

Abstract
Density functional theory (DFT) simulations of the oxygen evolution reaction (OER) are considered essential for understanding the limitations of water splitting. Most DFT calculations of the OER use an acidic reaction mechanism and the standard hydrogen electrode (SHE) as reference electrode. However, experimental studies are usually carried out under alkaline conditions using the reversible hydrogen electrode (RHE) as reference electrode. The difference between the conditions in experiment and calculations is then usually taken into account by applying a pH-dependent correction factor to the latter. As, however, the OER reaction mechanisms under acidic and under alkaline conditions are quite different, it is not clear a priori whether a simple correction factor can account for this difference. We derive in this paper step by step the theory to simulate the OER based on the alkaline reaction mechanism and explain the OER process with this mechanism and the RHE as reference electrode. We compare the mechanisms for alkaline and acidic OER catalysis and highlight the roles of the RHE and the SHE. Our detailed analysis validates current OER simulations in the literature and explains the differences in OER calculations with acidic and alkaline mechanisms.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 license. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
The oxygen evolution reaction (OER) is often regarded as the main bottleneck in water splitting due to its slow kinetics, which limits the efficiency of the energy conversion [1, 2]. During the last few decades, extensive studies have been devoted to the development and understanding of the OER [2–4]. Based on density functional theory (DFT) calculations, the OER mechanism under acidic conditions has been widely investigated [5–7]. However, most of the experimental studies are done in alkaline media instead of acidic media [8–10]. Although some computational papers consider OER calculations under alkaline conditions, the underlying theory from reaction mechanism to free energy equations is not made transparent and not derived explicitly [11–14]. Considering the dissimilarity between the computational and experimental approaches, it is crucial to derive the equations for DFT calculations of the OER with an alkaline mechanism explicitly and to compare the outcome to the acidic mechanism.
The widely accepted OER mechanism consists of four-electron/proton transfer steps in both acidic and alkaline media [3, 4, 14]. The OER is highly pH-sensitive; under acidic conditions, water molecules (H2O) are oxidized, and  pairs and oxygen molecules (O2) are released [3, 4]. In contrast, under alkaline conditions, hydroxyl groups (OH−) are oxidized to H2O and O2 with concomitant release of e− [14, 15]. In the literature, many theoretical studies assume an acidic mechanism for calculations of the catalytic activity of the OER of typical catalysts, such as Fe2O3 [5, 6], Co3O4 [10, 16], and Ni3S2 [17]. The catalytic activity for OER is typically characterized by calculating the Gibbs free energies of the individual reaction steps using the standard hydrogen electrode (SHE) as a reference electrode.
pairs and oxygen molecules (O2) are released [3, 4]. In contrast, under alkaline conditions, hydroxyl groups (OH−) are oxidized to H2O and O2 with concomitant release of e− [14, 15]. In the literature, many theoretical studies assume an acidic mechanism for calculations of the catalytic activity of the OER of typical catalysts, such as Fe2O3 [5, 6], Co3O4 [10, 16], and Ni3S2 [17]. The catalytic activity for OER is typically characterized by calculating the Gibbs free energies of the individual reaction steps using the standard hydrogen electrode (SHE) as a reference electrode.
The acidic mechanism involves the production of  pairs, and their Gibbs free energy is usually calculated implicitly by assuming the equilibrium
pairs, and their Gibbs free energy is usually calculated implicitly by assuming the equilibrium  ↔ ½H2 at standard conditions (pH = 0, pressure
↔ ½H2 at standard conditions (pH = 0, pressure  = 1 bar, and T = 298.15 K) and using the Gibbs free energy of hydrogen gas [3, 4]. At a pH different from 0, the Gibbs free energy of H+ ions can be corrected by the concentration dependence of the entropy,
= 1 bar, and T = 298.15 K) and using the Gibbs free energy of hydrogen gas [3, 4]. At a pH different from 0, the Gibbs free energy of H+ ions can be corrected by the concentration dependence of the entropy,  [3, 4]. The why and how of this correction factor has not been universally picked up in the literature and the step from the acidic to the alkaline reactions is not taken explicitly.
[3, 4]. The why and how of this correction factor has not been universally picked up in the literature and the step from the acidic to the alkaline reactions is not taken explicitly.
The alkaline mechanism involves the oxidation of OH− with concomitant release of  . Therefore, calculating the Gibbs free energies of OH− and
. Therefore, calculating the Gibbs free energies of OH− and  is crucial in order to characterize the OER under alkaline conditions. Some papers claimed that the calculation of the Gibbs free energies of OH− and
is crucial in order to characterize the OER under alkaline conditions. Some papers claimed that the calculation of the Gibbs free energies of OH− and  can be obtained from the Gibbs free energy of
can be obtained from the Gibbs free energy of  , but how to deal with the free energy of
, but how to deal with the free energy of  at a pH different from 0 is not thoroughly discussed.
at a pH different from 0 is not thoroughly discussed.
Using the computational hydrogen electrode (CHE) approach developed by Bagger et al [18], the calculation of the free energy of  at a pH different from 0 can formally be solved by taking the reversible hydrogen electrode (RHE) as the reference electrode. Besides, in contrast to the SHE, the RHE is also generally used as reference electrode in experiments [8, 15]. The main advantage of using the RHE as reference electrode is that it equals the Gibbs free energy of
at a pH different from 0 can formally be solved by taking the reversible hydrogen electrode (RHE) as the reference electrode. Besides, in contrast to the SHE, the RHE is also generally used as reference electrode in experiments [8, 15]. The main advantage of using the RHE as reference electrode is that it equals the Gibbs free energy of  to the energy of ½H2 at arbitrary pH (
to the energy of ½H2 at arbitrary pH ( = 1 bar and T = 298.15 K) [19].
= 1 bar and T = 298.15 K) [19].
In this paper, we compare the OER mechanism under acidic and alkaline conditions starting explicitly from the corresponding reactions and the species involved. We derive the theory required to couple the results of DFT calculations to experiment (but actual DFT calculations are not discussed). The reaction steps, their Gibbs free energies, and the overpotential are explained separately for both these cases. Besides, we demonstrate the connection between the OER calculations that use the RHE and the SHE reference electrodes and explain in detail how the correction to the Gibbs free energy of H+ ions at a pH different from 0 enters in the energy balance and in the overpotential.
2. OER using an acidic reaction mechanism
We consider a four-electron reaction mechanism for OER. Under acidic conditions, the overall water oxidation reaction is [3, 4, 20]

where (l) refers to the liquid phase. At p = 1 bar and T = 298.15 K, the Gibbs free energy difference  eV for this reaction. The reaction is generally believed to proceed in four steps [3, 4, 20]
eV for this reaction. The reaction is generally believed to proceed in four steps [3, 4, 20]


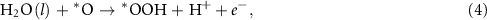
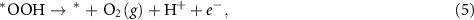
where * represents the active site of the catalyst, (g) refers to the gas phase, and *OH, *O, and *OOH represent the species adsorbed on the active site. As explained in the previous section, at standard conditions, the Gibbs free energy of  equals the Gibbs free energy of ½H2. The reaction Gibbs free energies, which provide the binding strength between the catalyst and OER intermediates, are affected by the electronic properties of the active site. The typical procedure widely used in the literature to calculate the reaction Gibbs free energies,
equals the Gibbs free energy of ½H2. The reaction Gibbs free energies, which provide the binding strength between the catalyst and OER intermediates, are affected by the electronic properties of the active site. The typical procedure widely used in the literature to calculate the reaction Gibbs free energies,  corresponding to equations (2)–(5) at standard conditions [6, 20], is shown in equations (6)–(9); more details about the derivation can be found in [20].
corresponding to equations (2)–(5) at standard conditions [6, 20], is shown in equations (6)–(9); more details about the derivation can be found in [20].
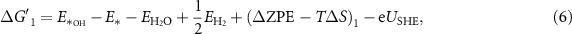

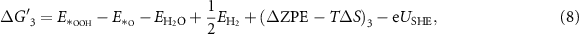
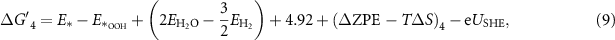
where  ,
,  ,
,  ,
,  are the total energies of the clean surface (*) and of surfaces with the single adsorbed species OH, O, and OOH, respectively, and
are the total energies of the clean surface (*) and of surfaces with the single adsorbed species OH, O, and OOH, respectively, and  and
and  are the total energies of the H2O and O2 molecules, all obtained from DFT calculations. ΔZPE and ΔS are the changes in vibrational zero-point energy and entropy from the initial state to the final state, respectively; T is temperature. In addition,
are the total energies of the H2O and O2 molecules, all obtained from DFT calculations. ΔZPE and ΔS are the changes in vibrational zero-point energy and entropy from the initial state to the final state, respectively; T is temperature. In addition,  contains the contributions of the gas and liquid phases used in reactions (2)–(5). USHE is the electrode potential relative to the SHE.
contains the contributions of the gas and liquid phases used in reactions (2)–(5). USHE is the electrode potential relative to the SHE.  (n = 1, 2, 3, 4) can be calculated as:
(n = 1, 2, 3, 4) can be calculated as:

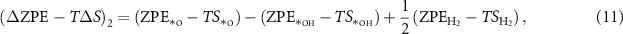
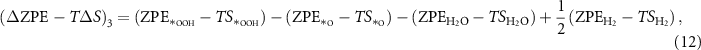

Therefore, the sum of change of the ZPE terms and the TS terms during the reaction cycle equals 0:
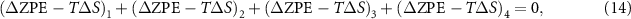
The reaction Gibbs free energies of equations (6)–(9) have to obey the sum rule.
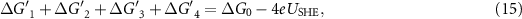
The overpotential is then given by

where  V is the equilibrium potential, which is independent of pH and defined at p = 1 bar and T = 298.15 K.
V is the equilibrium potential, which is independent of pH and defined at p = 1 bar and T = 298.15 K.  is the minimum potential required to run the reaction (1). Any reaction step (2)–(4) that has
is the minimum potential required to run the reaction (1). Any reaction step (2)–(4) that has  /e >
/e >  requires a higher potential to run it, and the maximum
requires a higher potential to run it, and the maximum  /e then defines the overpotential required for the whole four-step reaction to proceed.
/e then defines the overpotential required for the whole four-step reaction to proceed.
3. OER using an alkaline reaction mechanism
Unlike under acidic conditions, equation (1), the water oxidation reaction under alkaline conditions is given by [14, 15]
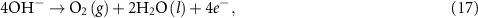
This reaction is usually assumed to proceed in four elementary steps [11]

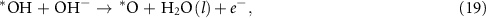

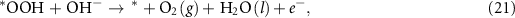
where the notation is the same as in equations (2)–(5). Different from the acidic mechanism, the Gibbs free energies of equations (18)–(21) are not commonly discussed. Thus, we would like to derive the theory for the OER under alkaline conditions step by step. The reaction Gibbs free energies corresponding to equations (18)–(21) can be expressed as
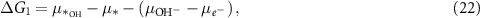
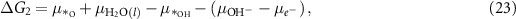
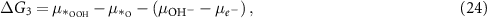
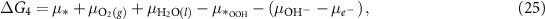
where  are the (electro)chemical potentials of the indicated species. These chemical potentials can be obtained from [20]
are the (electro)chemical potentials of the indicated species. These chemical potentials can be obtained from [20]





where  and
and  are the (DFT) total energies of the clean surface (*) and of surfaces with a single adsorbed species
are the (DFT) total energies of the clean surface (*) and of surfaces with a single adsorbed species  , respectively, and
, respectively, and  ,
,  are the corresponding vibrational zero point energy and entropy.
are the corresponding vibrational zero point energy and entropy.  is the total energy of the H2O molecule,
is the total energy of the H2O molecule,  and
and  the corresponding vibrational zero point energy and entropy contributions, where the latter also contains the free energy correction for the liquid state.
the corresponding vibrational zero point energy and entropy contributions, where the latter also contains the free energy correction for the liquid state.
For O2, the chemical potential is obtained from the experimental formation energy of O2 with respect to water, since the O2 molecule is not very well described within DFT. Assuming the equilibrium  , the chemical potential of O2 can be written as [20]
, the chemical potential of O2 can be written as [20]
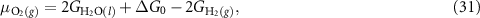
The only unknowns remaining in equations (22)–(25) are now  and
and  , the chemical potentials of the OH- species and the electron, respectively, where we actually only need the difference
, the chemical potentials of the OH- species and the electron, respectively, where we actually only need the difference  . To calculate this difference, we assume the equilibrium
. To calculate this difference, we assume the equilibrium

which relates the chemical potentials as

Using the trick
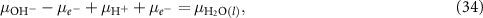
one can rewrite this as
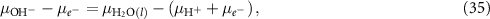
Here,  can be calculated according to equation (30) and
can be calculated according to equation (30) and  can be calculated using the CHE approach [4, 18], where one assumes the equilibrium
can be calculated using the CHE approach [4, 18], where one assumes the equilibrium

We describe this equilibrium using the RHE as reference electrode, which operates under standard conditions of hydrogen gas pressure  = 1 bar and T = 298.15 K (but with a pH of the actual experimental conditions, which is different from zero), so
= 1 bar and T = 298.15 K (but with a pH of the actual experimental conditions, which is different from zero), so

The remaining electrochemical potential of the electrons can then be expressed as

where  is the potential of the electrode relative to the RHE [18].
is the potential of the electrode relative to the RHE [18].
Combining equations (36)–(38), the equilibrium can then be expressed as
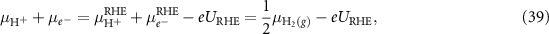
Substituting the equation (39) into equation (35), we finally have
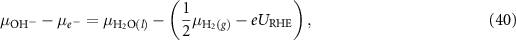
Using equation (40) in equations (22)–(25) we get
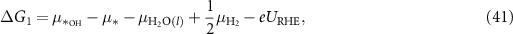
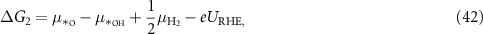
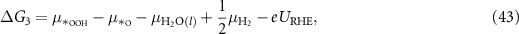
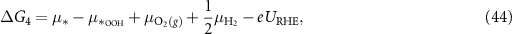
Applying equations (26)–(31) to equations (41)–(44), the final expressions for the reaction Gibbs free energies using an alkaline reaction mechanism become
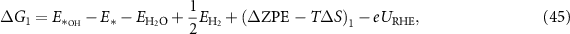
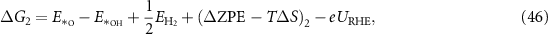

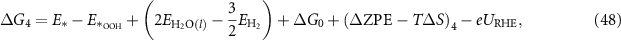
The reaction Gibbs free energies of equations (45)–(48) obey the sum rule
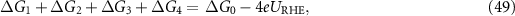
and the overpotential is given by

4. Comparison between the acidic and the alkaline reaction mechanism
Under acidic conditions, the Gibbs free energy of  is linked to the Gibbs free energy of ½H2 and the reaction Gibbs free energies are calculated from equations (6)–(9). Under alkaline conditions, the Gibbs free energy of
is linked to the Gibbs free energy of ½H2 and the reaction Gibbs free energies are calculated from equations (6)–(9). Under alkaline conditions, the Gibbs free energy of  can be related to the Gibbs free energy of
can be related to the Gibbs free energy of  according to equations (35) and (40). The reaction Gibbs free energies for the alkaline reaction mechanism can then be calculated from equations (45)–(48).
according to equations (35) and (40). The reaction Gibbs free energies for the alkaline reaction mechanism can then be calculated from equations (45)–(48).
Comparing the reaction Gibbs free energies of the acidic (equations (6)–(9)) and of the alkaline (equations (45)–(48)) reaction mechanisms, it can be seen that the only difference lies in the electrode potential. In the case of the acidic mechanism, the electrode potential is related to the SHE and in the case of the alkaline mechanism, it is related to the RHE. Hence, the reaction Gibbs free energies can be calculated with almost the same equations even though the reaction mechanisms for the acidic environment, equations (2)–(5), and for the alkaline environment, equations (18)–(21), are different.
The equations used for calculating the overpotential, equations (16) and (50) are exactly the same. The numerical results will of course be different, as two different reference potentials, SHE and RHE, are used to calculate the Gibbs free energies in the acidic and the alkaline cases. The electrode potential of the two reference potentials can be linked according to [18]

where  is the activity of the H+ ions in solution. Substituting equation (51) into equations (45)–(48), one obtains the reaction Gibbs free energies with the SHE as reference. At T = 298.15 K one has
is the activity of the H+ ions in solution. Substituting equation (51) into equations (45)–(48), one obtains the reaction Gibbs free energies with the SHE as reference. At T = 298.15 K one has  eV.
eV.
Substituting equation (51) into equations (45)–(48) for the alkaline reaction mechanism, one obtains the same expression as given in [3, 4, 20] for the acidic reaction mechanism with the correction factor  Therefore, at pH = 0, the reaction Gibbs free energies can be calculated according to equations (6)–(9), with the overpotential given by equations (16). At a pH different from 0, the reaction Gibbs free energy should be corrected with
Therefore, at pH = 0, the reaction Gibbs free energies can be calculated according to equations (6)–(9), with the overpotential given by equations (16). At a pH different from 0, the reaction Gibbs free energy should be corrected with  . Alternatively, using RHE as reference electrode, the reaction Gibbs free energy can be calculated by equations (45)–(48) at arbitrary pH and the overpotential can be calculated by equations (50).
. Alternatively, using RHE as reference electrode, the reaction Gibbs free energy can be calculated by equations (45)–(48) at arbitrary pH and the overpotential can be calculated by equations (50).
5. Summary
The OER mechanism under alkaline and acidic conditions are discussed and key parameters for the OER, such as the reaction Gibbs free energy and the overpotential, are derived for both reaction mechanisms. It is shown that—even though the single reactions look quite different—the same equations can be used for calculating these key parameters, provided the RHE is used as a universal reference. The effect of using different reference electrodes, i.e. RHE or SHE, on the equations for the key parameters is discussed. We suggest for the future to use for both acidic and alkaline mechanisms the RHE as a reference electrode. In that case, equations (45)–(50) can be used for calculating the reaction Gibbs free energies and the overpotential for the OER at all pH values for both acidic and alkaline mechanism.
Acknowledgments
Q Liang acknowledges funding from the China Scholarship Council (CSC) (No. 201708450082). Vivek Sinha is acknowledged for fruitful discussions.