

Abstract
Chemotherapy drugs are generally cytotoxic and can cause major side effects, including vomiting/nausea, fatigue, hair loss and pain. The use of targeted nanostructures to deliver drugs directly to tumours has the potential to reduce the side effects by decreasing the exposure of healthy cells and reducing the amount of drug needed. DNA can be used as a structural material to build drug-delivering nanorobots, but questions remain over the practicality of this approach. Here we show that it is potentially feasible for DNA nanostructure drug delivery to be more cost-effective than the drug-only approach. Our result suggests that the barriers to the development of DNA nanostructure-based drug delivery are likely to be primarily technical, regulatory and ethical rather than financial, as the potential exists for this to be a profitable therapeutic approach.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Background
In one study, 45% of chemotherapy patients questioned said that the side effects of chemotherapy affected their lives 'much' or 'very much' [1]. In another study, 86% of patients reported at least one side effect, and 27% reported a severe side effect [2]. In principle, using nanostructures (of size 1–100 nm) to deliver drugs to particular cells could allow tumours to be targeted specifically [3], reducing toxicity and side effects. Nanostructures tend to accumulate preferentially in tumours rather than 'normal' tissue, but the extent and mechanism are currently under debate [4]. Cancer cells can also be targeted actively [5], where nanostructures are modified with ligands or antibodies, enabling them to interact with particular biomarkers in the tumour. Some nanotherapeutics can self-deploy in response to changes characteristic of the tumour microenvironment, or be steered using external stimuli including magnetic fields, light, heat and ultrasound.
To date, use of nanomedicines has been limited, in part due to regulatory challenges. However, a few nanostructure-based chemotherapeutics have been approved for clinical use [6], the first being pegylated liposomal doxorubicin (PLD), which was approved in 1994 and is also known as Doxil, Caelyx or Lipodox [7]. Here, the well-established anti-cancer drug doxorubicin is encapsulated in a liposome, which is coated with PEG (Polyethylene glycol) to render it hydrophilic and reduce the chance that it will be removed rapidly by the body [8]. Pegylated liposomal doxorubicin is significantly less toxic than free doxorubicin, and survives much longer, giving time for the drug to accumulate in the tumour. However, liposomal doxorubicin has not supplanted free doxorubicin. The drug paclitaxel is also available in a nanotechnology-enabled formulation, as albumin-bound nanoparticles, and in Scotland this has been deemed to be a cost-effective monotherapy for metastatic breast cancer when anthracyclines are not to be used [9].
The principles of DNA nanotechnology [10] can be used to assemble drug delivery vehicles [11]. DNA nanostructures are made from synthetic DNA strands, the sequences of which are designed to ensure the nanostructure assembles into the desired configuration, through the mechanism of base-pairing. The earliest structure to be made was a DNA cube [12], followed by other cage-like polyhedra [13, 14]. Larger nanostructures are commonly assembled using the technique of DNA origami [15], which can produce gigadalton scale objects [16]. Origami involves folding a long single-stranded DNA 'scaffold' into a designed shape by hybridizing it with many short single-stranded DNA oligonucleotides ('staples'), which are designed to bind to specific domains [17]. An alternative technique is based on DNA configurations defined by a polyhedral mesh [18, 19].
DNA nanotherapeutics [20] show potential due to their biocompatibility and customizable morphology. Some chemotherapy agents can be loaded by intercalation between the base pairs, and the nanostructure is expected to accumulate in the tumour as a matter of course. The DNA nanostructure shape can affect the release characteristics of intercalating drugs [21], and not all potential intercalation sites are accessible for binding [22]. Doxorubicin-laden DNA nanostructures can be cytotoxic to resistant cancer cells that are not killed by free doxorubicin [23], and genes can be delivered with the drugs for combination therapy [24]. For active targeting, DNA origami nanorobots [25] can be loaded with therapeutic cargos such as thrombin [26] by specific modification of component strands, with the nanorobot being guided to the tumour by active targeting, using a recognition mechanism based on molecules such as aptamers.
When DNA nanostructures are used as drug delivery vehicles, cells are exposed to a large number of synthetic DNA sequences, and it is conceivable that this could have a significant effect, as the structure could potentially contain a complicated set of genetic instructions. Hence, it is usual to perform control experiments in which the cells are treated with a solution of nanostructures that contain zero chemotherapy agent or have been otherwise inactivated. Normally, such structures have minimal or no effect on the target cells [23–26]. However, p53 genes attached to the edge of a DNA origami nanostructure can have a significant effect on cell viability and this has been proposed as a gene therapy technique [24] Presumably, the information encoded in the constituent sequences of a compact nanostructure is relatively inaccessible, compared to the instructions found in an unencumbered DNA oligonucleotide tethered to the edge of such an object. Alternatively, it is possible that the quasi-random sequences in the nanostructures are meaningless, unlike the p53 gene. As the technology of DNA nanostructure-based drug delivery develops further, these two possibilities must be explored in detail, to ensure that potential off-target effects of the therapy can be identified. Interestingly, it has been shown that plain DNA nanostructures (without any drug at all) can alleviate symptoms of acute kidney injury in mice [27], but this was attributed to scavenging of reactive oxygen species rather than any genetic effect.
As evidenced above, many papers have been published on technical aspects of DNA nanostructure-based drug delivery. However, to the best of our knowledge no-one has yet published an economic assessment of the concept, despite the fact that increasing attention is being given to patenting DNA nanotechnology inventions and commercializing them [28]. The aim of this work is to ascertain whether DNA nanostructures are likely to be a cost-effective means of delivering chemotherapy drugs. At present, very limited information is available about the safety and efficacy of DNA-nanostructure-enabled treatments. Some experiments have been carried out in cell culture [21, 23, 25], and a few animal studies have been performed [24, 26]. However, to the best of our knowledge, no human studies have been conducted. No DNA nanostructure-based clinical trials are listed on the World Health Organization's International Clinical Trials Registry Platform (https://apps.who.int/trialsearch/), which includes data from American, EU and Chinese databases, among others. However, even at this early stage, it is important to evaluate the potential cost-effectiveness to establish potential for translation and value to industry. The problem must be framed such that a meaningful answer can be obtained despite the gaps in our information, which requires abstract assumptions.
In this study, we establish benchmarks for the performance of a DNA nanostructure-based therapy in comparison with the equivalent therapy without nanostructures, assuming that the use of DNA nanostructures enhances quality of life and/or provides more years of life without changing the amount of drug used. We also consider a scenario in which the nanostructures reduce the quantity of drug needed. We do not explicitly consider technical or regulatory viability, and we focus exclusively on costs. We note that our method could also be applied to the assessment of other early-stage technologies where information is limited. We use the concept of a quality-adjusted life year (QALY), which is used internationally to assess cost-effectiveness of new healthcare interventions, by appraising the performance of a new intervention in comparison with the status quo, ideally before the new measure is introduced [29].
The number of QALYs gained upon the introduction of a new treatment is defined as the product of the quality of life [30] and the number of additional years of life, where quality of life is defined by an assessment of wellbeing, based on a questionnaire. One year of perfect health is equivalent to one QALY. Treatments are often described as cost-effective if the cost per additional QALY gained lies below a given threshold, which varies depending on the healthcare system. Values quoted include £20–30 k for NICE (the National Institute for Health and Care Excellence, which approves the use of medicines within the English healthcare system) [30]. The same figure is used by the Scottish Medicines Consortium [31], and $50k is often used in other parts of the world [32]. QALYs are not the only factor considered by healthcare providers when they decide which interventions to deploy [32]. Some countries do not use QALYs, and particular challenges in the use of QALYs for evaluation of cancer treatments include fluctuations in the quality of life, switching between therapies and the lack of data on overall survival rates [33]. However, QALYs are still used frequently to assess cost-effectiveness of cancer drugs. For example, in one study pazopanib was compared to sunitinib for metastatic renal cell carcinoma, within the United Kingdom [34]. Overall, it was found that pazopanib was likely to be more cost-effective than sunitinib.
Methods
Estimating profit margin, p
We examined the company reports of 13 selected pharmaceutical companies, representing many of the larger players in the sector. From each report, we extracted the figure described as 'net earnings', 'net profit', 'net income', or equivalent. We also extracted the 'total revenue', 'total sales', 'sales', 'revenue' or 'turnover' where available. In some reports, only the 'net revenue' or 'net sales' figure was given. We expressed the net profit as a percentage of the revenue figure we had extracted, and obtained values ranging from 7.7% to 35.3%, with an average of 17.8%. In our model, we therefore assumed a range of profit margin p from 0.05 (i.e. 5%) to 0.40. This covers all the values obtained for the companies we examined. We present full details of our analysis in supplementary data 1, which is available online at stacks.iop.org/BPEX/6/065030/mmedia.
Manufacturing cost for DNA origami, cm,DNA
Traditionally, DNA origami is prepared using the M13mp18 bacteriophage genome as a scaffold. This can be acquired commercially; for instance tilibit nanosystems offers 800 picomoles of single-stranded DNA with this sequence for €635 (approximately £530 at exchange rate of €1 = £0.83, 14th December 2019). New England Biolabs provides 10 μg (4.2 pmol) for £32 (excl. VAT), excluding any institutional discounts. The staples are usually bought in the form of synthetic oligonucleotides in plates. Based on prices from a representative DNA synthesis company (Integrated DNA Technologies, again excluding institutional discounts and VAT) the price of a full set of staples, containing a total of 7249 nucleotides, at 25 nmole synthesis scale is £1300 (IDT: £0.18/base for 15–60 base oligos). 1 mg of DNA origami contains approximately 210 pmol of folded nanostructures. At a five-fold excess of staples, this means that 1050 pmol of staples are required, representing a fraction of 1.05/25 of the full set made, representing a cost of approximately £55. The corresponding quantity of scaffold from tilibit would cost approximately £139. Hence, with this approach the price of the raw materials to make 1 mg of DNA origami would be nearly £200. However, in a recent paper Praetorius et al estimated that their biotechnological approach could reduce the cost of DNA origami to as little as €0.18 per mg for production at scale, including labour and overheads [35]. In our model we assume for most calculations a fixed DNA origami manufacture cost cm,DNA equal to this value (£0.15 with exchange rate as at 14th December 2019).
Mass of drug needed, mdrug , and cost of drug, cdrug
The mass of drug needed is highly variable, depending on the drug, the patient and whether the drug is being used as a monotherapy or in combination with other treatments. In order to estimate the range of possible parameters, we considered 6 particular case studies of 'conventional' chemotherapy treatments. We selected the case studies to include examples of different drug types e.g. taxanes, anthracyclines and an antimetabolite. We considered mainly monotherapies, but also included a combination therapy based on paclitaxel and a platinum-containing compound. Where possible, we focused on drugs that are used for the most common types of cancer, which are identified in official UK statistics as breast, lung, prostate, colorectal [36, 37]. To calculate the mass and cost of drug, we combined information from official NICE (National Institute for Health and Care Excellence) 'evidence-based recommendations', entries in the British National Formulary (also known as MedicinesComplete) [38] and dosage suggestions on the electronic Medicines Compendium [39]. Dosage is often given in terms of mg per body surface area, which varies from patient to patient. We assumed an average value of body surface area of 1.79 m2, as given in [40]. Full details of our case studies are given in supplementary data 2. In clinical practice, treatment and dosage should always be determined by a qualified specialist in oncology and chemotherapy.
In the six case studies, the cumulative mass of chemotherapy drugs used per treatment ranged over more than three orders of magnitude, from 251 mg to 313 g (note units). However, the highest figure was an outlier, and if this is omitted, the values range from 251 mg to 2.7 g, differing by about one order of magnitude. The cost per mg ranged from less than a penny to £19.09. The cheapest price corresponded to the drug for which the anomalously high quantity was required, and when this was omitted the next lowest cost was £1.26/mg. For our model we therefore assumed a range of mdrug from 0.1 g to 3 g, and drug costs from £0.50 to £20 per mg.
Mass ratio, r
To establish the range of possible values for mass ratio we considered the results of several different studies. In the seminal paper on targeted delivery of cargo to specific cells, Douglas et al [25] attached fluorescently labelled antibody fragments to a DNA origami nanorobot. The origami structure comprised 7308 base pairs, with an approximate molecular weight of 4.8 MDa, while the mass of the antibody fragment was approximately 55 kDa. On average, there were three antibody fragments per nanorobot, which gives a total mass of 165 kDa of antibody for 4.5MDa of DNA and a mass ratio of 0.165/4.5 or 3.4%.
Some drugs are loaded into DNA nanostructures by intercalation between DNA bases. An early study of DNA origami examined the effect of ethidium bromide intercalation on origami structure [41] and we can use the results of that study to estimate the mass loading ratio for the case of ethidium bromide (EtBr). It was found that the most heavily loaded structure contained 53 EtBr intercalations per helix, with 191 base-pairs per helix. Hence, there were 53/191 EtBr molecules per base pair. The molar mass of EtBr is 394.3 Da and hence the mass loading ratio is (53 × 394.3)/(191 × 650), which is approximately 17%.
A recent study of the interactions between fluorescent intercalator YOYO-1 and DNA origami tiles suggested that there were 67 ± 25 YOYO molecules per tile [22]. Assuming that the tile contained 6480 bp, and that the molecular weight of YOYO-1 is 1271 Da, this gives a mass loading ratio of 67 × 1.271/4200 = 2%. In another study, an origami nanostructure was loaded with daunorubicin [42], which also intercalates between base pairs, it was observed that the treatment strategy was effective when there were 0.46 to 1 daunorubicin molecules per base pair. When the structure was overloaded (more than one molecule per base pair), aggregation and efficacy decline were observed. Given a daunorubicin molecular weight of 527.5 Da and an average base pair molecular weight of 650 Da, this gives a mass loading ratio of between 37% and 81%.
We may also consider the drug doxorubicin (Dox), which is similar to daunorubicin. Dox is used in numerous chemotherapy regimes (supplementary information, treatment case study 2) and it is a popular choice for DNA nanostructure drug delivery studies because it is fluorescent and readily available. However, as noted by Keller and Linko [43] and Ijäs et al [44], spectroscopic studies of Dox can yield peculiar or ambiguous results. Some studies have reported results suggesting extremely high drug loading, with multiple Dox molecules per base pair, giving mass loading ratios as high as 721% (for 8.6 Dox/bp) [24] or even 6448% (for 78 Dox/bp) [23] (details given in supplementary calculation 1). As Dox is an intercalator, steric considerations imply that this is impossible. Even a 1:1 Dox/base pair ratio is somewhat implausible, because this would be likely to induce significant disruption to the structure, but this value can be used as an upper limit. At a 1:1 ratio, the mass loading ratio would be 84%. Other studies have reported lower loading ratios. For instance, Raniolo et al loaded DNA octahedra with Dox and obtained a mass loading ratio of approximately 10% [45] (supplementary calculation 1). More recently, Ijäs et al obtained results that indicated a typical mass loading ratio of approximately 27% for Dox-loaded origami structures [44].
In the studies that we considered, excluding the implausible results, mass loading ratios ranged from 2% to just over 80%. Hence, in our model we used a range of 1% to 100%.
Total cost of R&D for new therapy, ΔCr&d
One estimate placed the cost of developing a new drug at $2.87bn (in 2013 dollars), when the costs of unsuccessful projects and post-approval R&D were included [46]. However, for the scenarios considered in this paper, the active agent is already known and the delivery method is changed. Clinical trials would still be required, and the cost of such trials can be hundreds of millions of dollars [47]. We therefore consider a range of development costs, from £50million to £4billion.
Number of treatments per year, t
Our analysis of pharmaceutical company reports confirmed that their annual revenues were often tens of billions of dollars. It is therefore reasonable to assume that a single therapy could yield annual income of $100 M. An average cost per treatment of around $2000 would be consistent with our analysis of the prices of various therapies (supplementary data 1). This suggests that the typical number of treatments could be around ($100 M/$2000) = 50 000. In our model we consider a range of number of treatments per year, from 10 000 to 200 000. The number of treatments will vary depending on the incidence of the disease being treated and relative efficacy of the treatment, among other factors.
Time to recoup cost, y
While a patent can theoretically protect a new drug for 20 years, in practice, the patent is likely to be filed at an early stage of the process, and a significant proportion of the patent lifetime will then be taken up with development and approval. Hence, it is desirable for costs to be recouped as quickly as possible. In our model we assume that the costs will be recouped in 1–8 years.
Range and spacing of parameters
For the purposes of our study, we evaluated the range of parameters shown in table 1. We used evenly spaced test values for all parameters, specified using the MATLAB command linspace. For example, we specified the possible values for m (mg of drug per treatment) with the command m = linspace(100,3000,x), where x = 20. This yielded the following range of values for m, rounded to the nearest whole number for the purposes of presentation here: (100, 253, 405, 558, 711, 863, 1016, 1168, 1321, 1474, 1626, 1779, 1932, 2084, 2237, 2389, 2542, 2695, 2847, 3000)
We applied similar statements for the other parameters, and tested every possible combination of the values defined in this way. This means that our model assumed no correlation between the parameters listed in table 1, and we treated them as independent variables. It is conceivable that some of these parameters could be coupled i.e. dependent on each other. For instance, the mass of drug required (m) and the mass loading ratio (r) could be affected by the underlying chemistry of the drug and mode of action and therefore might be related. There could also be a connection between the profit margin (p), the development cost (ΔCr&d ), the number of years (y) to recoup the cost and the number of treatments per year (t), depending on the business model of the manufacturer. Furthermore, the acceptable cost per QALY (CQ ) and the number of treatments per year (t) could be connected, as it might be deemed acceptable to increase the threshold cost for a particularly rare but devastating type of cancer. At this time, it is not possible to quantify potential correlations such as those noted in this paragraph and we therefore omitted them from the model. In this way, we investigated the broadest possible range of scenarios, and our model can be applied to more specific situations in the future, when these are fully defined by experimental and clinical studies.
Table 1. The parameters used in our model.
| Parameter | Value |
|---|---|
| p, profit margin | 0.05–0.40 (i.e. 5% to 40%) |
| cm,DNA, cost per mg of DNA nanostructure | For most of our calculations this was fixed at £0.15 per mg, except where otherwise stated |
| mdrug, the mass in mg of drug used in a chemotherapy treatment | 100–3000 mg |
| r, the mass loading ratio (number of mg of drug for each mg of DNA nanostructure) | 0.01–1 |
| ΔCr&d , total cost of research and development required to bring nanostructure-enabled treatment to market | £50million to £4billion |
| y, the number of years to recoup development costs | 1–8 years |
| t, the number of treatments per year | 10 000–200 000 |
| CQ , Threshold cost per QALY | £30 000 |
| Cost of drug | £0.50/mg to £20/mg |
Results
Total cost of treatments with and without nanostructures
Let the cost to a health service of treatment i be CT,i . If the profit of the manufacturer is ignored, this cost is the sum of the manufacture cost, CM , (including labour and 'parts'), a share, CD , of the development costs, and the cost of administering the treatment, CA (including the cost of personnel, hospital visits etc). Hence the cost in a break-even scenario is:

The actual cost will include an 'acceptable' profit for the manufacturer. Let the manufacturer's net profit margin be p, defined as net profit divided by revenue. The profit applies to the manufacture and development costs incurred by the manufacturer. Hence:

Scenario 1: nanostructures providing more QALYs
We first consider a scenario in which the same quantity of drug is used in the drug-only treatment (subscript 0) and the treatment with nanostructures (subscript n/s). Here, we assume that the use of the nanostructures improves the quality of life or lifespan of the patient at increased cost, where quality of life and lifespan are reflected by the number of QALYs provided, denoted by Qi for treatment i.
This assumption implies that: CT,n/s > CT,0 and Qn/s > Q0.
Let us define: ΔQ = Qn/s -Q0 and ΔC = CT,n/s -CT,0, where ΔQ is the number of QALYs gained by introducing DNA nanostructures for drug-delivery and ΔC is the additional cost.
In the worst-case scenario, the drug-only treatment is just about cost-effective. Here, the cost per QALY is given by
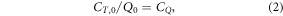
where CQ is the threshold cost per QALY.
By definition, the cost per QALY in the nanostructure-enabled case is:

If the drug-only treatment is only just cost-effective, and moving to the nanostructure-treatment is to be more cost-effective, we must have CT,n/s /Qn/s < CT,0/Q0 . Substituting using equation (3) yields:

Rearranging gives:

By substituting using equation (2), we obtain
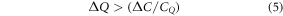
We can quantify the right-hand side of this equation. Using equation (1), we have:
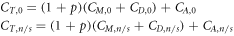
As we wish to consider differences in cost between the nanostructure-enabled and drug-only therapies, we can ignore any expenditure that would be comparable for different approaches. It is reasonable to assume that the cost of administering the two treatments will be identical, so that CA,0 = CA, n/s .
Thus:

Defining ΔCM as the difference in manufacturing cost and ΔCD as the additional research and development cost for the nanostructure-enabled treatment, we have

ΔCM includes the cost of the materials used to make the DNA nanostructure, the cost of the labour involved to make the nanostructure, and the cost of the labour involved in integrating the drug with the nanostructure. In most cases the latter will be negligible as drugs could potentially be integrated simply through incubation of the nanostructures with the drugs in question. If the cost of manufacturing one gram of DNA nanostructures (including both parts and labour) is cm,DNA , the mass of DNA nanostructure needed for the treatment is mDNA , and the cost of labour to integrate the drug is omitted, then:

We can express the mass of DNA nanostructure required in terms of the mass of drug required (mdrug ) and a mass ratio, r. The mass ratio is the number of grams of drug required for each gram of DNA nanostructure (so r = mdrug /mDNA ), and quantifies how heavily the nanostructure is loaded. Hence:

The additional development cost ΔCD is the share of research and development costs to be recouped from each individual treatment. If ΔCr&d is the total cost of research and development required to bring the nanostructure-enabled treatment to market, given prior approval of the drug-only treatment, and N is the number of treatments over which cost will be recouped, then

We can find N by estimating the number of treatments per year (t), and deciding on the number of years over which development costs must be recouped (y). Hence:


This is the additional cost of treating a single patient with the nanostructure-enabled therapy, as opposed to the drug-only therapy (figure 1(a)).

Figure 1. Scenario 1: using DNA nanostructures ('n/s') to deliver the drug provides more QALYs than the drug-only therapy. (a) Our model: the equation defining the additional cost for treating one patient with a nanostructured-enabled treatment, over and above the cost for the drug-only therapy. (b) and (c) Extra costs predicted by the model when all but two parameters are fixed. (b) Dependence on the mass loading ratio for five different values of the mass of drug used for the therapy, with the other parameters fixed. (c) Dependence on the profit margin for five different values of the mass loading ratio, with the other parameters fixed. (d) Histogram of predicted costs for 64 million sets of parameter values; the cost of manufacturing DNA was fixed but all other parameters were variable and we tested 20 different values for each. (e) Histogram showing the number of QALYs that must be gained for the situations specified by the parameters in part (d) to be cost effective, for the indicated threshold cost per QALY. Inset: results obtained when the analysis is repeated with greater costs for DNA nanostructure production. Graph shows the fraction of the 64 million parameter sets that give rise to cost-effective therapies for a gain of less than 0.25 additional QALYs (compared to the therapy without nanostructures).
Download figure:
Standard image High-resolution imageEquation (5) specified the condition for which the cost-effectiveness of the nanostructure-enabled treatment is greater than that of the drug-only treatment, when the drug-only treatment is only marginally cost-effective. Combining this with equation (9) gives us

Most of the parameters on the right-hand side of equation (10) are variable. We use the range of values shown in table 1 to evaluate the overall costs.
We find that the costs are particularly high when a large quantity of drug is needed (figure 1(b)). The additional cost depends only linearly on the manufacturer's profit margin (figure 1(c)), and the drug loading ratio is particularly important (figures 1(b), (c)). When the drug loading ratio is low, the cost is likely to be high even if the total mass of drug required is small and the manufacturer accepts low profit margins. We tested 64 million parameter sets, using 20 evenly spaced values for each of the 6 variables. The majority of these parameter sets produced nanostructure-enabled therapies that cost less than £20k more than their drug-only counterparts (figure 1(d)), but a small number of parameter sets give particularly high costs.
If £30k is an acceptable cost per QALY, just over half of the 64 million parameter sets describe therapies that would be cost-effective if they provided less than 0.25 additional QALYs (figure 1(e)). For this calculation, we assumed that the production cost of DNA nanostructures was £0.15/mg. This is considerably cheaper than inorganic nanostructures such as gold nanoparticles, which are available commercially at prices of approximately £50/mg. The present cost of DNA nanostructures for research is much higher (£200/mg, see Methods), but production volumes are low. The £0.15/mg value is based on estimates for synthesis at much larger scales [35], as required for pharmaceutical production lines. If such low costs prove to be unachievable, fewer of the parameter sets will yield cost-effective therapies (figure 1(e), inset). For example, if cm,DNA is set at £1.50/mg, only 22% of the 64 million parameter sets describe therapies that would be cost-effective if they provided less than 0.25 additional QALYs. If costs of DNA nanostructure production remain at present levels, none of the 64 million parameter sets describe therapies that would be cost-effective for a gain of less than 0.7 QALYs.
Scenario 2: same number of QALYs, reduced quantity of drug
We now consider the case in which the number of QALYs is unchanged but the mass of drug is decreased. Here, money is saved on the drug, but an additional cost is incurred in manufacturing the nanostructure. For some combinations of parameters, it will be cheaper to put the drug in a DNA nanostructure and in other cases it will be cheaper to use the drug alone. In scenario 2, the mass of drug used is mdrug *, and the mass loading ratio is now r = mdrug */mDNA .
The cost of adding the DNA nanostructure as a drug-delivery vehicle is a modified version of equation (9):

The cost saving due to the decrease in drug mass is

where cdrug is the cost per unit mass of the drug.
In scenario 2, the net change in cost between the nanostructure-enabled therapy and the conventional equivalent (for one patient) is:

By definition, mdrug * ≤ mdrug . Let us define mdrug * = Kmdrug , where 0 < K ≤ 1. Now:

In this scenario, the number of QALYs gained is the same for both treatments (they are equally effective). Hence cost-effectiveness is determined simply by the change in the cost, ΔCs2 . If the cost of adding the nanostructure exceeds the saving incurred by reducing the amount of drug required, the quantity defined by equation (11) (figure 2(a)) is positive and the nanostructure-enabled treatment is less cost-effective than the drug-only approach. The relationship between mdrug *, mdrug , mDNA , r and K is depicted graphically in figure 2(b).

{kind=link}
Figure 2. Scenario 2: using DNA nanostructures ('n/s') to deliver the drug provides the same number of QALYs as the drug-only therapy but changes the cost by reducing the amount of drug used. (a) Our model: the equation defining the change in the cost for treating one patient when nanostructures are introduced. (b) Sketch showing how the mass of drug (mdrug *, red) and DNA (mDNA , grey) used in this scenario depend on the parameters K and r. In our model, the mass of DNA used is defined with reference to the mass of drug used. K is smaller than or equal to 1 and strictly greater than zero. For K = 1 the use of DNA nanostructures does not enhance the potency of the drug and the quantity required is the same as if nanostructures are not used. Note that mdrug (no asterisk) is the mass of chemotherapy agent used in the drug-only case. (c)–(g) Histograms showing the change in cost for 170,859,375 sets of parameter values for six different values of K. The following 7 parameters were variable, and 15 linearly spaced values were tested for each, spanning the indicated range: p = 0.05–0.4, r = 0.01–1, mdrug : 100–3000 mg, ΔCr&d = £50M–£4B, y = 1–8 yrs, t = 10 × 103–200 × 103, cdrug = £0.50/mg–£20/mg. The DNA manufacture cost was fixed at £0.15/mg. The annotation shows the fraction of cases for which there is a cost saving (feff ) and the value of K.
Download figure:
Standard image High-resolution image{kind=link}
If the introduction of nanostructures enables the quantity of drug to be reduced by 80%, we find that 55.3% of the parameter sets correspond to situations in which money is saved, and the nanostructure-enabled therapy is cheaper (figure 2(c)). We see a decrease in the proportion of cases in which there is a net cost saving as the mass of drug increases (figures 2(d)–(f)). At K = 1, the same quantity of drug is used in the nanostructure-enabled and drug-only therapies. In this case, by definition, the nanostructure-enabled therapy is less cost-effective than the drug-only therapy for all possible values of the other model parameters (figure 2(g)). To elaborate: for K = 1, there is no cost saving on the chemotherapy agent (as the same amount is used as in the drug-only case). In addition, significant costs are incurred in manufacture of the DNA and in development (through research, testing and clinical trials). The spread of observed costs arises due to the range of values for the quantity of nanostructures required, the profit margin and all the other parameters. As K decreases, a smaller quantity of drug is required, and the nanostructures enhance the treatment to a greater extent.
Hypothetically, if the value of K were to be set equal to zero, this would imply that mdrug * = 0. However, we defined the required quantity of DNA nanostructures with reference to the mass of drug used, and hence a zero value for mdrug * implies that the mass of nanostructures used is also zero. Consequently, K = 0 would correspond to a scenario in which development costs are incurred, but no drug or nanostructure is administered, and the patient experiences the same benefit as if taking the drug. This scenario is absurd and hence we defined K to be strictly greater than zero.
It is not possible to compute costs for a 'nanostructure-only' scenario, because there is no basis on which to specify the dosage for such a therapy. In any case, the control experiments in published studies (see Introduction) suggest that a nanostructure-only treatment would be entirely ineffective against cancer.
Discussion
In scenario 1, the DNA nanostructures did not change the total mass of drug needed to treat the patient but did increase the number of QALYs provided. This could correspond to a decrease in side effects, yielding improved quality of life or more years of life. In this scenario, the drug-loading ratio was of critical importance, and it will be essential to optimize this as the technology is developed. We tested 64 million parameter sets, covering a range of drug loading ratios, total drug mass, profit margins etc, and we found that just over half of these cases (51.3%) described therapies that would be cost-effective if they provided less than 0.25 additional QALYs, given that the cost of DNA nanostructure production was £0.15/mg. While this is substantially lower than the present cost of DNA nanostructures in a research context, it is believed to be achievable. If major reductions in DNA nanostructure synthesis cost are not achieved, it is unlikely that DNA nanostructure-enabled therapies will be cost-effective. For chemotherapy applications, it will therefore be essential to demonstrate low-cost manufacture of DNA nanostructures at scale. As noted by Keller and Linko [43], this process must comply with the stringent regulations that apply for biologics and pharmaceuticals, and this may be particularly challenging in instances where the nanostructure is based on a genomic scaffold.
In the second scenario, the number of QALYs provided was unchanged by the use of DNA nanostructures, but the amount of drug was reduced. If the reduction was substantial (80%), over half of the parameter sets (55.3%) produced a net cost saving, implying that the nanostructure-enabled therapy would be more cost-effective.
In practice, the use of nanostructures to deliver drugs has the potential to reduce the amount of drug used and simultaneously increase the number of QALYs provided, but it would be difficult to model this scenario with the currently available information. Our analysis suggests that the use of DNA nanostructures to deliver chemotherapy drugs only needs to enhance the therapy by a reasonably small amount. This is a significant result, because it implies that the technology has the potential to be commercially viable, if DNA nanostructures can be manufactured cheaply at scale with appropriate quality. Our method could also be applied to other early-stage technologies to assess the likelihood of successful translation as they become more mature.
Acknowledgments
There is no applicable funding to acknowledge. The authors wish to thank the following people for useful discussions and input: Dr Peter Hall, Dr Michael Chen and Dr Aileen Neilson of the University of Edinburgh, and Prof Jon Timmis of the University of Sunderland.
Author contributions
E C: data curation, formal analysis, investigation, methodology, validation, writing—draft, review & editing. K D: conceptualization, data curation, formal analysis, investigation, methodology, project administration, software, supervision, validation, visualization, writing—draft, review & editing.
Competing interests
K D is in receipt of funding from a biopharmaceutical company in connection with another project; the funders were not involved in the present paper in any respect.