
Abstract
SARS-CoV-2 virus is the serious health concern throughout the world. A comprehensive investigation of binding of SARS-CoV-2 active site with host receptor protein hACE2 is important in designing effective drugs. In the present work, the major amino acid binding partners between the virus CTD and host receptor have been studied and are compared with SARS-CoV RBD binding with hACE2. Our investigation show that some unique hydrogen bond pairs which were not reported in previous work. Along with hydrogen bonding, salt-bridges, hydrophobic interactions and contributions of electrostatic and van der Waals contacts play significant role in binding mechanism. The binding affinity of SARS-CoV-2 CTD/hACE2 is greater than SARS-CoV RBD/hACE2. This outcome is also verified from the free energy estimation by using umbrella sampling.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Corona virus disease (COVID-19) pandemic, caused by severe acute respiratory syndrome (SARS)-like corona virus (SARS-CoV-2), is a serious health concern for the global community [1–4]. Although the origin of the virus is still unclear, it has been spread all over the world threatening the human civilizations after its initial outbreak from China in 2019. Till date, more than 52 millions infected population has been reported globally and more than 12 hundred thousands people have lost their lives [5]. There is no approved drug or vaccine against the COVID-19, even though several antiviral drugs have been proposed and are also in clinical trials [6]. Understanding more about interactions of this virus with human body is highly demanding at this pandamic time to design drugs. SARS-CoV and other viruses had also threatened the human society at different periods; however the influence of SARS-CoV-2 is significantly higher than other viruses throughout the globe.
SARS-CoV-2 has more than 70 percent of structural similarity with SARS-CoV; most of the residues at binding interface are similar [7, 8]. SARS-CoV-2 similar with SARS-CoV and other corona viruses utilize same human angiotensin converting enzyme 2 (hACE2) receptor to enter into human cell. This entry process is mediated by the spike(s) glycoprotein which are embedded in the capsid of SARS-CoV-2 [9]. The spike protein is subdivided into two receptor binding entities S1 and S2. S1 is responsible in the detection of receptor, whereas S2 plays important role in membrane fusion. Similar to SARS-CoV, C-Terminal Domain(CTD) of S1 subunit of spike protein in SARS-CoV-2 acts as receptor binding domain (RBD) [10, 11]. Even though both SARS-CoV and SARS-CoV-2 have same binding domain, the binding affinity of SARS-CoV-2 is different from that of SARS-CoV [12, 13].
Immediately after the COVID-19 outbreak, several researches have been carried out to identify the nature and location of binding of SARS-CoV-2 CTD with hACE2 using static crystal structure [14, 15]. Although, these researches have attempted to detect the entry process and binding mechanism of SARS-CoV-2 with hACE2, the breakthrough on drug designing is still challenging. Several works are in the way of hopeful future, exploration of detail binding mechanism is still being essential. Moreover, the detail dynamics of SARS-CoV-2 and hACE2 can be very important to design the drug. When we were independently working on the binding mechanism of SARS-CoV-2 with host receptor hACE2, in the mean time similar type of research works have been published [12]. However our technique as well as some results are different from previous works.
We focus on the estimation of free energy difference of virus protein and hACE2 complex. The free energy calculation provides in-depth insight on the binding mechanism between the protein molecules [16]. There are several experimental techniques of measuring binding free energy such as isothermal titration calorimetry (ITC) [17], fluorescence resonance energy transfer (FRET) [18], nuclear magnetic resonance (NMR) [19], surface plasmon resonance (SPR) [20] and many others. The computational approach can be the best complement for large scale investigations [21–23]. Out of many computational approach, umbrella sampling is one of the widely used method for the estimation of free energy in large molecular systems [24, 25]. It improves the sampling system by designing and implementing the biasing potentials as a function of reaction coordinates [26, 27]. If an energy barrier exists in between two regions of configuration states, there may be poor sampling, despite the long simulation run being carried out. The applied biasing potential bridges such configuration states and makes it easier in searching local or global minima, which can be considered as the structurally favorable state in the molecular complex [28]. Besides these techniques, free energy can be calculated directly from steered molecular dynamics (SMD) [29, 30].
In the present work, we have carried out molecular dynamics (MD) simulations for the comprehensive study of binding mechanism of SARS-CoV-2 CTD with hACE2 and also compared with SARS-CoV-RBD/hACE2. In addition, umbrella sampling method has been executed to estimate the binding free energy of SARS-CoV-2 CTD/hACE2. Required windows for the umbrella sampling have been taken from steered molecular dynamics (SMD) [31] simulations. In SMD, SARS-CoV-2 CTD has been translated taking the hACE2 as the reference molecule. The quantitative estimation of binding affinity between the targeted molecules facilitates in silico-drug designing. We have also performed comparative study of various interactions such as hydrogen bonding, salt bridges, hydrophobic, electrostatic and van der Waals interactions at the binding interface of SARS-CoV-2 and SARS-CoV with hACE2. Furthermore, the contact surface area of these complexes have been estimated and compared to investigate the stability.
2. Methods
2.1. System set up
Two molecular structures, PDB IDs 6LZG and 2AJF, were taken for the molecular dynamics simulations. The PDB ID 6LZG contains the complex of SARS-CoV-2 CTD and hACE2 receptor protein (i.e., SARS-CoV-2 CTD/hACE2 complex) and that of PDB ID 2AJF contains the complex of SARS-CoV RBD and hACE2 receptor protein (i.e., SARS-CoV RBD/hACE2 complex) [32]. CHARMM-GUI [33] was used to create the pdb and psf files of these complexes. Then, both the complex structures were solvated using TIP3P [34] water and electrically neutralized by adding NaCl. We have added the NaCl in the system with concentration 0.15 M by using CHARMM-GUI. In SARS-CoV-2 CTD/hACE2 complex system 220 Na+ ions and 197 Cl− ions were added to neutralize the system. Similarly in SARS-CoV RBD/hACE2 complex system 214 Na+ and 188 Cl− ions were added so that the system became neutral. A cubical box of dimensions 144 × 144 × 144 Å3 was prepared for NPT simulation of the complex SARS-CoV-2 CTD/hACE2 and another cubical box of dimensions 131 × 131 × 131 Å3 was prepared for NPT simulation of the complex SARS-CoV RBD/hACE2. Further, two equal sized orthorhombic simulation boxes were prepared in order to estimate the free energy differences of above complexes by changing the dimensions to 250 × 90 × 90 Å3 and electrically neutralized by adding NaCl with concentration 0.15 M.
2.2. Molecular dynamics simulation
All atom molecular dynamics (MD) simulations were performed using NAMD [31] simulation package. The CHARMM36m [35] force field was used for each simulations. The Particle Mesh Ewald (PME) [36] was used for the long-range interactions with a 12.0 Å non-bonded cut-off. The energy minimization was performed for 10,000 steps, using the conjugate gradient and line search algorithm [37, 38]. The system was then equilibrated at 310 K for 10 ns with harmonically restrained heavy atoms taking 1 fs time step. Finally, the production run was propagated for 250 ns simulation under NPT conditions by using Langevin dynamics with a damping constant of 1 ps−1 taking time step of 2 fs.
2.3. Molecular dynamics and umbrella sampling
To perform the umbrella sampling, sample windows were chosen from steered molecular dynamics (SMD) trajectories. During SMD, CTD/RBD of SARS-CoV-2 CTD/SARS-CoV RBD were pulled correspondingly towards the negative x-direction with constant velocity pulling method of velocity 0.00005 Å/fs. In this process, the alpha carbons of hACE2 protein were taken as the fixed atoms and alpha carbons in CTD/RBD part of spike protein of the systems were taken as the dummy atoms. CTD/RBD of spikes were pulled from their center of mass (COM) along the negative x-direction with constant velocity  in water and ions environment. Then the SMD atom experiences the force
in water and ions environment. Then the SMD atom experiences the force  , providing the external potential energy,
, providing the external potential energy,
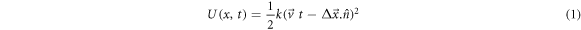
where, k (=5 kcal mol−1 Å−2) is the spring constant and gives the stiffness of the applied harmonic restraining force, and  , is the displacement of SMD molecules from initial position
, is the displacement of SMD molecules from initial position  to instantaneous position
to instantaneous position  and
and  is the unit vector along the direction of pulling.
is the unit vector along the direction of pulling.
Out of many other techniques of free energy calculations [39], umbrella sampling was performed to investigate the free energy difference during the translation of SARS-CoV-2 CTD from hACE2 protein for system SARS-CoV-2 CTD/hACE2; and identical condition is applied for system SARS-CoV RBD/hACE2. SMD trajectories were used to select the appropriate windows. Identifying the information on the termination of molecular interactions from SMD, we estimated the number of umbrella windows in both the systems. Ten windows were prepared in SARS-CoV-2 CTD/hACE2 and six windows were prepared for SARS-CoV RBD/hACE2 complexes. Every successive window was taken from the SMD trajectories during the translation of 1 Å along the negative x-direction. The window size ensures the sufficient overlapping of successive windows to cover the entire reaction coordinate space. The reaction coordinate was chosen as the distance between the center of mass (COM) of hACE2 and CTD/RBD spike along the negative x-axis. To make the necessary overlapping reaction coordinates, a bias potential of the ith window Vi (x) was used to force the system to fluctuate in coordinate space, which is given by,
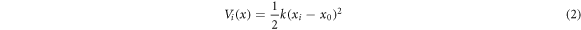
where x0 is the harmonic constraint defining a center of window i (i=1 to 10 for for SARS-CoV-2 and 1 to 6 for SARS-CoV), and force constant k is the window width. We used the force constant of 1.5 kcal mol−1 Å−2.
2.4. Data analysis
Visual Molecular Dynamics (VMD) [40] and Pymol [41] were used to visualize as well as generate images of the complex structures . VMD analysis tools were used to identify and analyze non-bonded interactions by using the simulation trajectories. The NAMD energy plugin, available in VMD, was used to calculate the non-bonded interaction energy contributions. Pycontact [42] software package was used to analyzed the hydrophobic interactions and salt bridges between the targeted protein residues in CTD/RBD of spike protein and ACE2. Weighted Histogram Analysis Method (WHAM) program [43] was used to estimate the free energy from umbrella sampling simulation. The free energy calculation of large molecular system is generally computationally demanding. This method minimizes the statistical errors as well as increases the efficiency of computational simulation. Moreover, it has the advantage of multiple overlapping of probability distributions for obtaining better estimation of free energy calculations.
3. Results
3.1. Conformational variation in complexes
To examine the conformational variation during the dynamics, we have estimated the root mean square deviation (RMSD) of each molecule on SARS-CoV-2 CTD/hACE2 and SARS-CoV RBD/hACE2 complexes. Besides RMSD, contact surface area between the molecules within the each complex has also been calculated for both complexes.
We have calculated the RMSD for all atoms of proteins backbone without taking hydrogen atoms. The structure from first step of the simulation was taken as the reference to calculate the RMSD. The RMSD of hACE2 and spike CTD/RBD have been compared separately to evaluate the structural integrity of the molecules. Figures 1(a) shows the RMSD of hACE2 molecule in SARS-CoV-2 CTD/hACE2 and SARS-CoV RBD/hACE2 complexes and figure 1(b) represents the RMSD of spike CTD/RBD. From the figure, it is observed that RMSD of hACE2 of SARS-CoV-2 CTD/hACE2 is smaller than that of SARS-CoV RBD/hACE2. RMSD of both the systems are stable with the values below 3.0 Å and 4.5 Å for CoV-2 and CoV respectively. SARS-CoV-2 CTD has the RMSD of 2.5 Å, whereas the RMSD of SARS-CoV RBD is 4.0 Å. This shows the large rearrangements of structure in SARS-CoV RBD, while SARS-CoV-2 CTD structure remains relatively stable.

Figure 1. RMSD plot of SARS-CoV-2 CTD/hACE2 and SARS-CoV RBD/hACE2. RMSD plot in blue color represents the SARS-CoV-2 and red color represents the SARS-CoV. The graphs reveal the stable structure in SARS-CoV-2 CTD than that of SARS-CoV RBD.
Download figure:
Standard image High-resolution imageTo get more insight into stability, we also analyzed the contact surface area between the spike protein CTD/RBD and hACE2 receptor using MD trajectories. Contact area is the surface buried at the interface between two proteins which contributes to bind and stabilize the protein-protein complexes. Larger contact surface indicates greater stability of the structure [44]. The estimated values of contact surface area are presented in table 1. From the table 1, it has been observed that SARS-CoV-2 CTD/hACE2 has larger contact area than SARS-CoV RBD/hACE2 by 77.02 ± 2.46 Å2. The contact surface area for SARS-CoV-2 CTD is more in comparison to SARS-CoV RBD indicating the greater binding affinity of SARS-CoV-2 with receptor.
Table 1. Calculation of contact surface area; A1, A2 & A3 are the average solvent accessible surface area (SASA) of hACE2, SARS-CoV-2 CTD and their complexes; and  are the average solvent accessible surface area (SASA) of hACE2, SARS-CoV RBD and their complexes.
are the average solvent accessible surface area (SASA) of hACE2, SARS-CoV RBD and their complexes.
| Complex | SASA (Å2) for | Net contact area (A)[44] | |||
|---|---|---|---|---|---|
| SARS-CoV-2 CTD/hACE2 | hACE2 (A1) | SARS-CoV-2 CTD (A2) | complex (A3) | A1 + A2 − A3 | (A1 + A2 − A3)/2 |
| 29 100.36 | 11227.97 | 38 549.02 | 1779.31 | 889.66 ± 1.07 | |
| SARS-CoV RBD/hACE2 | hACE2 
| SARS-CoV RBD 
| complex 
|
| Net contact area ( ) ) |
| 29 092.62 | 10859.54 | 38326.88 | 1625.27 | 812.64 ± 2.22 | |
3.2. Non-bonded interactions
Furthermore, we studied in details the hydrogen bonds, salt-bridges, hydrophobic, electrostatic and vdw interactions between the residues residing at the interface between SARS-CoV-2 CTD/hACE2 and obtained results are compared with SARS-CoV RBD/hACE2.
3.2.1. Hydrogen bonds
At the interface region, hydrogen bonds play pivotal role in binding the molecules to form a stable complex. Wang et al (2020) and Lan et al (2020) have studied the atomic interactions at the interface of static crystal structure of SARS-CoV-2 CTD/hACE2 complex [14, 15], whereas we have investigated the hydrogen bonds at the interface of two complexes by analyzing the MD simulations trajectories. The cut-off distance for hydrogen bond was taken to be 3.5 Å [14]. We monitored the time evolution of number of hydrogen bonds formed at the interface between SARS-CoV-2 CTD/hACE2 and also compared with that of SARS-CoV RBD/hACE2 as shown in figure 2 (also see supplementary table S 1). Hydrogen bonds were found to be consistently existing in both complexes. Total hydrogen bonds formed during the simulations were seen to be more in case of SARS-CoV; however the strength and life time of potential hydrogen bonds were found to be greater in case of SARS-CoV-2.

Figure 2. Comparison of time evolution of number of hydrogen bonds at the interfaces of SARS-CoV-2 CTD/hACE2 and SARS-CoV RBD/hACE2.
Download figure:
Standard image High-resolution imageMany research works have been published by analyzing the hydrogen bonds pair partners between the molecules in the complexes. Even though our investigations regarding the hydrogen bonds in the complexes are in consistent with those published papers, some pair partners are not consistent with these previously published outcomes. Ali et al reported three unique hydrogen bonds in SARS-CoV-2 CTD/hACE2 complex, which were not detected in SARS-CoV RBD/hACE2 complex [12]. We found consistent result in GLU35-TYR449 pair, however the result is not consistent with other two pair partners: TYR449-ASP38 and GLN498-LYS353. We have clearly observed ASP38-TYR436 and LYS353-GLY488 pairs in SARS-CoV-RBD/hACE2. Furthermore, a strong hydrogen bond has been detected between GLN498 with GLN42. In static structures, no hydrogen bond was formed by SER19 of hACE2 with ASP463 residue of SARS-CoV RBD [14, 15]. Our investigation shows two potential hydrogen bonds formed between main-main & main-side chains of SER19 of hACE2 with side chain of ASP463 of SARS-CoV RBD and similar type of bond has been detected between SER19 and ALA475 in SARS-CoV-2 CTD/hACE2 complex (see figure 4(a) and supplementary figures S1 and S2).
In the present work, interactions of molecules in each complex has been observed considering three main regions where the interfacial residues of hACE2 take part actively in binding with the spike CTD/RBD section of the virus molecule as shown in supplementary figures S1(a) and S2(a). The hydrogen bonds formed at three key regions of interface between SARS-CoV RBD/hACE2 in the begining of the simulation run are shown in figures (see supplementary figures: S1(a), S1(b) & S1(c)). Some residue pairs whose hydrogen bond (Hbond) occupancy percentage greater than 20% is shown in figure 3 and detail of Hbond occupancy percentage in three key regions are shown in figures 4(a), (b) and (c). We have observed some differences in the atomic interactions at the interface of both virus proteins and hACE2 than that of static crystal structure. The two different approaches might be the reason of variation in the number of interactions.

Figure 3. The arrangement of major interaction residues at the interface of SARS-CoV RBD/hACE2 complex. Three subdivided parts of the interface region are circled in 3(a). Figure 3(a) represents the molecular structure of the complex in the beginning of the simulations and that of at the end of the simulation is represented by figure 3(b). The contact pairs in color contrast are SER19-ASP463 (red), ASP30-TYR442 (green), LYS31-ASN479 (blue), ASP355-THR486 (magenta), and LYS353-GLY488 (violet).
Download figure:
Standard image High-resolution image
Figure 4. Hbond occupancy percentage details of major interacting residues in SARS-CoV RBD/hACE2 complex. The double bars for SER19-ASP463 in bar diagram shows the significant bonding of these residues in side chain—main chain and main chain—main chain interactions. 4(a), 4(b) and 4(c) are the Hbond occupancy percentage for three key regions respectively.
Download figure:
Standard image High-resolution imageThe rearrangement of aminoacids at the binding vicinity have been detected in both the complexes. Different color contrast have been used to indentify the pair partners as shown in figures 3 and 5. The binding affinity of the complex was observed increasing due to the allignments of pair partners at the interface.
At the binding interface of SARS-CoV-2 CTD and hACE2 receptor, three key regions where most of the polar contacts are actively participated to form the hydrogen bonds. The interactions at the interface in which Hbond occupancy greater than 20% are shown in figure 5, and also the details of Hbond occupancy in these regions are shown in figures 6(a), (b) and (c). In region 1, SER19, GLN24 and TYR83 of hACE2 form hydrogen bonds with ALA475 and ASN487 of SARS-CoV-2 CTD as in figures (see supplementary figures: S2(a), S3(a), S4(a), S5(a) & S6(a)). In region 2, there are interactions between the residues LYS417, TYR453 and GLN493 of SARS-CoV-2 CTD forming hydrogen bonds with ASP30, LYS31 and GLU35 of hACE2 (see supplementary figures: S2(b), S3(b), S4(b), S5(b) & S6(b)). Similarly, in region 3, there is extensive network of hydrogen bonds between SARS-CoV-2 CTD residues TYR449, GLN498, THR500, ASN501, GLY502 and TYR505 with the hACE2 residues GLU37, ASP38, TYR41, GLN42, LYS353 and ASP355 (see supplementary figures: S2(c), S3(c), S4(c), S5(c) & S6(c)). Because of dynamical nature of our system, there is continuous formation, breaking and reformation of hydrogen bonds during the simulations. The variation of hydrogen bonds formed during the five representative frames of 250 ns simulation of SARS-CoV-2 has been shown in supplementary figures S2–S6.

Figure 5. Major interacting residues at the interface of SARS-CoV-2 CTD/hACE2 complex. Three subdivided regions are circled in which 4(a) represents the molecular complex at the beginning stage of the simulation run and 4(b) represents the structure rearrangement of residues of molecular complex at the end of simulation run. The pairs in color contrast are TYR83-ASN487 (red), ASP30-LYS417 (green), GLU35-CLN493 (violet), LYS353-GLY502 (lime), TYR41-THR500 (orange) and ASP355-THR500 (magenta).
Download figure:
Standard image High-resolution image
Figure 6. Details of Hbond occupancy percentage of major interacting residues in SARS-CoV-2 CTD/hACE2 complex. 6(a), 6(b) and 6(c) are the Hbond occupancy percentage for three key regions respectively.
Download figure:
Standard image High-resolution image3.2.2. Salt-bridges
In addition to extensive network of interfacial hydrogen bonds, another important contributions to protein-protein binding comes from salt-bridge interactions. MD trajectory analysis has shown three salt-bridges, having different bond length and strength, formed at the interface of SARS-CoV-2 CTD/hACE2. The salt-bridge formed between the residue LYS417 of SARS-CoV-2 CTD with ASP30 of hACE2 is found to be the strongest one among them owing to its short bond length. The remaining residues GLU484 and LYS458 of SARS-CoV-2 CTD have formed salt-bridges with LYS31 and GLU23 of hACE2 respectively. In contrast, we found only one salt-bridge formed between ARG426 of SARS-CoV RBD and GLU329 of hACE2 which is weaker than that of SARS-CoV-2 because of larger bond length as in figure 7.

Figure 7. Comparison of time evolution of salt-bridge bond length at the interface of SARS-CoV RBD/hACE2 and SARS-CoV-2 CTD/hACE2. The residue pair ASP30-LYS417 corresponds to the SARS-CoV-2 CTD/hACE2 system and GLU329-ARG426 corresponds to the SARS-CoV RBD/hACE2 system.
Download figure:
Standard image High-resolution image3.2.3. Hydrophobic interactions
3.2.4. Electrostatic and van der Waals (vdw) interactions
The electrostatic and van der Waals (vdw) interactions in two complexes SARS-CoV RBD/hACE2 and SARS-CoV-2 CTD/hACE2 have been studied. Supplementary figure S7 depicts the comparative analysis of energy due to electrostatic and vdw interactions as a function of time. In the beginning of simulations, the electrostatic contributions of SARS-CoV-2 CTD/hACE2 was distinctly higher than SARS-CoV RBD/hACE2, however most of the simulation time, the contributions were almost equal. In addition, the potential energy contributed by vdw interactions were consistently almost equal for both the systems throughout the simulations. It reveals that electrostatic and vdw interactions are almost equally contributed in binding both the complexes.
3.3. Free energy
To investigate the energetic difference in binding of hACE2 with SARS-CoV-2 CTD and SARS-CoV RBD, the free energy differences have been estimated using umbrella sampling technique. Umbrella windows were taken from the trajectories of SMD simulations. The interactions between the molecules in SARS-CoV-2 CTD/hACE2 were found terminated after traversing 9 Å distance away from the original position. To incorporate all interacting pathways, ten windows with 1 Å distance separation were taken for every successive window. On the other hand, the interactions between the molecules in SAR-CoV RBD/hACE2 were found ceased after traversing 5 Å distance from the original position. Therefore, six windows were prepared separating 1 Å distance away for every successive window. To ensure the overlapping of consequent data sets in umbrella sampling, we have plotted the distributions of data obtained from the simulations. We found sufficient overlapping of data sets. The graphs for distribution versus COM distance have been included in the supplementary figures S8 and S9. Figure 8 shows the change in free energy during the translation of virus CTD/RBD from active pocket of hACE2 for both complexes. The center of mass (COM) distance as a reaction coordinate allows us to track the free energy changes for SARS-CoV-2 CTD in complex with hACE2 and SAR-CoV RBD in complex with hACE2. Free energy has also been used to compare the differences in the binding affinity for the two complexes. The SARS-CoV-2 CTD in complex with hACE2 is found to have the greater binding free energy of ∼1.91 kcal/mol compared to the SAR-CoV RBD in complex with hACE2. This, as well as the nature of the free-energy curve, provides an insight on binding mechanisms of the complexes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 8. Free energy curve for SARS-CoV-2 CTD/hACE2 and SARS-CoV RBD/hACE2 during the translation of spikes CTD/RBD from hACE2.
Download figure:
Standard image High-resolution image{kind=link}
4. Discussion
COVID-19 pandemic has seriously threatened public health throughout the globe. Since there is no approved drug till date to combat against the SARS-CoV-2 virus, more comprehensive study is essential through the various aspects at molecular level. The fundamental necessity is to understand the entry mechanism of the virus into the human cell, which is really helpful to discover the drug against the virus. To deal the entry mechanisms and dynamical characteristics of the virus cell in complex with hACE2 receptor, we used various computational techniques. C-Terminal Domain (CTD) of S1 subunit of spike protein, being the active interacting region, has been taken into consideration in SARS-CoV-2. We performed the comparative analysis of the key residues and atomic interactions responsible for the binding of the SARS-CoV-2 CTD and SARS-CoV RBD with human ACE2 receptor.
Estimation of structural variation during the simulation is the foremost judgement of molecular stability in molecular dynamics study. RMSD is the measure of stability of molecular structure in the cellular environment. Well equilibrated system with consistent RMSD ensures us to proceed for the further study of binding affinity and energy variations of the molecular complexes. Moreover, contact surface area between the molecules identifies the binding strength of the complex. Therefore, we have obtained the contact surface area of both complexes calculating the solvent accessible surface area (SASA). SASA has been determined from time evolution data generated from the 250 ns NPT run. Then, average value of contact area for both the systems have been presented in table 1 and are interpreted graphically in figure 2. Larger contact surface area in SARS-CoV-2 CTD/hACE2 complex depicts the stronger binding of this complex than that of SARS-CoV RBD/hACE2 [45].
Our results show considerable similarity in the binding sites, interfacial residues and important atomic interactions in both viral protein receptor binding domain (i.e., SARS-CoV-2 CTD and SARS-CoV RBD). However, there are some variations in loop between two structures in the binding region and some residues at the binding sites are different. This facilitates more and stronger atomic contacts between SARS-CoV-2 CTD and hACE2 interface and thereby enhancing its binding affinity. Polar residues residing at the interface form an extensive network of hydrogen bonds and salt-bridge interactions [46–49]. Our study reveals that interfacial hydrogen bonds, salt-bridges and hydrophobic interactions play an important role in the binding of SARS-CoV-2 CTD to host cell receptor. Furthermore, comparative analysis of the binding mechanism of two viral proteins with hACE2 show that binding affinity of SARS-CoV-2 is greater than that of SARS-CoV. Notably, more residues are engaged in the binding of SARS-CoV-2 CTD with hACE2. We find the greater number of potential hydrogen bonds formed in the case of SARS-CoV-2 CTD which contributes to higher binding affinity. More and stronger salt-bridges formed in case of SARS-CoV-2 CTD establish stronger binding to the receptor than SARS-CoV RBD. Additionally, we observe hydrophobic interactions are stronger in case of SARS-CoV-2 which also contribute to enhanced binding.
The contributions of electrostatic and vdw contacts are significant to form a stable protein-protein complex [50, 51]. The potential energy in binding the virus CTD/RBD and host receptor are compared in both the systems. Though, initially the electrostatic energy is observed relatively larger in SARS-CoV-2 CTD/hACE2 than that of SARS-CoV RBD/hACE2, the dynamical results show almost equal contributions in both the complexes. This shows that the contributions of hydrogen bonds, salt bridges and hydrophobic interactions are responsible to provide the greater binding strength in SARS-CoV-2 CTD/hACE2.
The binding mechanisms of the complexes are further analyzed to estimate the free energy differences from umbrella sampling method. SMD trajectories are taken for the appropriate samples that ensure the sufficient overlapping on windows [52]. In SMD, the virus CTD/RBD are pulled upto that distance, beyond which no interactions persists. We find the interactions of molecules in complex SARS-CoV RBD/hACE2 have been terminated after the displacement of RBD by 5 Å from host receptor, whereas the interactions sustain upto 9 Å displacement from the initial position in SARS-CoV-2 CTD/hACE2. Comparisons of free energy of two complexes have provided the insight of bonding affinity between the virus CTD/RBD and hACE2 molecules. The greater free energy difference between SARS-CoV-2 CTD in complex with hACE2 depicts the stronger binding strength than the complex of SARS-CoV RBD and hACE2. As the further investigation, we plan to calculate the solvation free energy of SARS-CoV-2 and SARS-CoV molecule in the aqueous environment.
Acknowledgments
RPK & SPK acknowledge the partial financial support from Nepal Academy of Science and Technology (NAST).JP acknowledges the partial financial support from university grant commission (UGC). NPA acknowledges the UGC Award no. CRG-73/74-S&T-01 and TWAS research grants RG 20-316. We acknowledge the computing facilities of Supercomputer Centre Kathmandu University, which was established with equipment donated by CERN and We thank Prof. Prem Chapagain and Prabin Baral at Florida International University for their helpwith computing facilities for MD simulations.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).