
Abstract
Alumina (α- and γ-Al2O3) particles are formed in liquid Al-Mg alloys during the liquid dealing and cast processes. These native oxide particles have non-trivial influences on the microstructures and properties of the solidified parts, and may act as potential heterogenous nucleation sites during solidification. At present there is still a lack of understanding about the interaction and atomic arrangements at the interfaces between liquid-Al and γ-Al2O3 substrates. Here we investigate the liquid-Al/γ-Al2O3{1 1 1} interfaces by means of ab initio molecular dynamics simulations and electronic structure calculations. We found that the interfacial interaction at the interfaces leads to formation of an ordered terminating Al layer. This newly formed terminating Al layer is positively charged and chemically bonded to the substrate and thus, becomes part of the substrate. Analysis showed that the terminating Al layer contains vacancies and displacements, being atomically rough. The newly-formed Al layer is also structurally coupled with the substrates. These γ-Al2O3 particles are weak templates for nearby liquid to nucleate. The present study sheds some light on the role of alumina particles in grain refinement of Al-based alloys during solidification processing.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Introduction
Alumina particles are formed inevitably during the liquid-dealing and casting of Al-based melts [1–7]. Oxidation reactions in the Al-based melts produce γ-Al2O3 plates at 750 °C [2, 5] and α-Al2O3 particles at 920 °C [5], respectively. Images of high resolution transmission electron microscopy (HR-TEM) revealed that the γ-Al2O3 plates exhibit hexagonal shapes and are {1 1 1}-faceted [5, 7]. These native alumina particles usually exist in the melt as oxide films that have negative influences on the performance of solidified Al parts, but they may be harnessed as potential nucleation sites during the solidification processes of Al-based alloys [5, 7–9].
Recent study revealed that the early stage of solidification processes contains several steps [10, 11]. The initiating step is prenucleation which refers to the atomic ordering in liquid atoms near a solid substrate at temperatures above the nucleation temperature [10–13]. The epitaxial nucleation model [14] suggested that heterogeneous nucleation builds on the ordering created by prenucleation and proceeds in a layer-by-layer growth mechanism. The substrate surface provides a structural template to induce atomic ordering in the liquid or prenucleation. Prenucleation provides a precursor at the nucleation temperature for heterogeneous nucleation of the solid phase. Therefore, knowledge about prenucleation at the interfaces between liquid Al and γ-A2O3{1 1 1} surfaces is crucial to obtain insight into the role of the alumina particles in heterogeneous nucleation during solidification of Al-based alloys.
Chemically, alumina is an ionic compound due to the electronegativity difference between Al (1.61 in Pauling scale) and O (3.44). Crystallographically, α- and γ-Al2O3 phases are very different. α-Al2O3 exhibits a trigonal lattice, in which the Al ions are in distorted octahedral coordination of O ions [15, 16]. Meanwhile, γ-Al2O3 has a cubic lattice with the Al ions being coordinated both tetragonally and octahedrally by O [17–20]. However, in common along the γ-Al2O3 [1 1 1] and the α-Al2O3 [0 0 0 1] axis, both structures are composed alternatively of an O layer and an Al layer, as shown in figure 1. The O ions form two-dimensional (2D) distorted hexagonal sublattices as exampled in figures 1(b) and 1(c). The Al ions occupy the interstitial sites of the neighboring O layers in different ways. The Al ions occupy two thirds of the octahedral sites orderly, forming two sublayers along its [0 0 0 1] axis in α-Al2O3 (figure 1(d)). The Al arrangements in γ-Al2O3 are based on the replacement of the Mg sites in spinel (MgAl2O4) by Al. In order to satisfy the charge balance, part of the Al octahedral sites in spinel become unoccupied in γ-Al2O3 [18, 20]. Along its [1 1 1] axis, there are two types of Al layers (figure 1(a)). At the Al2 layer which is below the O1 layer, the Al ions occupy two thirds of the octahedral sites [10]. The Al1 layer below the O2 layer (figure 1(a)) is composed of three sublayers: a sublayer of octahedrally coordinated Al being sandwiched by two tetragonally coordinated Al sublayers (figure 1(a)). Such rich Al arrangements in the alumina phases shall have impacts on the prenucleation at the interfaces between liquid aluminum and alumina.
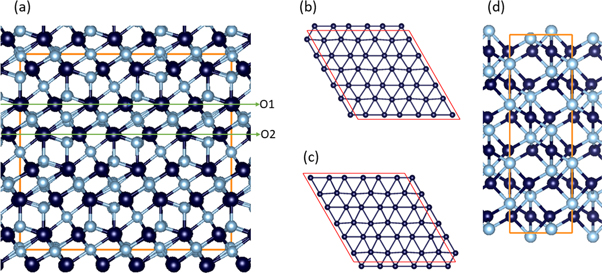
Figure 1. (a) Schematic structure of γ-Al2O3 in a hexagonal 3ah×3ah×1ch supercell (ah = (√2/2)ao, ch = √3ao with ao being the lattice parameter of the conventional cubic unit cell [18]) projected along its [1 0 0] axis. O ions occupy two distinctive types of lattice sites marked as O1 an O2; (b) atomic arrangement of the O1 layer; (c) atomic arrangement of the O2 layer; and (d) schematic structure of α-Al2O3 along the [1 0 0 0] projection. The silvery spheres represent Al, dark blue O. The red lines represent the axis of the unit cell with the horizontal line representing the b-axis and vertical c-axis in (a) and (d).
Download figure:
Standard image High-resolution imageMany experimental investigations have been conducted to study the stability, structural and physical properties of the alumina phases [16–20]. To get some insight into heterogeneous nucleation at the liquid-Al/alumina (L-Al/alumina, in short) interfaces, experiments were performed on wetting of liquid Al on single-crystalline α-Al2O3{0 0 0 1} surfaces (L-Al/α-Al2O3{0 0 0 1}) using various techniques [21–27]. Theoretical approaches, including parameter-free ab initio methods have also been applied to investigate the α-Al2O3{0 0 0 1} and γ-Al2O3{1 1 1} surfaces [28–32], as well as the interfaces between solid aluminum and α-Al2O3{0 0 0 1} [33]. Semiempirical molecular dynamics simulations were performed to investigate the atomic arrangements at liquid-metal/solid-metal interfaces [34, 35], effect of lattice misfit [12] and atomic level surface roughness on prenucleation [35], as well as the atomic ordering at the liquid-Al/α-Al2O3 interfaces [36]. Parameter-free ab initio molecular dynamics simulation techniques have been applied to study the atomic arrangements at the interfaces between liquid aluminum and TiB2{0 0 0 1} [27, 37, 38] and effect of substrate chemistry on prenucleation [13]. Recently, ab initio study focuses on prenucleation at the interfaces between liquid metal and oxide substrates, such as L-Mg/MgO [39, 40], L-Al/MgO [40], L-Al/α-Al2O3 [22, 24, 25, 41] and L-Al/MgAl2O4 [42]. Here we investigate the atomic ordering at the L-Al/γ-Al2O3{1 1 1} interfaces using an ab initio molecular dynamics simulation technique. This study reveals formation of an ordered Al layer terminating the substrates. This newly formed Al layer exhibits unusual atomic arrangements, determining the prenucleation at the interfaces.
2. Methods
2.1. Supercells for simulating the L-Al/γ-Al2O3{1 1 1} interfaces
The structural model of γ-Al2O3 was built based on the cubic spinel with a chemical (ionic) formula Mg2+(Al3+)2(O2-)4 via replacement of Mg by Al. To maintain charge balance, Al vacancies must be introduced. The recent studies showed that Al vacancies distribute at the octahedral sites preferably in a homogeneous way [18, 20]. The built L-Al/γ-Al2O3{1 1 1} interfaces have a hexagonal supercell with a = (3√2/2)a0, where a0 is the lattice parameter of the cubic γ-Al2O3 phase with consideration of the thermal expansion at the simulation temperature [16, 18]; while the length of the c-axis is determined by the γ-Al2O3 slab and the number of Al atoms with the density at the simulation temperature [43]. Thus, a hexagonal supercell with a = 17.06 Å, c = 40.58 Å which contains 144 O and 72 Al atoms in the substrates and 450 liquid Al atoms was used. The selected substrates are O-terminated (figure 1), one with O1-type (denoted as L-Al/γ-Al2O3{1 1 1}O1) and the other with O2-type (denoted as L-Al/γ-Al2O3{1 1 1}O2). We chose the two O-terminating substrates as the previous simulations [39–42] indicated that at thermal equilibrium, different inputs converge into one of the two independent L-Al/γ-Al2O3{1 1 1} interfaces. The supercells are deliberately large for avoiding risk of artificial crystallization of the Al liquid.
2.2. Simulation techniques and settings
For the ab initio molecular dynamics simulations and electronic structure calculations, we employed a pseudo-potential plane-wave approach based on the density-functional theory (DFT), which was implanted into the code VASP (Vienna ab initio simulation package) [44, 45]. This code permits variable fractional occupation numbers, working well for insulating/metallic interfaces [22, 24, 39–42]. The molecular dynamics simulation uses the finite-temperature density functional theory of one-electron states, the exact energy minimization and calculation of the exact Hellmann-Feynman forces after each MD step using the preconditioned conjugate techniques, and the Nosé dynamics for generating a canonical NVT ensemble [44]. The Gaussian smearing was employed with the width of smearing, SIGMA = 0.1eV. The code also utilizes the projector augmented-wave (PAW) method [46] within the generalized gradient approximation [47]. The atomic electronic configurations used are Al ([Ne] 3s2 3p1) and O ([He] 2s2 2p4).
For electronic structure calculations, we used cut-off energies of 400.0eV for the wave functions and 550.0 eV for the augmentation functions. The default energies of the potentials are Enmax/Eaug = 240.3 eV/291.1 eV for Al and 400.0eV/605.4eV for O. Reasonably dense k-meshes were used for sampling the electronic wave functions, e.g. a 2 × 2 × 1 (8 k-points) in the Brillouin zone (BZ) of the supercell of the interfaces [48]. For the ab initio MD simulations of the interfaces, we employed a cut-off energy of 320 eV, and the Γ-point in the BZ with considering the lack of periodicity of the whole system in such liquid/solid interfaces [37–42]. Test simulations using different cut-off energies ranging from 200.0 eV to 400.0 eV demonstrated that the settings are reasonable.
We prepared liquid Al samples by equilibrating at 3000K for 2000 steps (1.5 fs per step). Then the obtained liquid was cooled to the desired temperature. We used the obtained liquid Al samples together with the oxide substrates for building the L-Al/γ-Al2O3{1 1 1} interfaces. A two-step approach was applied in our simulations. We first performed ab initio molecular dynamics simulations with the substrate O atoms pinned for about 2ps (1.5fs per step). Then, we equilibrated further the systems with full relaxation of the substrate atoms for another 4,000 to 7,000 steps (figure 2). The two-step approach avoids risk of collective atomic movements occurring often when we relax all atoms from start. The time-averaged method was used to sample the interfaces over 3.0–4.5 ps to ensure statistically meaningful results [39–42, 49].
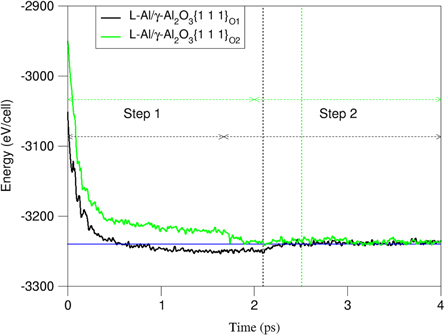
Figure 2. The total-valence electrons energies of of the L-Al/γ-Al2O3{1 1 1} system as a function of simulation time at 1000K. The black and green dotted lines represent the beginning of full relaxation (step 2) for L-Al/γ-Al2O3{1 1 1}O1 and L-Al/γ- Al2O3{1 1 1}O2, respectively. The horizontal blue line represents the averaged total-valence-electron energy of the systems at equilibrium.
Download figure:
Standard image High-resolution image2.3. Parameters describing atomic ordering at the L-Al/oxide interfaces
In order to assess the atomic ordering at the equilibrated L-Al/γ-Al2O3{1 1 1} interfaces three coefficients are employed as follows.
2.3.1. Atomic density profile
In order to provide a quantitative measure of atomic layering at a liquid/solid interface, we use the atomic density profile, ρ(z) which is defined as [12, 13, 34, 50, 39–42]:
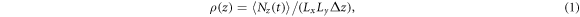
here, Lx and Ly are the in-plane x and y dimensions of the unit cell, respectively, and z the dimension perpendicular to the interface. Δz is the bin width, and Nz (t) is the number of atoms between z − (Δz/2) and z + (Δz/2) at time t. 〈Nz (t)〉 means a time-averaged number of atoms in the duration. The unit of the atomic density profile is (Å−3).
2.3.2. In-plane (atomic) ordering coefficient
In-plane ordering coefficient, S(z) can assess the atomic ordering in an individual layer [34, 50]. It is defined as:
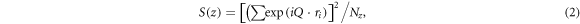
here, the summation is over all atoms within a layer with a bin of width, Δz = z − (Δz/2) and z + (Δz/2). Q is the reciprocal lattice vector in the x-y plane, r i is the Cartesian coordinates of the ith atom, and Nz is the number of atoms in the layer, respectively.
2.3.3. Atomic roughness of an individual layer
Atomic roughness (R) of an individual layer [39, 42] is quantified as:
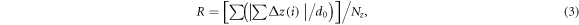
where ∆z(i) is the deviation of the ith atom from the atomic plane along the z-axis, d0(>0) is the interlayer spacing of the metal, and Nz is the total number of atoms in the layer. When an atom is located in the lattice of the plane, ∆z(i)/d0 = 0, when an atomic site is unoccupied, ∣∆z(i)∣/d0 = 1.0.
Ab initio molecular dynamics simulations are performed at elevated temperature at which atoms move around their equilibrium positions. Therefore, we use the atomic density profiles for estimation of the atomic roughness. The base-plane is set to be the peak/center at the atomic density profile.
3. Results
3.1. Atomic evolution at the L-Al/γ-Al2O3{1 1 1} interfaces
During the ab initio molecular dynamics simulations, some liquid Al atoms moved quickly towards the O ions terminating the substrates. Correspondingly, the total energies of the systems decrease sharply at the first 0.4ps, and then level off slowly with time (figure 2). Full relaxation of all atoms in the system causes some changes in atomic rearrangement as indicated by the energy changes (figure 2). After about 1ps, the systems reached thermal equilibrium and the total energy curves of the two systems overlap with each other, being consistent with the fact that these two systems have the same atomic species and numbers in the unit cells of the same dimensions. This also indicates the similar stability of the two γ-Al2O3{1 1 1} surfaces in liquid Al. Furthermore, the simulations showed that the two-step approach provided quick and steady convergence and avoided collective movements of the atoms. The latter happened using one step approach [38–41]. The simulations also showed that at thermal equilibrium, the liquid Al atoms at the terminating layer exhibit ordering and are more solid-like (details in next section). Meanwhile, the Al atoms adjacent to the substrates were moving around and even moved to neighboring layers. But, the numbers of Al atoms at each layer keep statistically constant.
3.2. Atomic layering at the L-Al/γ-Al2O3{1 1 1} interfaces
Figure 3 shows snapshots of the thermally equilibrated L-Al/γ-Al2O3{1 1 1} interfaces at 1000 K. The snapshots provide us with information about the atomic ordering at the interfaces. Clearly, the O and the Al ions in the substrates are positioned orderly, similar to those in the solid (figure 1), and the Al atoms in the liquid away from the interfaces remain disordered. A closer examination at the snapshots (figure 3) shows that: (i) there is an Al layer terminating the substrates; (ii) the liquid Al atoms close to the substrates display density variation perpendicular to the substrates (layering); (iii) the newly formed Al layer is smooth at L-Al/γ-Al2O3{1 1 1}O2, while the Al atoms terminating the substrate form an uneven layer at the L-Al/γ-Al2O3{1 1 1}O1 interface; and (iv) the liquid Al atoms are well separated from the substrate at the L-Al/γ-Al2O3{1 1 1}O2 interface, whereas there is no clear border between the terminating Al and the liquid Al atoms at L-Al/γ-Al2O3{1 1 1}O1.
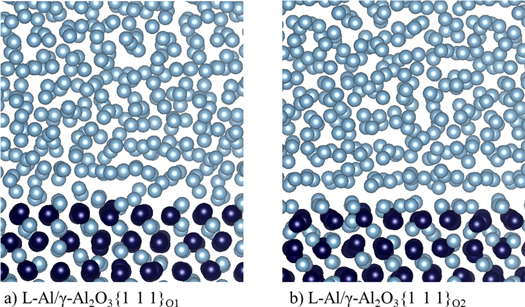
Figure 3. Snapshots of the equilibrated L-Al/γ-Al2O3{1 1 1} interfaces. (a) L-Al/γ-Al2O3{1 1 1}O1; and (b) L-Al/γ-Al2O3{1 1 1}O2. Dark blue spheres represent O, the silver spheres Al.
Download figure:
Standard image High-resolution imageThe atomic density profiles for the L-Al/γ-Al2O3{1 1 1} interfaces were obtained for the configurations summed over 3.0ps via equation (1). The results are plotted in figure 4.

Figure 4. Atomic density profiles for (a) the L-Al/γ-Al2O3{1 1 1}O1 and (b) L-Al/γ-Al2O3{1 1 1}O2 interface. The dashed line represents the center of the new terminating Al layer, and the dotted lines present the positions of the liquid Al peaks.
Download figure:
Standard image High-resolution imageThe atomic density profiles (figure 4) confirmed the layering at the L-Al/γ-Al2O3{1 1 1} interfaces (figure 3). The terminating Al atoms are close to the outmost O peak of the substrates. The two interfaces exhibit different atomic arrangements at the terminating Al layer. At L-Al/γ-Al2O3{1 1 1}O2, the terminating Al atoms form a peak which is well separated from the 1st Al layer, whereas at the L-Al/γ-Al2O3{1 1 1}O1 interface the terminating Al atoms form an uneven layer consisting of two or three sublayers. There is no clear border between the 1st Al and the substrate (figure 4(a)), which is similar to that at the L-Al/α-Al2O3{0 0 0 1} interface, where the terminating Al layer is composed of two Al subpeaks and admixed with the 1st Al layer [36, 40].
Figure 4 shows structural coupling between the terminating Al atoms and those at the subsurface Al layer in the substrate below the outmost O layer. At L-Al/γ-Al2O3{1 1 1}O1 the multiply peaked terminating Al layer is coupled to the single peak at the subsurface Al layer (figure 4(a)), whereas at L-Al/γ-Al2O3{1 1 1}O2 the single-peaked Al terminating layer is accompanied by the multiply peaked subsurface Al layer in the substrate. From another angle, the atomic arrangements of the terminating Al atoms are similar to those at its 3rd Al layer in the substrate at each L-Al/ γ-Al2O3{1 1 1} interface, respectively. (figure 4(b)).
The number of recognizable Al layers, n = 3 for L-Al/γ-Al2O3{1 1 1}O1 and n = 4 for L-Al/γ-Al2O3{1 1 1}O2 with a flatted peak at about 9.0Å (figure 4(b)), respectively. Therefore, the layering phenomenon at L-Al/γ-Al2O3{1 1 1}O2 is more pronounced than that at L-Al/γ-Al2O3{1 1 1}O1.
3.3. In-plane ordering at the L-Al/γ-Al2O3{1 1 1} interfaces
The epitaxial nucleation model suggested that the substrate surface provides a structural template for nucleation of the solid phase [14]. The prenucleation at the interface relates to the capability of the substrate to nucleate the solid phase in liquid. Figure 5 presents snapshots for the terminating Al, the 1st and 2nd Al layers at the L-Al/γ-Al2O3{1 1 1} interfaces.

Figure 5. Atomic arrangements in the new terminating Al layer, the 1st and 2nd Al layers at (a) the L-Al/γ-Al2O3{1 1 1}O1 and (b) the L-Al/γ-Al2O3{1 1 1}O2 interface. The orange lines represent the in-plane axis.
Download figure:
Standard image High-resolution imageThe terminating Al layers at both interfaces contain vacancies, and displacements (figure 5). There are moderate atomic ordering at the 1st Al layer but little at the 2nd Al layer at L-Al/γ-Al2O3{1 1 1}O2. At L-Al/γ-Al2O3{1 1 1}O1 even the 1st Al layer seems hardly any atomic ordering.
The in-plane ordering coefficients for the atomic layers near the interfaces were obtained using the configurations summed over 3ps via equation (2). The results are plotted in figure 6.

Figure 6. In-plane atomic ordering coefficients at the L-Al/γ-Al2O3{1 1 1} interfaces as a function of position of the atomic layers.
Download figure:
Standard image High-resolution imageThe in-plane atomic ordering coefficients of the terminating Al layer and the 1st Al layer at L-Al/γ-Al2O3{1 1 1}O2 are higher than the corresponding values at the L-Al/γ-Al2O3{1 1 1}O1 interface as shown in figure 6. This agrees with the conclusions from the snapshots (figure 3), the atomic density profiles (figure 4) and the snapshots of the layer-resolved atomic arrangements (figure 5). The prenucleation at L-Al/γ-Al2O3{1 1 1}O2 is more pronounced than that at L-Al/γ-Al2O3{1 1 1}O1. The S(z) values of the terminating Al layer at L-Al/γ-Al2O3{1 1 1}O2 (0.32) and that at L-Al/γ-Al2O3{1 1 1}O2 (0.17) are smaller than that at the L-Al/α-Al2O3{1 1 1} interface (0.37) [40, 41].
We analyzed the occupation rates of the octahedral sites at the terminating layer using the statistics over the summed configurations. The occupation rate at the octahedral sites of the terminating layer by Al is 54.0% L-Al/γ-Al2O3{1 1 1}O1 and 58.1% at the L-Al/γ-Al2O3{1 1 1}O2 interface, respectively. These values are lower than that in the bulk of alumina (66.7%), but they are comparable with that for L-Al/α-Al2O3{1 1 1} (55.9%) [36, 40, 41].
3.4. Atomic roughness of the terminating Al layer
For the L-Al/γ-Al2O3{1 1 1} interfaces, one issue is the occupation of metallic sites at the termination metal layer [42]. Charge balance in bulk Al3+ 2O2− 3 requires a Nmetal/NO ratio to be 66.7% (two thirds). The triple nature of the metal atoms and the related ordering at the terminating metal layer provides a constant free electron density at the substrate surfaces to interact with the nearby liquid Al atoms. Nz is the same as that in a substrate metal layer.
Using equation (3) with Nz /N0 = 2/3 (N0 is the number of sites produced by the outmost O layer), we can estimate R values for the terminating Al layers at the L-Al/alumina interfaces. The terminating Al layer at L-Al/γ-Al2O3{1 1 1}O2 is flat with an occupation of 58.1% and therefore, R = 12.9%. The terminating Al layer at L-Al/γ-Al2O3{1 1 1}O1 has an Al occupation of 54.0% which contributes 19.0% to R. Moreover, the Al atoms also display splitting along the z-axis and analysis produces another 14.5%. Overall, the R = 33.5% for the terminating Al layer at L-Al/γ-Al2O3{1 1 1}O1. Similar analysis produced R = 33.6% for the terminating Al layer at L-Al/α-Al2O3{0 0 0 1} [40, 41].
3.5. Chemical interaction at the L-Al/γ-Al2O3{1 1 1} interfaces
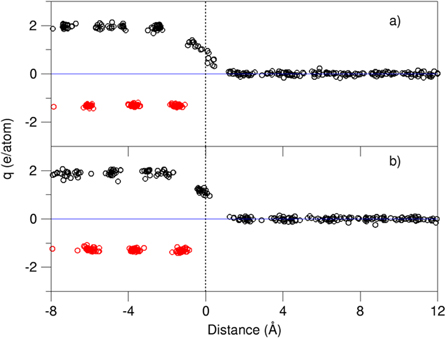
The chemical interaction between the substrate and liquid affects the atomic ordering of the liquid adjacent to the substrate [10, 13]. Here, we employed Bader charge model [51] to analyze the atomic charges at the interfaces. The results for the systems are plotted in figure 7.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 7. Charges at the atomic sites at the L-Al/γ-Al2O3{1 1 1}O1 (a) and L-Al/γ-Al2O3{1 1 1}O2 (b) interface from Bader charge analysis on the calculated electron density distributions. The red spheres represent the charges at the O ions, the black at the Al atoms/ions. The dotted line represents the central position of the terminating Al layer.
Download figure:
Standard image High-resolution image{kind=link}
The oxygen ions in both substrates have an average charge value of −1.3e/O, whereas the Al in the substrates are positively charged with an averaged value of +2.0e/Al. Therefore, the substrates can be described with the formula, (Al2.0+)2(O1.3−)3. These charge values are smaller than the ionic model with Al3+ and O2− at the atomic sites, indicating some covalent nature of alumina. Meanwhile, the Al atoms away from the interface are electronically neutral.
Figure 7 shows that the terminating Al atoms are charged partially. There is charge transfer from the terminating Al atoms to the outmost O atoms, indicating strong chemical bonding between the terminating Al atoms and the substrates. Therefore, the terminating Al atoms are better regarded as part of the substrate. The charge at a terminating Al-layer decreases strongly with the distance from the outmost O atoms, agreeing well with the chemical bonding theory [52].
4. Discussion
The present ab initio molecular dynamics simulations revealed the formation of an ordered Al layer terminating the γ-Al2O3{1 1 1} substrates in liquid Al. Charge transfer occurs from the terminating Al to the outmost O. Consequently, the terminating Al atoms are positively charged and chemically bonded to the outmost O. The terminating Al atoms/ions exhibit ordering and are more solid-like, and therefore, belong to the substrates. The simulations also discovered that the terminating Al are structurally coupled with the Al ions at the subsurface. The origin of the structural coupling comes from the Coulomb repulsion between the Al ions at the terminating layer and the subsurface cross the outmost O layer, similar to that at the L-Al/MgAl2O4{1 1 1} interfaces [42]. Such structural coupling is a general phenomenon, found at the other interfaces between liquid-metal and oxide substrates [39–42]. This unusual structural coupling between the newly formed terminating metal layer to the oxide substrate helps us to obtain some insight into the growth of the oxide particles. Here we try to make a scenario of γ-Al2O3{1 1 1} growth in liquid Al. In liquid Al, the solute O ions/atoms aggregate at the space between the terminating Al layer and the 1st Al layer due to the Coulomb interaction, forming a new O outmost later at L-Al/MgAl2O4{1 1 1}. During the formation of the new O outmost layer, the composition and atomic arrangements at the original terminating layer are adjusted into those in the corresponding bulk. Consequently, a new Al terminating layer with structural coupling to the substrate will be formed to terminating this new O outmost layer. This process continues until the O ions/atoms in the liquid are consumed up. A detailed discussion is beyond the scope of the present manuscript.
Prenucleation is related to the intrinsic capability of the substrate surface to induce atomic ordering in the liquid adjacent to the interface. Therefore, it corresponds to the potency of the substrate for nucleation of the solid [10, 11, 53]. Recent studies revealed three factors affecting prenucleation at a liquid-metal/solid-substrate interface [10]. Firstly, at a flat substrate, the lattice misfit between a metal and a substrate (f) hinders strongly the in-plane ordering, but affects little on the atomic layering (structural factor, f) [12]. Secondly, the chemical interaction between liquid metal and a flat substrate influences the atomic ordering at the interface. A chemically affinitive substrate promotes atomic ordering at the interface, whereas a repulsive substrate weakens prenucleation (chemical factor) [13]. Furthermore, the atomic roughness of a substrate surface deteriorates both layering and in-plane ordering at the interface (effect of atomic roughness, R) [35, 42].
The present study revealed charging of the terminating Al at the L-Al/γ-Al2O3{1 1 1} interfaces (q), which can be considered as a measure of the interfacial chemical interaction. We summarize the factors, lattice misfits (f), the atomic roughness of the terminating metal layer (R), and the charges at the interfacial atomic sites (q) in table 1. These factors affecting prenucleation at the L-Al/γ-Al2O3{1 1 1} interface are listed in table 1 in comparison with those at L-Al/α-Al2O3{0 0 0 1} from the literature [40, 41]. Table 1 shows that all the terminating Al atoms at the L-Al/alumina interfaces are charged. Moreover, the lattice misfits were moderate for Al/α-Al2O3{0 0 0 1} (4.8%) [5] and Al/γ-Al2O3{1 1 1} (5.6%). Table 1 shows overall weak prenucleation at the at the three L-Al/alumina interfaces as compared with the liquid-metal/solid-metal interfaces [12, 13]. The observed layering at Al/α-Al2O3{0 0 0 1} is in line with the experimental observations [21–27].The atomic roughness of the terminating layer is different at the three interfaces: R = 12.9% for L-Al/γ-Al2O3{1 1 1}O2, which is notably smaller than those of L-Al/γ-Al2O3{1 1 1}O1 and L-Al/α-Al2O3{0 0 0 1}. Consistently, the prenucleation at the L-Al/γ-Al2O3{1 1 1}O2 interface is more pronounced than that at the other two interfaces. This indicates the atomic roughness is the dominating factor at these interfaces. However, the moderate/weak prenucleation at the L-Al/alumina interfaces is not determined by a sole factor, such as atomic roughness, but a combination of the lattice misfit, chemical interaction (charging) and atomic roughness.
Table 1. Characteristics of the equilibrated L-Al/γ-Al2O3{1 1 1} interfaces with comparison with those at L-Al/α-Al2O3{1 1 1} [40, 41]. f = (dAl − dsub.)/dAl ×100 represents the lattice misfit between an oxide substrate and the Al, here dAl is the lattice spacing of Al, dsub the spacing of substrate.
| Interface | f (%) | Char. Al layer Nm /NO (%) | R(%) | q(e/Al) | No. Layers | S(z) 1st Al |
|---|---|---|---|---|---|---|
| L-Al/γ-Al2O3{1 1 1}O1 | +4.8 [5] | Split, Vacan.54.0% | 33.5 | 0.4–1.5 | 3 | 0.2% |
| L-Al/γ-Al2O3{1 1 1}O2 | +4.8 a [5] | Flat Vacan.58.1% | 12.9 | 0.8–1.2 | 3 ∼ 4 | 2.6% |
| L-Al/α-Al2O3{0 0 0 1} | +5.6 b | SplitVacan. 55.9% | 33.6 | 0.5–1.5 [41] | 3 ∼ 4 [41, 42] | 1.0% [40, 41] |
a Orientational relation is {111} [100]Al//{111}[100]γ-Al2O3. b Orientational relation is {111} [110]Al//{0001}[1000]α-Al2O3.
The poor prenucleation at the L-Al/Al2O3 interfaces indicates requirements of large drive forces (undercoolings) to nucleate the solid Al phase. The nucleation temperatures at these interfaces might be even lower than the corresponding grain initiation temperatures. In this case, as soon as the grain initiation temperature is reached, heterogeneous nucleation and grain initiation occur in a narrow time interval, leading to explosive grain initiation [10, 11]. This indicates that more alumina particles can act as potential sites for nucleation and growth of solid aluminum. This explosive nucleation may help to materialize fine particles of solidified metals if no other more potent particles of significance exist [10, 11].
5. Conclusions
We performed ab initio molecular dynamics simulations with a two-step approach and electronic structure calculations for the L-Al/γ-Al2O3{1 1 1} interfaces. The simulations showed that the two-step approach is helpful in avoiding risk of collective movements of atoms during ab initio MD simulations. This study revealed the formation of an ordered Al layer terminating the γ-Al2O3{1 1 1} substrates in liquid Al at thermal equilibrium. The terminating Al are electronically charged and chemically bonded to the outmost O ions, becoming part of the substrates. Moreover, the terminating Al atoms/ions at the L-Al/γ-Al2O3{1 1 1} interfaces are structurally ordered but are topologically rough. This leads to its weak templating capability and therefore, poor prenucleation at the interfaces. Moreover, the investigations also discovered structural coupling between the terminating Al ions with the those at the subsurface layer in the substrates. This is helpful to understand the growth mechanism of oxide particles in the liquid metals.
Acknowledgments
We thank Prof. Brian Cantor and Dr Y Wang (BCAST) for useful discussions. Financial support from EPSRC (UK) under grant number EP/N007638/1 is gratefully acknowledged.
Data availability statement
No new data were created or analysed in this study.