

Abstract
The dioxygen molecule has two bound states, singlet and triplet, which are different in energy, lifetime, and reactivity. In the context of oxygen electrocatalysis as applied to fuel cells and water splitting the involved O2 is typically considered to be exclusively in its triplet ground state. However, applying spin-conservation rules for the transformation between triplet O2 and singlet OH−/H2O reaction intermediates predicts an additional free energy barrier associated with the required spin flip. As a result, for conditions under which both can form, the formation of triplet dioxygen from the singlet OH−/H2O might be slower than the formation of singlet O2. Correspondingly, singlet O2 might be more active than triplet O2 in the oxygen reduction reaction. Here, we discuss the possible existence and influence of singlet oxygen in oxygen electrocatalysis. Some perspectives for studying singlet oxygen in oxygen electrocatalysis are also provided.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Oxygen electrocatalytic reactions, including the oxygen evolution reaction (OER) and the oxygen reduction reaction (ORR), have attracted great attentions in recent years [1–3]. Equation (1) shows the OER and ORR in acidic environment and equation (2) shows them in alkaline
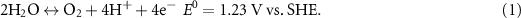
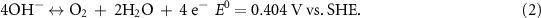
OER and ORR are, respectively, rate determining for water electrolysis and hydrogen fuel cells. In recent years, new insights into the reaction mechanisms have been developed for the design of better oxygen electrocatalysts. For example, (a) some highly active catalysts have been identified as pre-catalysts due to surface reconstruction under the OER condition. The observed high OER activity is given by the reconstructed surface [4–6]. (b) The observation of lattice oxygen involvement leads to the setup of a lattice oxygen mechanism (LOM) in addition to the conventional adsorbate evolution mechanism (AEM). LOM routes enable the direct O–O coupling, which breaks the adsorption-energy scaling relation between adsorbed OH and OOH intermediates, and thus better OER performance [7, 8]. (c) There is a close relationship between the spin state of active metal cations and catalytic activity [9–12]. On one hand, the adsorption free energies of key intermediates are highly sensitive to the spin state of cations at the surface [10]. On the other hand, charge transport may be more efficient in catalysts with spin channels [11, 12].
From an orbital point of view, the oxygen molecules have two spin states (figure 1(a)), the triplet state (3O2, ground state) and singlet state (1O2, metastable state). These figures are approximate 'cartoons' that do not adequately represent the required antisymmetric wavefunctions [13, 14]. In the triplet ground state (3Σg −), there are two unpaired and spin-parallel electrons in π* orbitals, which makes the molecule paramagnetic. A reduced electronic repulsion stabilizes this open-shell orbital configuration due to quantum spin exchange interactions (QSEI) [15]. In singlet oxygen, all the energy levels are closed shells, where the frontier electrons can share the same space (orbital) and instead, the Coulomb attractions are optimized (see the Fermi heap in the center of the 1Δg O2 molecule in figure 1(b) right). According to the difference in the occupancy of π* orbitals, singlet oxygen can be further divided into two types, 1Δg and 1Σg + (figure 1(a)). The 1Σg + singlet state oxygen is 0.65 eV higher in energy than the 1Δg state) and can be quickly (in microseconds or nanoseconds) converted to the low-energy 1Δg state. The 1Σg + singlet state oxygen is always shorter-lived than the 1Δg singlet state oxygen [16]. Therefore, unless otherwise stated, all the singlet oxygen mentioned in the following part refers to the 1Δg state oxygen. Compared to the ground state triplet oxygen, excited singlet oxygen 1Δg is still 0.98 eV higher in energy and is metastable, which can exist for about 1 h in the gas phase [17]. It can be easily transformed into the triplet oxygen in solvents within milliseconds and microseconds [17]. The reactivity of singlet oxygen is different from that of triplet oxygen [18]. Singlet oxygen is regarded as one of the 'reactive oxygen species' [19].

Figure 1. (a) Molecular orbital diagram of triplet and singlet oxygen; (b) distinctive reshaping of the wavefunction for the 3Σg − (left) states and 1Δg (right). Because of the Fermi hole that mitigates electronic repulsions, unpaired electrons do not share the center of the 3Σg − O2 molecule. Therefore, triplet oxygen (yellow shading, left) is much more stable than singlet oxygen (blue shading, right).
Download figure:
Standard image High-resolution imageAs the energy of triplet oxygen is 0.98 eV lower than that of singlet oxygen, in most cases, it is considered that only the triplet O2 is involved in oxygen electrocatalysis. The reversible potential of the four-electron transferred triplet oxygen generation is 1.23 V, while the reversible potential of singlet oxygen evolution can be calculated to be 1.23 + 0.98/4 = 1.475 V based on the Gibbs free energy ΔG = −nEF. Since the potential gap between anode and cathode could be up to ∼2 V in water electrolysis, it is thus energetically possible to produce singlet oxygen during the OER. According to the spin-conservation [20], the reaction rate might slow down if the spin polarization (i.e. the number of spins 'up' minus the number of spins 'down') of the products differs from that of the reactants [21]. For instance, if electron transfer must wait for an incoherent step, a slow spin relaxation mechanism occurs via a collision of paths that are against the foreseen spin-rules for optimal electrocatalysis [22, 23]. In this regard, singlet oxygen is kinetically less challenging, more desirable for OER and ORR in reaction kinetics, because electron transfers are easily all coherent in the transformation between singlet oxygen and singlet OH−/H2O. Considering the highly active feature of singlet oxygen, attentions need to be paid to detecting its existence and exploring its possible functions in oxygen electrocatalysis.
Some experiments have shown that not all oxygen generated by electrochemical reactions is in the triplet state. Recently, the generation of singlet oxygen was detected in Li (Na)-oxygen batteries [24, 25] and Li-ion batteries [26]. Specifically, 4% of the oxygen generated in the cathode of a Li-oxygen battery is singlet oxygen, which increases to 6% in the presence of trace water [24]. The singlet oxygen not only decays to triplet oxygen but also reacts with electrolyte to form reactive intermediates, such as ROOH, R*, and ROO* [24]. The active singlet oxygen has been demonstrated as an oxidant to react with the carbon-contained electrolytes. The parasitic reaction products (i.e. Li2CO3, Li acetate, and Li formate) severely limit the cycle life of the Li-oxygen batteries [24]. Generation of singlet oxygen in Na-oxygen batteries has also been shown to negatively affect stability [25]. In a study of Li-ion batteries Wandt et al reported direct generation of singlet oxygen from the lattice of layered LiNix Coy Mn2O2 when charging it to a high state of charge [26]. The highly active singlet oxygen was proved to be responsible for the electrolyte degradation in this Li-ion battery. One possible route for the release of singlet oxygen in metal-air and Li-ion batteries, as proposed by Houchins et al, is the disproportionation of superoxide (O2 − + O2 − → 1O2 + O2 2−) [27]. On the basis of the Marcus theory analysis, they predicted that the reaction rate constant for the production of 1O2 is three orders of magnitude higher than that of the 3O2, suggesting nonnegligible possibilities of singlet oxygen production. Furthermore, the production of singlet oxygen has also been found in photocatalytic water splitting [28, 29], and its generation is correlated to the oxidation of superoxide anion radical (˙O2 −), which needs additional energy inputs (compared to the direct generation of triplet oxygen) [30]. Generally, a minimum energy of 237.24 kJ mol−1 is needed for triplet OER [31]. As the singlet oxygen is 94.29 kJ mol−1 higher in energy than that of triplet oxygen, when the input energy is higher than 331.54 kJ mol−1, the production of singlet oxygen is thermodynamically possible in electrocatalytic water splitting.
While the formation of singlet O2 is energetically possible at a high overpotential employed for OER, no direct observation of singlet oxygen has been reported to date. However, some studies experimentally proved that some of the reaction intermediates have singlet character. By studying the Co4O4 cubane OER catalyst, Nocera et al proposed that the in situ formed antiferromagnetic (AFM) coupled Co(IV)2 dimer may drive singlet O–O bond formation by conveying singlet character to the oxygen radicals to promote direct coupling and bond formation with no spin barrier (figure 2) [32]. The preferable production of triplet oxygen on ferromagnetic (FM) catalysts has been successfully demonstrated by Garcés-Pineda et al [33]. The magnetic field favors the parallel alignment of oxygen radicals on the FM catalysts during the formation of the triplet O–O bond. In several of our co-author's works, it was observed that catalysts with dominant FM interactions, between the active metal centers with unpaired electrons, facilitate the 3O2 formation. These works strongly indicate that OER activity is spin-dependent. A hypothesis may be made that the catalysts with dominant AFM interactions (cooperative interactions) can favor the formation of 1O2 to avoid the spin barrier.

Figure 2. Electronic considerations for the mechanism of singlet oxygen bond formation (red line) by intramolecular coupling of oxyl radicals. The double-head arrows link the resonance structure of Co(IV)=O and Co(III)–O˙. Adapted with permission [32].
Download figure:
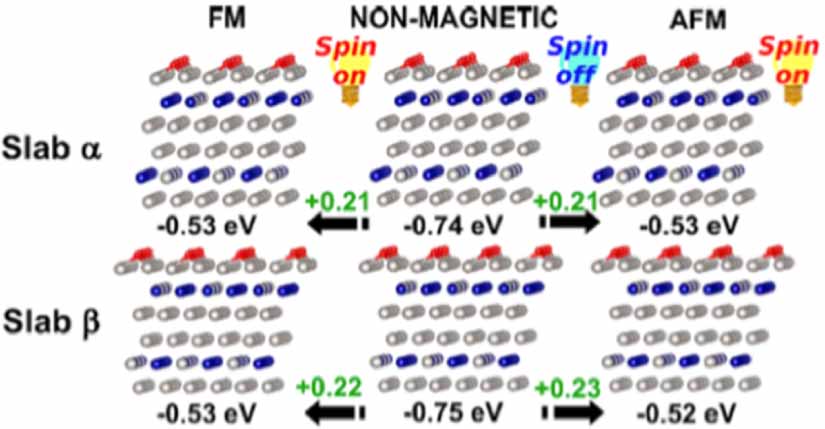
Standard image High-resolution imageThe electrochemical triplet ORR activity can be related to magnetic property of the electrocatalysts [34]. Jose et al performed comprehensive density functional theory (DFT) simulation to unveil the correlation between the ORR activity and magnetic property of the Pt3Co materials (figure 3) [34] Due to the QSEI character of the open-shell FM and AFM Pt3Co catalysts, both FM and AFM Pt3Co catalysts have milder chemisorption energies than the non-magnetic Pt3Co alloy. This milder chemisorption energy is beneficial for the electron delocalization and thus improving the ORR activity.

Figure 3. Oxygen chemisorption comparison between spin polarized Pt3Co (111) slab α (FM) and slab β (A-type AFM) and the equivalent hypothetical non-spin polarized (nonmagnetic) nanostructures. Reprinted with permission from [34]. Copyright (2020) American Chemical Society.
Download figure:
Standard image High-resolution imageFor electrochemical ORR, there is no experimental report involving singlet oxygen to date. This is because the lifetime of singlet oxygen is milliseconds or even microseconds in solution. It is difficult to experimentally compare the influences of triplet and singlet oxygen to the ORR performance. In contrast, theoretical studies have predicted that singlet oxygen may be easier to be reduced as compared to triplet oxygen. Using DFT calculations, Nazmutdinov et al have theoretically analyzed the spin effects in ORR [35]. The rate-determining step (RDS) for ORR is calculated to be the first electron transfer to form the adsorbed O2 − on the surface of Pt. The energy of Coulomb repulsion (U) of electrons on the two different oxygen orbitals was compared. During the reduction of triplet oxygen, the first electron is transferred from an electrode to a half-occupied π* orbital (U > 0); while in the case of singlet oxygen, the first electron is transferred to an empty π* orbital (U = 0). Therefore, relatively, a more negative potential is needed for the first electron transfer from electrode to triplet oxygen. It was therefore raised that singlet oxygen should facilitate the RDS and reduce the overpotential of ORR, versus triplet O2.
Inspired by above pioneering works, we propose a possible mechanism for triplet and singlet oxygen generation by considering the spin direction of the four transferred electrons in OER. Our discussions are based on the recognized AEM [3, 36, 37] and LOM [7, 8] routes. AEM path has two subtypes, namely, the Eley–Rideal (ER)-type (figure 4(a)) and the Langmuir–Hinshelwood (LH)-type (figure 4(b)) [37]. For OER in base, each of the four steps involves the orbital overlap between the lone pair on the OH− and an empty orbital on the active site. Due to the strong spatial overlap between metal eg and O 2p orbitals (for example, 3dz2 and 2pz orbitals) in octahedral site, the oxygen intermediates are adsorbed on the metal site through the formation of σ-like bond between O 2p and metal eg during the entire process [38]. The transferred spin direction is then dominated by the partially occupied metal t2g orbitals via exchange interactions through a space-separated π-type tunneling between t2g and px /py orbitals [15]. According to previous study [11], it is more favorable to transfer electrons with opposite spin direction to that of the unpaired t2g electron. In this way, the spin direction of the 1st transferred electron should be antiparallel to that of the unpaired t2g electron.

Figure 4. The adsorbate evolution mechanism (AEM) mechanisms of OER by considering the spin of transferred electrons. (a) The ER-type route and (b) LH-type route. M means the metal site and the arrow beside M represents the spin direction of unpaired t2g electrons; the blue O and wine O represent the lattice oxygen and adsorbed oxygen species, respectively. SSS refers to the spin selective step. The singlet oxygen formation is highlighted in yellow.
Download figure:
Standard image High-resolution imageThrough the ER-type route, an unoccupied orbital on the adsorbed oxygen is favorable for the coupling of the next OH−. Thus, the 2nd transferred electron should be the one in the same orbital with opposite spin to that of the 1st transferred electron (figure 4(a)). Subsequently, with the transition of adsorbed O to adsorbed OOH, the electron in the fully occupied π* orbitals could transfer in the 3rd elementary step. Similar to the 1st transferred electron, the spin direction of the 3rd transferred electron is also dominated by the spin direction of unpaired t2g electrons, which means the 1st and 3rd transferred electrons possess the same spin direction. In the last step, which is the spin selection step, if the spin state of the 4th transferred electron is the same as that in the 3rd step, 3O2 will form; otherwise, 1O2 will form. In this regard, catalysts with unpaired t2g electrons as well as chiral molecules with spin selectivity should be favorable for the transfer of electrons with a specific spin direction, thus facilitating the release of 3O2 [39].
The LH-type route occurs when the distance between two neighboring metal sites is relatively short (figure 4(b)). For the successful formation of a σ O–O bond between the two adjacently adsorbed OH−, the spin direction of the 1st and 2nd electrons should be opposite, indicating an AFM configuration of metal sites is preferable. The 2nd electron transfer is the spin selection step. The spin directions of the 3rd and 4th transferred electrons are also dominated by the AFM configuration. In the LH-type route, the generation of 1O2 might be more favorable (figure 4(b)).
LOM may occur when the O p-band of lattice oxygen is higher than the d-band center of metal [40]. Oxygen molecules generated through LOM path have two possible origins: (a) both two oxygen atoms of the produced dioxygen molecule are from the lattice of the oxides; and (b) one from lattice oxygen while the other from the electrolyte (figures 5(a) and (b)). In the former case (figure 5(a)), to form the σ O–O bond, each of the two adjacent oxygens needs a half-filled 2p orbital and the two unpaired electrons need to have opposite spins. Thus, two spin-antiparallel electrons are extracted in the 1st and 2nd steps. The spin direction of the 3rd transferred electron should be antiparallel to the unpaired t2g electron of the center metal site for optimizing the quantum spin-exchange interactions with the metal site. The last step determines whether the generated oxygen molecule is a singlet or triplet state, which is similar to the case of the ER-type route. However, if the 1st and 2nd transferred electrons possess the same spin direction, it is difficult for the σ O–O bond formation. Under this circumstance, the latter case will take place (figure 5(b)), wherein one lattice oxygen couples an adsorbed OH [7], instead of another lattice oxygen, to form an O–O bond. In this one-lattice-oxygen-participated route, similar to the case of the ER-type route, the generation of 3O2 or 1O2 is also dependent on the spin direction of the 4th transferred electron. Thus, from the spin-selective point of view, OER via the LOM route could also selectively produce 3O2 or 1O2 depending on the spin direction of 4th electron transferred.

Figure 5. Lattice oxygen mechanism (LOM) mechanisms of OER by considering the spin of transferred electrons. (a) Both two oxygen atoms of the produced dioxygen molecule are from the oxide lattice; (b) one from lattice oxygen while the other from the electrolyte. M refers the metal site; the blue O and wine O represent the lattice oxygen and adsorbed oxygen species, respectively. The singlet oxygen formation is highlighted in yellow.
Download figure:
Standard image High-resolution imageLikewise, the electrocatalytic triplet and singlet O2 reduction reactions (denoted as 3O2RR and 1O2RR) are compared by considering the spin direction of the four transferred electrons. The ORR mechanism can be regarded as a reverse OER route [41]. Here, the reverse ER-type mechanism is selected as an example to show the electron transfer in the 3O2RR (figure 6(a)) and 1O2RR (figure 6(b)). In the case of 3O2RR, due to the two unpaired and spin-parallel electrons in π* orbitals of 3O2, the 1st and 2nd transferred electrons should be anti-parallel to the existing electrons. Assuming the electrons in the π* orbitals of 3O2 are spin up, then the 1st and 2nd electrons should be spin down. After accepting the 1st and 2nd electrons, the two π* orbitals of adsorbed OOH intermediates are fully occupied, so that the 3rd electron could only fill the higher-energy σ* orbital of adsorbed OOH intermediates. The electron filling in the higher-energy anti-bonding makes the O–O bond unstable, so that the adsorbed OOH transforms into adsorbed O intermediates and OH−. The 3rd electron should fill the 2p orbital of adsorbed O intermediates, while its spin is most probably anti-parallel to the unpaired t2g electron. In the end, the last spin-down electron transfers to pair with the 3rd electron. As for 1O2RR, because of the existing of an empty π* orbital, the transfer of the 1st electron faces no Coulomb repulsion. The spin direction of the 1st electron is most probably anti-parallel to the unpaired t2g electron, which is under the same determining factor as mentioned in the OER part. Then the 2nd to the last electron transfer should be the same to corresponding steps of 3O2RR.

Figure 6. The mechanisms of (a) 3O2RR and (b) 1O2RR by considering the spin of transferred electrons.
Download figure:
Standard image High-resolution imageSummarizing, we have proposed several pathways for singlet oxygen generation during OER within both the AEM and LOM reaction mechanisms. LH-type route on AFM-configuration catalysts most likely favor singlet oxygen formation. To further promote the investigations of the possible roles of singlet oxygen in oxygen electrocatalysis, some perspectives are raised below.
- (a)The detection of singlet oxygen during OER. Singlet oxygen in the gas phase with a pressure of 104.3 kPa at 296 K is relatively long-lived (72 min) [42], but contact with a liquid reduces its lifetime to milliseconds or even microseconds. The transition of 1O2 to the ground state gives the emission of photons at 1270 or 633 nm, which provides an optical method to detect the singlet oxygen which has been used in lithium ion batteries [26]. The 1270 nm radiation (phosphorescence) is caused by the spin/parity-forbidden radiative transition from 1O2 to 3O2 [43], and the 633 nm emission (luminescence) is due to the formation of a 1O2 dimer followed by the spin-allowed transition to the 3O2 [26]. For quantifying the singlet oxygen, the 633 nm emission is more sensitive than 1270 nm radiation, even though the yield of 1270 nm is generally much higher than that of 633 nm emission, which depends on the square of the steady-state 1O2 concentration [26]. Another route is to use chemical probes. For example, the fluorophore and trap probes will become fluorescing or electron paramagnetic resonance active, respectively, upon reacting with the singlet oxygen [17]. Considering the possible oxidation of chemical probes under the OER conditions, detecting the 1270 nm and 633 nm emissions originated from the transition of singlet oxygen to triplet oxygen may be more promising. One should keep in mind that these two radiative emissions' yields are very small [26], which brings challenges to the detection of singlet oxygen.
- (b)ORR with singlet oxygen. Compared to reducing triplet oxygen, reducing singlet oxygen is expected to facilitate the first electron transfer in ORR. However, the singlet oxygen has very limited lifetime when contacting a liquid like water. It is thus challenging to provide a singlet oxygen saturated electrolyte for ORR. According to previous report [17], the lifetime of singlet oxygen in D2O electrolyte is relatively longer (∼67 μs) than in H2O (∼3.5 μs). It may be possible to use a properly designed electrochemical cell with D2O electrolyte to investigate 1O2 reduction. In addition, an impinging jet flow cell with a rotating valve for accurately controlling oxygen gas dosing might be another possible tool to test the ORR with singlet oxygen (figure 7) [44]. In order to enable 1O2 participated ORR before its decay in the electrolyte, the pipeline length divided by the electrolyte flow rate should be less than 67 μs. The 1O2RR and 3O2RR activity can be evaluated by cyclic voltammetry measurements. Conventionally, ORR kinetic activity is measured by rotating disk electrode (RDE), which allows us to conduct mass-transport-correction to obtain the ORR kinetic current given by the Koutecky–Levich equation [45]. However, the recommended setup (figure 7) for studying 1O2RR vs. 3O2RR is not equipped with RDE; the working electrode does not rotate. Thus, we recommend the activity analysis to be performed within the kinetic-controlled region (for example, >0.95 V vs. reversible hydrogen electrode for Pt-based catalysts), where the influence of mass transport is negligible, and the measured current is approximately kinetic current. With this method, the activity measured in the dosing period of 1O2 and 3O2-saturated electrolytes represent the 1O2RR and 3O2RR activity, respectively, and comparing these two activities allow us to identify the effect of singlet and triplet oxygen on ORR.
- (c)Spin-polarized DFT calculations. Spin-polarized DFT calculations can describe molecules with unpaired electrons. Orbitals with paired electrons or unpaired electron have different energies [46]. Properly using this strategy, the reaction mechanisms and free energy change for singlet and triplet oxygen production on a given catalyst can be systematically predicted. It may also facilitate the design of appropriate catalysts for generation of either 3O2 or 1O2. Here, the spin-polarized DFT calculations is performed on Pt (111) to compare the relative adsorption energy of singlet and triplet oxygen (figure 8). The adsorption energy of the 1st step for 1O2RR is approximately 1.1 eV lower than that of the 3O2RR, indicating better thermodynamics for singlet oxygen capture.
- (d)The opportunities and challenges brought by the highly active 1O2. Considering the distinct properties of singlet oxygen, strategies to make full use of its advantage and to avoid its negative effects can be proposed. On one hand, the generation of 1 O2 with high reactivity may facilitate some parasitic oxidation reactions (for example, nitrogen oxidation reaction [47], electrochemical degradation of polluted water [48] et al) by oxidizing reaction intermediates. On the other hand, since singlet oxygen is highly active, there are possible interactions between singlet oxygen and catalysts or additive agents (i.e. conductive carbon and binder). The influence by such interactions on the stability of the catalyst surfaces probably need to be considered. For example, active 1O2 may directly lead to the degradation of carbon-based materials [49]. The electrode stability may be also influenced by 1O2 through indirectly reacting with additive agents such as binder.

Figure 7. Sketch of the impinging jet flow system for the study of singlet oxygen reduction reaction. WE, RE, and CE refer to the working electrode, reference electrode, and counter electrode, respectively. For a fair comparison, the pipeline's length for 1O2 and 3O2-saturated electrolyte should keep identical, so does the electrolyte's speed. Adapted from [44], Copyright (1998), with permission from Elsevier.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Calculated adsorption energy of singlet and triplet oxygen on Pt (111).
Download figure:
Standard image High-resolution image{kind=link}
Acknowledgments
This work was supported by the Singapore National Research Foundation under its Campus for Research Excellence And Technological Enterprise (CREATE) programme, through The Cambridge Center for Carbon Reduction in Chemical Technology (C4T) and eCO2EP programmes. This work was also supported by the Singapore Ministry of Education Tier 2 Grant (MOE2018-T2-2-027) and Tier 1 Grant (2019-T1-002-125).