) or shortly TPXTM, in order to improve the cycling stability of titanium(III) chloride-aluminum chloride added 2LiH + MgB2. The presence of TPXTM does not affect the reaction pathyway of the RHC, which is also supported with determined rate-limiting reactions, and it improves the cycling stability. Decrement in the capacity after ten hydrogenation/dehydrogenation cycles is only 0.05 wt%, while it is 0.32 wt% for the system without TPXTM. Prevention of the phase segregation between the two main phases, due to the presence of scaffolding polymer surrounding the particles, is thought to be the reason for this improvement. One of the main challenges in the design of RHC based hydrogen storage systems is to develop reliable kinetic models to describe sorption reacitons. These models are beneficial to save time and evaluate the feasibility of the system. To ensure the potential of RHCs for the real applications, thermodynamic and kinetic investigations are carried out by Neves et al [345]. In this study, TiCl3 added LiBH4/MgH2 is examined under absorption conditions. Enthalpy and entropy are found to be 34 kJ mol−1 H2 and 70 J K−1 mol−1 H2. Separable variable method is applied to investigate absorption conditions of the developed Li–RHC. Applying this model, the effects of temperature, pressure and transformation of the hydride forming material are considered in the rate expression. According to the results, transformation of the forming hydride is limited by the movement of the not-hydrogenated/hydrogenated material interface, described by the one-dimensional interfacecontrolled model with a fixed number of nuclei and constant interface velocity, also known as Johnson–Mehl–Avrami–Erofeyev–Kolmogorov (JMAEK) with n = 1 model. After taking the driving force component into account, the apparent activation energy Ea and pre-exponential factor A are, respectively, 1.8 × 108 s−1 and 146 kJ mol−1 H2. Developed models could be applied to model the hydrogen storage tanks with RHC materials through FEM simulations, or combined with novel computational approaches as machine learning, in order to better understand and optimize their designs. Modification of the RHCs with an appropriate additive is also an important step towards designing materials with fast reaction kinetics. In a recent study, catalytic role of NbF5 and ScCl3 during the hydrogenation and dehydrogenation reactions were investigated [340, 346]. Addition of NbF5 reduces the dehydrogenation time required for the LiBH4/MgH2 composite from 45 h to 1.5 h. During ball milling, NbF5 transforms to NbB2, which remains stable in the subsequent cycles. In details, Nb reacts with LiBH4 generating NbB2, while F is partially substituting hydrogen in LiH and LiBH4. While in larger amounts the latter leads to a favourable thermodynamic change in the reaction enthalpy, in smaller amounts this effect is negligible [3, 167, 168]. Nano-sized NbB2 particles in the grain boundries act as nucleation seeds for the nucleation and growth of MgB2. Presence of NbB2 nanoparticles considerably lowers the interfacial tension between the matrix and MgB2 nuclei, which can lead to a smaller energy barrier for the nucleation and growth process for MgB2 phase in the doped LiBH4/MgH2 system. Similar to NbF5, ScCl3 transforms to ScB2 nanoparticles during the ball milling. Formation of ScB2 nanoparticles with a stable nanostructure serving as grain refiner and acting as nucleation agent, providing nucleation sites and reducing the energy barrier for nucleation and growth of the MgB2 phase. The resulting smaller nanostructures result in a larger surface area and shorter diffusion path ways, leading to improved reaction kinetics. In order to investigate the affect of additives on RHCs in detail, advance characterization techniques; such as small angle x-ray scattering/ultra small angle x-ray scattering, anomalous x-ray scattering, small angle neutron scattering, in-situ SR-PXD, high resolution transmission electron microscopy, can provide the key informations related to the role of additive in the matrix. With the outcome from these advance characterization techniques with experimental results and computer simulations, current research is still ongoing to further reduce the operating temperatures and improve the reaction kinetics of the RHCs.
3. New trends and properties
As illustrated in the present review, complex hydride-based materials and systems have been extensively investigated in academia to define the best synthesis and processing methods, structural and physical properties, thermodynamics, and kinetics of involved reactions, and developing combined experimental, theoretical, modelling, and calculation methods to further understand and tailor these materials towards possible applications.
Research was first boosted by a great interest in hydrogen storage applications in the solid state, owing to their elevated gravimetric and volumetric content of hydrogen, mainly bonded in the complex anion. High hydrogen density can guarantee low weight and modest volume of storage, as in the case of borohydrides and alanates, advantageous for mobile applications. On the other hand, modest hydrogen density, typical of transition metal complex hydrides, can also be considered for stationary hydrogen storage in compact volumes. Extreme hydrogen densities have been reached in e.g. M(BH4)n ċxNH3, NH4BH4 and NH4–M–BH4 systems [98, 129, 238, 329].
However, the drawback evidenced by basic research on these systems was their limited reversibility and cycling properties, related to harsh conditions of pressure and temperature for hydrogen release and uptake, in addition with a material sensitivity to moisture and water, which make them relatively unusable in integrated systems that include hydrogen production from water by electrolysis and its final use in a fuel cell in mild conditions [79]. For this reason, advanced feasibility analysis of integrated systems or life cycle assessment studies based on complex hydrides are limited [78] and they are instead usually focused on metal hydrides [347–349]. In the case of real applications for hydrogen storage, magnesium based materials [67, 350, 351] or metal hydrides have been explored [352–356], considering complex hydrides only when included in the formulation of RHCs mixtures [77, 182, 334, 357, 358].
The wide possibility of applications for which complex hydrides have been explored underline how chemical and structural diversity is key for developing new multifunctional materials. Basic research evidenced new trends as it will be described hereafter, however, further applicative studies involving complex hydrides should be considered to further prove their properties on a large scale.
Recent reviews extensively present the advances of complex hydride based research [2, 72, 359–361], evidencing how the focus is shifting towards alternative applications beside hydrogen storage [43–45].
Applications in the field of solar thermal or heat storage require cycling properties and adequate thermodynamics, which once again are more advantageous in the case of metal hydrides or limited to few examples in transition metal complex hydrides [49, 362–372].
A reinassance of elemental properties studies on complex hydrides has started together with the exploration of their ionic conductivity to be used as solid state electrolytes in batteries or as electrodes [71, 285, 295, 301, 360, 373–380].
Due to the high number of Li-ion conducting borohydrides reported in literature, a statistical analysis has been performed for the most conductive and studied ones, e.g. LiBH4, Li2BH4NH2, Li4BH4(NH2)3, and Li5(BH4)3NH, to obtain the average values [375]. In addition, the energy limits of a certain migration map periodicities have been obtained with DFT calculations. A clear correlation between the experimental and calculated values for Li-ion mobility was obtained, suggesting that DFT and topological analysis can adequately explain ion conductivity in complex hydrides.
The effect of the composition and the structure dimensions of the h-Li(BH4)1−α−β (Br)α(Cl)β solid solution on the Li-ion conductivity has been investigated, showing that the highest value of RT Li-ion conductivity in the ternary solid solution is reached for h-Li(BH4)0.7(Br)0.2(Cl)0.1 sample (1.3 × 10−5 S cm−1 at 30 °C) [155].
Recently, a total of 11 polymorphs of the compositions M(HCB11H5X6), M = Li, Na, X = Cl, Br were reported representing a new series of halogenated closo-carborates. These exhibit order-disorder polymorphic transitions in the temperature range 203 °C and 305 °C. The high temperature polymorphs of these materials have superionic Li+ or Na+ conductivity, which is associated with the disordered structures [381]. Theoretical investigations reveal that the cation mobility in these systems is associated with the strength of the interaction with the anionic network and the dynamics of the anions, which open new routes for design of functional materials. A possible new type of proton conductor was also recently reported among the closo-borates, with a potential as solid-state ionic H+ conductors. The structures containing the complex ammonium-ammonia cation N2H7 +, with synthesis, crystal structures, and properties of (NH4)2B10H10·xNH3, x = ½, 1 (α and β) and (NH4)2B12H12·xNH3 (x = 1 and 2) are reported in ref [240]. The significant recent interest in this class of materials is mainly due to the possible design of fast ion conductors for solid state batteries. In particular, entropy effects, i.e. anion dynamics, have proven a powerful phenomenon for tuning ion conductivity. There are several studies of metal borates using static solid-state NMR or neutron spectroscopy that show a correlation between cation translational diffusion and dynamics of the anion lattice [219, 382–386].
It has been theoretically demonstrated that Li5MoH11 containing MoH9 3− exhibits an unprecedentedly high lithium-ion conductivity at room temperature [387]. The key to the high ionic conductivity lies in the peculiar dynamics of MoH9 3−, called 'pseudorotation' [388]. In fact, unlike rigid-body rotation, pseudorotation only requires small displacements of highly mobile hydrogen atoms, and thus is easily activated, leading to an order-disorder phase transition with significant cation diffusion at low temperature. The pseudorotation occurs due to the presence of energetically competing metastable coordination geometries that can easily be interconverted, especially for hydride complexes with high hydrogen coordination number exceeding six. This mechanism is quite general and may be applicable to a wide range of complex transition metal hydrides containing hydride complexes with high hydrogen coordination.
The use of complex hydrides as catalyst or electrocatalyst in reactions mainly involved studies on ammonia synthesis and decomposition [294, 299, 389], or CO2 storage and conversion [390, 391]. In particular, the use of metal amides/imides as catalyst or co-catalyst with transition metals is one of the latest developments in the low temperature/pressure ammonia synthesis [297–300] and several new catalysts based on amide/imide materials has been recently reported [294].
Recently, new types of physical properties have been explored in lanthanide-bearing borohydrides related to solid state phosphors and magnetic refrigeration. Furthermore optical and magnetic properties have been reported, but they should be further explored [123, 124, 392].
REBs have received much attention over the years [2, 121] with respect to hydrogen storage materials [393, 394] or as solid state ion conductors for battery applications [92, 163, 395]. However, investigation of RE(BH4)2(THF)2 (RE = Eu, Yb) revealed the interesting property of bright blue luminescence from Eu(BH4)2(THF)2 with a quantum yield of 75% [396]. A few perovskite-type metal borohydrides were investigated with respect to luminescent properties too, and it was found that CsEu(BH4)3 exhibits a 20 nm red shift compared to pristine Eu(BH4)2 [106]. Interestingly, closo-borates were also investigated regarding their photoluminescence, where ultraviolet emission was observable from Sc, Y, and La as well as Li, Na, and Eu-based B10H10 2− and B12H12 2− compounds, while visible light emission was postulated from arachno-borates [397].
Finally, magnetic properties have been investigated for most of the REBs with K2Gd(BH4)5 standing out as the most promising with a magnetic entropy change of 54.6 J kg−1 K−1 at 7 T, which is among the highest reported for inorganic materials [97, 398, 399]. The entropy change may be advantageous in application such as magnetic refrigerants.
The presence of unpaired f-electrons in Tb(BH4)3 and Tb(BH4)3ċS(CH3)2 allowed for an investigation of the local structure of the Tb3+ ion using luminescence spectroscopy, where the latter resulted in an increased splitting of the f-f transitions [140]. Obtained data were compared to Density Functional Density calculations coupled with the Hubbard correction (DFT + U), which revealed that the calculations overestimate both the free ion electron-electron repulsion, the spin-orbit parameters as well as the crystal field parameters.
The rare earth metal borohydrides α-Pr(BH4)3 and LiCe(BH4)3Cl are both paramagnetic compounds as Pr3+ and Ce3+ have two and one unpaired electrons respectively. This gives rise to paramagnetic electron spin-nuclear spin interactions, which significantly affect the 11B MAS NMR spectra of these compounds [400]. Even at a spinning frequency of 12.0 kHz the observed manifolds of spinning sidebands (ssb) in the 11B MAS NMR spectra are highly asymmetric and resemble the ssb patterns for the chemical shift anisotropy interaction for spin-½ nuclei. The observed isotropic chemical shifts,  , are dominated by the contribution from the isotropic hyperfine shift,
, are dominated by the contribution from the isotropic hyperfine shift,  , representing the contribution from the unpaired electrons.
, representing the contribution from the unpaired electrons.  and the paramagnetic shift anisotropy, estimated by comparison with the isostructural and diamagnetic compounds α-Y(BH4)3 and LiLa(BH4)3Cl, are approximately 5 and 4 times larger, respectively, for the 11B site in α-Pr(BH4)3 as compared to LiCe(BH4)3Cl [400]. The great difference in both hyperfine shift and paramagnetic shift anisotropy are ascribed to the metal-borohydride contacts, i.e. two Pr3+ ions are in the vicinity of
and the paramagnetic shift anisotropy, estimated by comparison with the isostructural and diamagnetic compounds α-Y(BH4)3 and LiLa(BH4)3Cl, are approximately 5 and 4 times larger, respectively, for the 11B site in α-Pr(BH4)3 as compared to LiCe(BH4)3Cl [400]. The great difference in both hyperfine shift and paramagnetic shift anisotropy are ascribed to the metal-borohydride contacts, i.e. two Pr3+ ions are in the vicinity of  in α-Pr(BH4)3, whereas only one Ce3+ is in the vicinity of
in α-Pr(BH4)3, whereas only one Ce3+ is in the vicinity of  in LiCe(BH4)3Cl, but it is also ascribed to the electronic spin state of the metal ions as the coupling parameters scale with S(S + 1), where S is the electronic spin state. Finally, the anisotropic dipolar interactions show a 1 r−3 dependency, where r is the 11B nuclear spin-paramagnetic electron spin distance. It may be possible to utilize this dependency for the determination of internuclear distances in similar systems [400].
in LiCe(BH4)3Cl, but it is also ascribed to the electronic spin state of the metal ions as the coupling parameters scale with S(S + 1), where S is the electronic spin state. Finally, the anisotropic dipolar interactions show a 1 r−3 dependency, where r is the 11B nuclear spin-paramagnetic electron spin distance. It may be possible to utilize this dependency for the determination of internuclear distances in similar systems [400].
Finally, a niche application that has been evidenced recently is the use of hydrides for radiation shielding in space [401, 402], which should be considered and investigated for complex hydrides too.
4. Outlooks and conclusions
The proficient synthesis of new compounds, chemical availability and flexibility of complex hydrides has been evidenced by the prolific literature publications and the detailed study of their crystal structure [3, 97, 112, 217, 403, 404].
Synthesis routes include wet chemistry [95, 405] and especially mechano-chemistry [101, 102, 207, 406], that can be easily scaled up, while structural investigations include the use of either x-ray or neutron diffraction and spectroscopies (e.g. Raman, NMR, FT-IR (Fourier transform infrared) etc) [158, 228, 388–398].
The interest has moved to a deeper analysis and understanding of the local structure of complex hydrides to understand the role of amorphous phase or defects on hydrogen storage and especially on ion migration path to enhance ion conductivity [115]. The study of local structure by PDF should be further expanded in the future, focusing on both solid and liquid phases, as in the case of borohydrides, borates and eutectic mixtures.
As previously introduced, the research on complex hydrides has spotlighted some general trends based on chemical bonds between different elements and hydrogen in these complex anions, which evidenced dihydrogen bonds as the interaction of the future. Before, it was suggested that this bond strength can determine the hydrogen release, however it has been shown that there is no relation between the bond strength and the decomposition temperature, nevertheless dihydrogen bonds strongly contribute to structural flexibility in the solid state and cation mobility and possibly also other interesting properties.
The direct correlation of decomposition temperature with Pauling electronegativity was reported but in the case of RE(BH4)n , the opposite trend has been observed. These correlations and trends have been widely exploited to tailor hydrogenation properties of complex hydrides, especially by cation or anion substitutions. Furthermore, nanoscaling and nanoconfinement have emerged as a particularly promising strategy for improving kinetics of complex hydrides, as illustrated in several of the aforementioned examples [52–55, 57, 58, 60, 64, 211, 212, 216, 306, 407]. Strategies can leverage 'non-innocent' chemical interactions with the confining medium, reduced diffusion lengths, confinement stress effects, lowered nucleation barriers, and surface/interface energy penalties to speed reaction rates or alter reaction pathways [52, 54, 60, 64, 212, 306]. Many of these strategies were recently reviewed in a comprehensive article on nanostructured metal hydrides for hydrogen storage [407]. Further research along these lines, including improved fundamental mechanistic understanding of nanoconfinement, may aid in the selection of ideal complex hydride formulations and host matrices for practical low-pressure, high-capacity storage solutions.
The future opportunities in tailoring these systems must involve a massive exploitation of thermodynamics evaluation and modelling efforts. Challenges and opportunities in modelling and simulations are based on a proficient determination of thermo-chemical properties of newly discovered materials, which requires huge calculations and theoretical thermodynamic modelling to build up a comprehensive database, coupled with a massive experimental effort. It has been demonstrated that the CALPHAD approach is beneficial to create and expand thermodynamic calculations on complex hydrides systems, but it needs to be updated and integrated while new materials are discovered and investigated [16, 75, 161, 408]. A continuous flow of thermodynamic data will also allow the development of machine learning techniques to be applied to complex hydrides, as it has been demonstrated on metal hydrides [409–412]. This approach will combine an ocean of data on these systems and reveal new trends and correlations to further develop a rational design of target-based materials and new promising compositions or mixtures.
It is worth noting that high throughput ab-initio calculations can be a huge support to thermodynamic assessments, and they can be integrated and compared with available experimental data owing to the CALPHAD approach, which leads to a comprehensive modelling of investigated system. To our knowledge no other possible approaches have been proposed for modelling thermodynamics of hydrides systems.
Recent advances in modelling and simulation have provided additional insight into complex hydride reactions [60, 64, 211–213, 215, 225, 306, 407, 413–415]. These solid-state reactions are notoriously complex, thus simulations must consider non-idealities that alter thermodynamics, as well as impacting reaction and diffusion kinetics [415]. Recent computational activities have incorporated interfaces and interfacial energy penalties more directly in thermodynamic and kinetic investigations of reaction pathways in complex hydrides, with a focus on nanoscale materials [60, 64, 211, 306, 415]. It is also increasingly being realized that diffusion in complex hydrides is likely dominated by interfacial, surface, and grain boundary pathways, rather than bulk defect dynamics. Investigations are underway to quantify the contributions of these diffusion pathways more directly.
The role of phase nucleation is also being considered more prominently. Nucleation is a multi-step process for complex hydrides, which involves transformations of chemical species in addition to phase-level reorganizations. Recently, a model was developed to account for this multi-step reactions within a classical nucleation theory context [413]. A separate study was also published detailing how local morphology and dimensionality of complex hydride clusters, surfaces, and bulk crystals play a role in determining reaction pathways and nucleation barriers in the solid state [225]. These calculations can be the basis for a more quantitative approach to nucleation prediction.
For nanoconfined complex hydrides, an emerging area of interest for computation is the development of approaches for predicting interaction effects with the confining host for different particle sizes. Recent investigations have addressed this challenge by combining advanced free-energy sampling methods with direct computation of hydride-host interactions [60, 64]. This methodology provides an avenue for predicting both size-dependent and host-dependent reaction enthalpies, while simultaneously providing insights into kinetic effects in confined systems. The approach has been successfully deployed to study carbon-confined nanoscale Li–Al–H [60] and Al–H [64] systems and it is currently being applied to the Li–N–H system. An additional investigation pathway has focused on the integration of spectroscopic techniques and ab-initio modelling to investigate the chemical interaction with the confining medium and the impact on phase behaviour [60, 64, 212, 215, 414]. Examples include recent work on borohydrides, which invoked computational models and direct spectroscopic calculations to interpret experiments and elucidate the interfacial chemistry [212, 213, 215]. An analogous approach combining spectroscopy and simulations was also recently raised to investigate the effects of native surface oxidation on NaAlH4 dehydrogenation kinetics [66].
A recent paper by Wood et al [415] highlighted four areas where advances beyond idealized models have helped to elucidate key phenomena in metal hydrides, with an emphasis on nanoscale and nanoconfined systems. These included the role of surfaces and interfaces in determining enthalpy and reaction pathways; the role of anharmonicity at surfaces and interfaces in determining entropy-enthalpy compensation effects; the role of solid mechanics in altering properties of confined systems in stiff matrices; and the role of surface contamination in altering chemical pathways and barriers for dissociation and diffusion. Although not exhaustive, these examples nicely illustrate the types of advances that are required to achieve parity between theoretical predictions and experimental realities, and they can be also applied in the future on complex hydride systems.
Acknowledgments
This paper was realised within the framework of the Hydrogen Technology Collaboration Programme (TCP) of the International Energy Agency (IEA) in Task 40 'Energy storage and conversion based on hydrogen'. Work at Pacific Northwest National Laboratory (PNNL) and Lawrence Livermore National Laboratory (LLNL) was supported by the Hydrogen Materials Advanced Research Consortium (HyMARC), established as part of the Energy Materials Network under the U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy, Fuel Cell Technologies Office. PNNL is a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy (DOE). Work at LLNL was performed under the auspices of the DOE under Contract DE-AC52-07NA27344. Kasper T Møller would like to acknowledge the Carlsberg Foundation for a Reintegration Fellowship (CF19-0465). Funding from the Danish Ministry of Higher Education and Science through the SMART Lighthouse, the independent research fund Denmark for technology and production (Grants 9041-00226B and 0217-00327B) and the Carlsberg Foundation are gratefully acknowledged. C E Buckley (CEB), Mark Paskevicius (MP) and T D Humphries (TDH) acknowledge financial support from the Australian Research Council for Grants LP120101848, LP150100730 (CEB, TDH), DP150101708 (CEB), FT160100303 (MP), LE140100075 (CEB, MP), LE0989180 (CEB) and LE0775551 (CEB). Shin-ichi Orimo acknowledges funding from JSPS KAKENHI (Hydrogenomics, JP18H05513). Shigeyuki Takagi acknowledges funding from JSPS KAKENHI (19H05514, 20K20438). P Ngene and P E de Jongh acknowledge funding from NWO (Dutch Research Council) Materials for sustainability (739.017.009) and NWO ECHO (712.015.005) grants. Y W Cho and Y-S Lee acknowledge funding from National Research Foundation of Korea (NRF-2020M1A2A2080881).
Data availability statement
No new data were created or analysed in this study.