
Abstract
Structural, electronic, vibrational and thermodynamic properties have been tested when Hubbard parameter U is implemented in density functional theory calculations for TiO2 polymorphs: anatase, rutile, TiO2–B and for hydrogen titanate (H2Ti3O7) bulk. Optimum U parameter values were found for each system, balancing geometric changes and electronic properties, namely, U = 4 eV for anatase and TiO2–B, U = 5 eV for rutile and hydrogen titanate. Although the addition of this parameter improves the prediction of electronic properties, with no significant structural changes, we found that it would not be adequate for predicting vibrational properties.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Titanium oxide (TiO2), has been widely studied both experimentally and theoretically [1–5] due to its potential applications like high-efficiency solar cells, photocatalysts, and storage capacitors in dynamic random access memories [6–8]. TiO2 presents various polymorphs among which the most studied ones are anatase and rutile. They exhibit exceptional physical and chemical properties, specially for photochemical and photoelectrochemical applications. Sunlight can be used to produce electricity or to drive chemical reactions by photocatalysis and photovoltaics. In order to find efficient materials for solar energy conversion we need to study their properties i.e. structure, surfaces and interfaces, light absorption, charge transport, electron and hole trapping, among others.
The polymorph TiO2–B has been studied to a lesser extent and currently, only few theoretical and experimental studies of their properties exist [9–11]. Fewer reports are found regarding hydrogen titanate (H2Ti3O7) [12–14].
In systems with d- and f- localized electrons the overestimation of electron delocalization is a known weakness in DFT methodology. In the 90 s the DFT + U method was developed, which consists in an explicit treatment of electronic correlation with a Hubbard-like model for a subset of states in the system [15]. The essential idea is to treat the strong on-site Coulomb interaction of localized electrons using an additional Hubbard-like term. The strength of the on-site interaction is described by two parameters U and J, the on-site Coulomb and the on-site exchange interaction, respectively. The DFT + U method has been widely applied in several systems [16–20]. This implementation improves the results in the calculation of energetic, electronic and magnetic properties of insulating and semi-conducting materials which contain transition metals. Nevertheless, in early transition metal compounds (like Ti) the more extended orbitals decrease the electron correlation, so it is needed to study the performances of the DFT + U method. In the last decade, several studies have considered the U-term in predicting the properties of anatase and rutile [4, 21–23].
Here, we report how the structural, electronic, vibrational and thermodynamic properties behave when the U-term is taken into account during calculation. As it is known, theoretical modeling plays a very important role in materials research, since it provides information at an atomic level which is inaccessible via experiments, helping to identify which factors control the behavior of a specific material.
Methodology
Electronic structure and energy calculations have been performed within the frame of the density functional theory (DFT) [24] implemented in the Vienna ab initio simulation package (VASP) [25]. The projector augmented wave pseudopotential [26] was used to account for the electron−ion core interaction, using the Perdew–Burke–Ernzerhof functional as generalized gradient approximation (GGA) [27] for the exchange−correlation term. A cutoff energy of 400 eV was used to expand the Kohn–Sham orbitals into plane wave basis sets. The employed pseudopotentials correspond to the following configuration: 3s2 3p6 3d2 4s2 for Ti, 2s2 2p4 for O and 1s1 for H. This choice amounts to a valence of 12 for Ti, 6 for O and 1 for H. In every case the K-point mesh taken was equivalent to 4 × 4 × 4 Monkhorst–Pack grid [28] for the full (reducible) Brillouin zone, allowing total convergence. We have implemented the Hubbard U model to improve the calculated band gap width [15, 29–31]. In this implementation, the on-site coulombic (U) and exchange (J) terms are combined into a single effective U parameter (Ueff) to account for errors in exchange correlation on Ti 3d orbitals.
We have modeled the following TiO2 polymorphs: anatase (unit cell containing 12 atoms), rutile (6 atoms) and TiO2–B (24 atoms), as well as hydrogen titanate (21 atoms), as it can be seen in figure 1 (for better visualization the unit cell was multiplied in some directions).

Figure 1. Crystal structure of the studied TiO2 polymorphs: (a) anatase, (b) rutile, (c) TiO2–B and (d) hydrogen titanate (H2Ti3O7). Blue, red and white spheres represent titanium, oxygen and hydrogen atoms, respectively.
Download figure:
Standard image High-resolution imageAs a first step the bulk structures (i.e. the cell parameters and atomic positions) were fully relaxed in the absence of the U parameter (U = 0 eV). Then, from these relaxed structures, the bulks were allowed to relax again, but within the GGA + U approximation, setting the U term which takes the following values: U = 3, 4 and 5 eV.
In order to proceed with the vibrational and thermodynamics analysis, we performed first a phonon analysis by means of density functional perturbation theory as implemented in VASP. For these calculations, we increased the accuracy of the grids calculations by selecting a better grid for the FFT-grid and the fine FFT-grid, corresponding to 2 × Gcut and 4 × Gcut, correspondingly (PREC = accurate and ADDGRID). Additionally, we doubled the k-point sampling in each direction, assuring an 8 × 8 × 8 grid sampling. It guarantees a better accuracy for phonons and second derivatives calculations. With this purpose, we have utilized the pristine unit cell (see table 1) for all the considered polymorphs. Due to possible long range dipole–dipole interactions, the non-analytical term correction was considered in the calculation. In every case the absence of soft modes were checked by the analysis of the phonon dispersion diagrams. The phonon analysis and the further thermodynamical properties were determined by using the post-processing code Phonopy [32].
Table 1. Lattice parameters of TiO2 polymorphs and hydrogen titanate calculated in a set of U values (eV).
| a(Å) | c(Å) | c/a | |||
|---|---|---|---|---|---|
| Anatase | GGA | U = 0 | 3.784 | 9.531 | 2.519 |
| GGA + U | U = 3 | 3.807 | 9.565 | 2.512 | |
| I41/amd | U = 4 | 3.823 | 9.554 | 2.499 | |
| U = 5 | 3.826 | 9.602 | 2.510 | ||
| a | c | c/a | |||
| Rutile | GGA | U = 0 | 4.592 | 2.954 | 0.643 |
| GGA + U | U = 3 | 4.598 | 2.994 | 0.651 | |
| P42/mnm | U = 4 | 4.619 | 2.991 | 0.647 | |
| U = 5 | 4.612 | 3.012 | 0.653 | ||
| a | c | c/a | |||
| TiO2–B | GGA | U = 0 | 12.157 | 6.555 | 0.539 |
| GGA + U | U = 3 | 12.240 | 6.566 | 0.536 | |
| C12/m1 | U = 4 | 12.269 | 6.572 | 0.536 | |
| U = 5 | 12.311 | 6.576 | 0.534 | ||
| a | c | c/a | |||
| H2Ti3O7 | GGA | U = 0 | 7.871 | 9.399 | 1.194 |
| GGA + U | U = 3 | 7.879 | 9.442 | 1.198 | |
| C2/m | U = 4 | 7.899 | 9.467 | 1.198 | |
| U = 5 | 7.887 | 9.469 | 1.201 | ||
Results and discussion
Geometric optimization
As it was said above, anatase, rutile, TiO2–B and hydrogen titanate were relaxed in order to find their optimum geometries. In table 1 the lattice parameters a, c and c/a ratio at different U values are listed.
The incorporation of U term affects the lattice parameters, expanding the cell while the U value increases. That is the reason why it is needed to find the right U value, which balances the accuracy of the predicted geometrical structure and electronic properties for each system. In table 2 it can be seen that Ti–O bond lengths in every system increase when U takes higher values. However, the changes are not significant or drastic.
Table 2. Shorter Ti–O bond length (in Å) at different U term values (eV), for each studied bulk system.
| U = 0 | U = 3 | U = 4 | U = 5 | |
|---|---|---|---|---|
| Anatase | 1.932 | 1.944 | 1.951 | 1.953 |
| Rutile | 1.949 | 1.966 | 1.970 | 1.975 |
| TiO2–B | 1.852 | 1.879 | 1.888 | 1.895 |
| H2Ti3O7 | 1.745 | 1.764 | 1.772 | 1.778 |
Electronic properties
In table 3 a comparison of the band gap widths at different U term values and experimental results obtained from literature are summarized [33–35]. As the U term value increases, the band gap increases as well.
Table 3. Comparison of band gap values (eV) calculated at GGA and GGA + U level with experimental values for the four cases under study.
| GGA + U | |||||
|---|---|---|---|---|---|
| GGA | U = 3 eV | U = 4 eV | U = 5 eV | Exp | |
| Anatase | 2.12 | 2.58 | 2.61 | 2.76 | 3.20 [33] |
| Rutile | 1.84 | 2.16 | 2.28 | 2.41 | 3.00 [33] |
| TiO2–B | 2.82 | 3.12 | 3.22 | 3.35 | 3.22 [34] |
| H2Ti3O7 | 3.10 | 3.20 | 3.25 | 3.30 | 3.27 [35] |
The density of states is plotted in figure 2, each system at promising optimum U term value, namely, U = 4 eV for anatase and TiO2–B, U = 5 eV for rutile and hydrogen titanate. Band gap energies are in good agreement with those found in previously cited literature for TiO2–B and hydrogen titanate. However for the cases of anatase and rutile a selection of higher U values leads to a major structural deviation, that is why we have selected values of U = 4 eV for anatase and U = 5 eV for rutile (see table 4). Despite the band gap energies are far from experimental reported data an improvement can be seen when U term in taken into account in the calculation.

Figure 2. Density of states for the four systems at promising U values.
Download figure:
Standard image High-resolution imageTable 4. Percentage deviation of band gap width and c/a lattice parameter for the four studied systems.
| Percentage deviation | ||||
|---|---|---|---|---|
| Anatase U = 4 eV | Rutile U = 5 eV | TiO2–B U = 4 eV | H2Ti3O7 U = 5 eV | |
| Egap | 18.44 [33] | 19.67 [33] | 0.00 [34] | 0.92 [35] |
| c/a | 0.56 [33] | 1.40 [33] | 0.56 (U = 0 eV) | 0.59 (U = 0 eV) |
In these curves a significant contribution of O 2p states to the valence band, and an important contribution of Ti 3d states in the conduction band, can be seen. This is a well-known feature for bulk TiO2 polymorphs.
In order to determine the appropriate values of U for each system, we have compared the calculated band gap widths for a set of U values (3, 4 and 5 eV) with experimental band gap values. However, it is not only necessary to fit the band gap width but also it is needed to maintain the experimental lattice parameters. This information can be seen in table 4, for TiO2–B polymorph and hydrogen titanate there is no structural experimental information available and for this reason the values calculated for different U parameters were compared with the results calculated using U = 0 eV.
Vibrational properties
In order to obtain the zone-center phonons of the TiO2 polymorphs and hydrogen titanate, the dynamical matrix should be diagonalized for q = 0. Through factor analysis [36], in the I41amd, P42/mnm, C12/m1 and C2/m space groups for anatase, rutile, TiO2–B and hydrogen titanate bulks respectively, the irreducible representations of the optical vibrations are as follows:
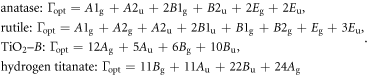
The modes with subscript g (gerade) are Raman-active and those with subscript u (ungerade) are infrared-active, while the modes represented with the letter E are degenerate.
In all the cases, the phonons density of states have been obtained, and Ti, O and H atoms are projected in black, red and blue curves, respectively, as it can be seen in figure 3. In TiO2 polymorphs the low values of frequency correspond mainly to titanium atoms, and high values to oxygen atoms, anatase and rutile plots agree with those from literature [37]. In the case of hydrogen titanate we can see a wide gap among curves, there are two picks at high energy values which correspond to the light hydrogen atoms.

Figure 3. Projected phonon density of states versus frequency for four studied bulk systems.
Download figure:
Standard image High-resolution imageIn figure 4 the histograms of each bulk system are presented, the bars represent the group of vibration modes in a specific range of frequency. The vertical black lines are the vibration modes calculated at DFT U = 0 eV level for comparison. From these charts, it can be noticed that the implementation of the U term has an effect on the vibrational properties, developing different vibration modes at different frequencies. How this influences the thermodynamic properties will be analyzed in the following section.

Figure 4. Histograms of frequency modes for the studied systems, vertical black lines represent the frequencies of the systems without considering the U parameter.
Download figure:
Standard image High-resolution imageFor bulk rutile, there are lots of data reported: in table 5 a comparison of this information and our results is shown [38–42]. Here it can be seen clearly how the Hubbard implementation changes the vibrational properties, frequency values of vibrational modes differ and shift significantly when different values of U term are taken into account in the calculation. Reported theoretical results without U term approach better to experimental information.
Table 5. Comparison of frequency values (cm−1) for rutile polymorph with literature.
| Modes | This work (U = 5 eV) | Theory | Theory | Exp | Exp |
|---|---|---|---|---|---|
| B1u | 121.0 | 116.7 | 117.7 | 113 | |
| B1g | 175.1 | 125.2 | 132 | 142 | 143 |
| A2u | 249.3 | 176.1 | 191.3 | 173 | 167 |
| Eu | 249.5 | 164.8 | 143.7 | 189 | 183 |
| Eu | 318.1 | 351.5 | 352.3 | 375 | 373 |
| Eu | 405.1 | 391.3 | 393 | 388 | |
| B1u | 405.4 | 407.5 | 417.3 | 406 | |
| A2g | 420.0 | 415.5 | 412.7 | ||
| Eu | 460.4 | 441.7 | 434.7 | 429 | 458 |
| Eg | 465.3 | 471.5 | 472 | 445 | 447 |
| Eu | 466.6 | 492.8 | 498.3 | 494 | 500 |
| A1g | 525.0 | 622.5 | 615.3 | 610 | 612 |
| A2u | 525.1 | 769.3 | 800.7 | 769 | |
| Eu | 613.9 | 808.4 | 787.7 | 812 | |
| B2g | 809.3 | 828 | 800.7 | 825 | 827 |
Thermodynamic properties
Once the phonon frequencies set is obtained, thermodynamic properties of the systems can be calculated by quasi harmonic approximation. According to the following expressions, entropy, vibration energy and Helmholtz free energy can be determined:
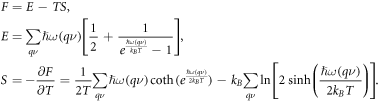
Also, specific heat at constant volume can be calculated as follows:
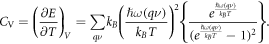
Properties of solids can be extrapolated at temperatures higher than 0 K by vibration harmonic approximations. Based on these quasi-harmonic approximations, we have obtained the phonon frequencies at five different volumes, namely, 3% and 1.5% reduced volume, 0%, and 3% and 1.5% expanded volume. In this manner, we have calculated a set of thermodynamic parameters for TiO2 polymorphs and hydrogen titanate bulks. In figures 5–8 we have plotted enthalpy and entropy versus temperature for the four systems as well as Cp (specific heat at constant pressure) and Cv (specific heat at constant volume) versus temperature, respectively. In black crosses experimental data for anatase and rutile from literature was added [43].

Figure 5. Enthalpy versus temperature at DFT and DFT + U level for anatase, rutile, TiO2–B and hydrogen titanate bulks.
Download figure:
Standard image High-resolution image
Figure 6. Entropy versus temperature at DFT and DFT + U level for anatase, rutile, TiO2–B and hydrogen titanate bulks.
Download figure:
Standard image High-resolution image
Figure 7. Cp versus temperature at DFT and DFT + U level for anatase, rutile, TiO2–B and hydrogen titanate bulks.
Download figure:
Standard image High-resolution image
Figure 8. Cv versus temperature at DFT and DFT + U level for anatase, rutile, TiO2–B and hydrogen titanate bulks.
Download figure:
Standard image High-resolution imageIt can be seen in these four figures that the three TiO2 polymorphs behave in a similar way, while hydrogen titanate takes higher values in every thermodynamic property studied here. In the enthalpy versus temperature plot (figure 5), we can see that up to 200 K the three TiO2 polymorphs take very similar values at standard DFT, however at DFT + U level the enthalpy values are overestimated. In the case of entropy (figure 6) the curves of each system can now be distinguished, nevertheless the curves fit better with experimental information when rutile takes a U = 5 eV value and anatase U = 4 eV than DFT standard calculation. In Cv (and Cp) versus temperature plot (figures 7 and 8), from 400 K up once again, the three TiO2 polymorphs behave similarly, approaching an asymptotic behavior at higher temperatures. Differences in these three TiO2 polymorphs are not significant.
Thermal volumetric expansion can be determined and are plotted as shown in figure 9. TiO2 polymorphs have the same behavior; hydrogen titanate does not experience any significant volumetric change when temperature is increased. Instead, a volume contraction is observed near 200 K which is correlated to the presence of several imaginary vibrational modes, that only appear for the cell volumes associated to that temperature, but that does not appear for the equilibrium values associated at 0 K. This fact could be connected to a possible phase transformation from hydrogen titanate to TiO2(B) in the same range of temperatures [44]. Anatase and rutile lead to bigger volumetric variations.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 9. Volume variation versus temperature for anatase, rutile, TiO2–B and hydrogen titanate bulks.
Download figure:
Standard image High-resolution image{kind=link}
Thermodynamic properties fluctuate significantly when the Hubbard term is considered in our analysis. The previous differences noticed for phonon frequencies have an effect on the results of thermodynamic properties calculation.
In table 6 bulk modulus and volume variation using DFT and DFT + U at 0 K and 298 K is listed to compare the effect of addition of Hubbard parameter in our calculations [13, 45, 46].
Table 6. Comparison of DFT and DFT + U calculation in bulk modulus and volume, at 0 and 298 K (U in eV)
| T (K) | B0 (Gpa) | B'0 | V (Å3/TiO2) | ||
|---|---|---|---|---|---|
| Anatase | 0 | DFT [43] | 185 | 33.58 | |
| DFT U = 4 | 189 | 4.36 | 36.37 | ||
| 298 | DFT [43] | 175 | 4.50 | 33.86 | |
| DFT U = 4 | 176 | 4.59 | 36.51 | ||
| Rutile | 0 | DFT [43] | 250 | 30.64 | |
| DFT U = 5 | 212 | 4.46 | 33.34 | ||
| 298 | DFT [43] | 241 | 6.50 | 30.96 | |
| DFT U = 5 | 195 | 5.48 | 33.51 | ||
| TiO2–B | 0 | DFT [44] | 182 | 37.39 | |
| DFT U = 4 | 184 | 4.27 | 37.86 | ||
| 298 | DFT U = 0 | 174 | 4.85 | 36.95 | |
| DFT U = 4 | 177 | 4.12 | 37.95 | ||
| H2Ti3O7 | 0 | DFT U = 0 | 164 | 3.97 | 47.00 |
| DFT U = 5 | 155 | 4.82 | 47.09 | ||
| 298 | DFT [13] | 46.01 | |||
| DFT U = 5 | 154 | 6.12 | 47.08 |
The addition of Hubbard parameter leads to an increase in cell volume being less significant for TiO2–B and hydrogen titanate. The bulk modulus varies when U term is considered, being larger for anatase and TiO2–B, and smaller for rutile and hydrogen titanate. In the cases of TiO2–B and hydrogen titanate few data in literature is found, in order to compare our results, we have added two rows called 'DFT U = 0' which correspond to our results at a DFT standard level.
Conclusions
DFT-based calculations have been performed in order to study a set of properties of TiO2 polymorphs and hydrogen titanate bulks and how these properties change when the Hubbard parameter U is implemented in the calculation. It was found that this addition improves the electronic properties prediction, shows small changes in structural properties, but leads to larger variations in vibration frequencies, making the use of U term not suitable for vibrational properties calculation. Thermodynamic properties are affected by implementation of the Hubbard U term in GGA calculations, showing larger deviations from experiment of the calculated enthalpy, entropy, Cp and Cv quantities.
Acknowledgments
EG is a member of CONICET. Authors acknowledge SGCyT (UNS), IFISUR-CONICET, CIC-Buenos Aires, PICT 1770. EG would like to thank Agencia Nacional de Investigación e Innovación (ANII) for providing the opportunity to undertake this research by supporting her Post-doc 'Fondo Profesor Dr Roberto Caldeyro Barcia' code PD_NAC_2015_1_108133. A W M and R F acknowledge ANII, PEDECIBA and CSIC.