

Abstract
Providing better biodegradable materials for medical applications has always been an important premise for improving the therapeutic effect of clinical diseases. The Poly (butylene succinate) (PBS) and Sodium alginate (SA) composites were prepared using melt blending technique. Fourier transform infrared spectroscopy (FTIR), scanning electron microscope-energy dispersion spectrum (SEM-EDS), mechanical properties, water contact angle, thermal properties, and in vitro degradation and cytotoxicity tests were determined to evaluate the properties of the composites with a varied SA proportion of 5%, 10%, 15%, and 20%. The FTIR and SEM-EDS results confirmed the successful preparation and microphase distribution of the composites. With the increasing in SA loading, the distribution of the filler became unevenly gradually from evenly, the Young's modulus increased first and then decreased, the tensile strength and elongation at break decreased gradually, the hydrophilicity, in vitro biodegradability increased, thermostability decreased, and the Tm, Tg, and crystallinity of the composites did not change significantly. The composite with 10% SA loading has uniform dispersion of the filler, the highest Young's modulus (1091.21 MPa), mild hydrophilicity (θ = 88.40°), an adequate thermal processing temperature range (110 °C–200 °C), and has good biodegradability and biocompatibility with no significant deleterious impact on the cell membrane, lysosomal membrane, cell proliferation, cell apoptosis, cytoskeleton, or intracellular reactive oxygen species levels. It can be used as a biodegradable material for medical applications such as suture anchors.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 4.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
1. Instruction
Biodegradable materials have been the focus of medical studies in recent decades because they can degrade gradually in the body, leaving no foreign matter. Aliphatic polyesters, such as Polylactic acid (PLA), Polyglycolic acid (PGA), Poly ε-caprolactone (PCL), and Poly (p-dioxanone) (PPDO), are frequently employed in biomedical fields and clinical practice due to their biocompatibility and biodegradability [1–4]. Poly (butylene succinate) (PBS), which is also an aliphatic polyester, has become more interesting in recent years because it might have uses in medicine [5].
PBS is a biodegradable thermoplastic polyester synthesized by the condensation of succinic acid and 1,4-butanediol. It is also a sustainable, and biobased polyester since its two components can be manufactured by the biological fermentation of sugarcane, cassava, and maize. Due to its good mechanical property, thermostability, and thermal processing capability, PBS has been widely used in industry and daily life, and has been the subject of a growing number of medical application studies, such as bone repair, cartilage tissue engineering, cardiac tissue engineering, drug delivery, and antibacterial function [6–8].
In order to meet the requirements of a certain medical application, the performance of PBS needs to be optimized and modified in many cases. Blending filler into the PBS matrix for creating composites is a common method of modification. The interaction of components in composites can modify the mechanical properties, thermostability, and crystallinity, as well as promote protein adsorption, cell proliferation, and tissue healing [9–12].
The suture anchor is a medical implant that is used to suture torn tendon to bone in the treatment of rotator cuff tears and other sports injuries [13]. The tendon-bone interface is predominantly repaired by inflammatory infiltration, collagen production, and regeneration between soft tissue and bone, which is composed of four consecutive layers: tendon fibres, uncalcified fibrocartilage, calcified fibrocartilage, and bone [14]. Therefore, it is beneficial for patients' rehabilitation if the material used to manufacture suture anchors has biodegradability, good mechanical qualities, and the function of promoting tendon, cartilage, and bone regeneration.
Chitosan (CS), hydroxyapatite (HA), and tricalcium phosphate (TCP) are the most studied fillers in the medical applications of PBS based composites. It has been shown that PBS-based composites using CS as the filler can enhance the proliferation and osteogenic differentiation of human mesenchymal stem cells and bovine articular chondrocytes in vitro and bone growth in vivo [8, 15]. Suphakit Arphavasin et al prepared PBS-based composites with HA and TCP as fillers, which improved hydrophilicity and the function of supporting the proliferation and osteogenic differentiation of human mesenchymal stem cells [16]. These studies utilized the properties of CS, HA, and TCP that promote bone or cartilage regeneration [11, 17]. However, to the best of our knowledge, no research has shown that CS, HA, or TCP can improve tendon regeneration.
Sodium alginate (SA) is a kind of biosynthesized anionic polysaccharide mainly extracted from brown seaweed, which is the second most abundant biopolymer in the natural world. SA consists of two monomers bonded by (1–4)-linkage: β-D-mannuronic acid and α-L-guluronic acid, both of which contain hydroxyl (HO−) and carboxyl (−COO−), making it a good hydrophilicity [18, 19]. In addition, the molecular structure of SA resembles glycosaminoglycan, a principal component of the extracellular matrix that regulates the activity of growth factors and promotes the growth of local tissues [12]. Furthermore, SA has good bioactivity and biocompatibility and has been approved by the US Food and Drug Administration (FDA) for medical applications [20–22]. SA has been employed as a drug carrier, wound dressing, and a scaffold for tissue regeneration in arteries, pancreas, muscle, liver, and peripheral nerves [23]. Additionally, it promotes the regeneration of bone, cartilage, and tendons. Data have shown that SA can support the growth of bone marrow stromal cells, as well as increase calcium content and osteocalcin mRNA expression in mouse osteoblasts [24, 25]. In animal implantation experiments, SA showed the effect of promoting the differentiation of induced pluripotent stem cells and the generation of chondrocytes [26]. SA can also promote the activity and differentiation of tendon-derived stem cells by improving local inflammatory infiltrates and the arrangement of tendon cells and collagen [27]. SA, which may induce regeneration of bone, cartilage, and tendons, is appropriate for manufacturing medical implants to repair the bone-tendon interface. However, SA is unsuitable for the fixation of tendon and bone directly due to its insufficient mechanical properties [23]. Therefore, by manufacturing the composite of PBS and SA, we integrated the benefits of both materials and gave a new option for the bone-tendon interface repair material.
In this work, PBS based composites with SA as a filler were prepared by melt blending. By measuring the microstructure, mechanical characteristics, water contact angle, and thermal properties of the composites, the ratio with the best overall attributes was identified. The biodegradability and biosafety were validated using in vitro degradation and cell tests to prove the biocompatibility for medicinal applications.
2. Materials and methods
2.1. Experimental reagents, cell lines, and equipment
PBS particles (Macklin Biochemical Co. Ltd., Shanghai, China); SA powders (Macklin Biochemical Co. Ltd., Shanghai, China); Sorensen buffer solution (0.2 M, pH 7.4) (Macklin Biochemical Co. Ltd., Shanghai, China); DMEM high-glucose medium (Solarbio, Beijing, China); 0.25% trypsin (Gibco, Waltham, Massachusetts USA); neutral red staining solution (Beyotime Institute of Biotechnology, Jiangsu, China); phosphate buffer solution (Beyotime Institute of Biotechnology, Jiangsu, China); CCK-8 kit (Solarbio, Beijing, China); Phalloidin-iFluor 555 reagent (Abcam, Cambridge, UK); antifading (with DAPI) (Solarbio, Beijing, China); Annexin V-FITC apoptosis detection kit (Beyotime Institute of Biotechnology, Jiangsu, China); 0.25% trypsin without EDTA (Solarbio, Beijing, China); reactive oxygen species (ROS) assay kit (Beyotime Institute of Biotechnology, Jiangsu, China); 0.25% trypsin (Gibco, Waltham, Massachusetts USA).
NIH/3T3 and BALB/c 3T3 cells, clone A31 (Procell CL-0477), were obtained from Procell Life Science & Technology Co., Ltd. (Wuhan, Hubei, China).
RM-200C torque rheometer (Hapro, Harbin, China); Nicolet iN10MX Fourier transform infrared spectrometer (FTIR) (Thermo Fisher Scientific, Waltham, Massachusetts, USA); JSM-7610F ultra-high-resolution thermal field emission scanning electron microscopy (SEM-EDS) (JEOL Ltd., Tokyo, Japan); SPM-9700 atomic force microscope (AFM) (Shimadzu, Kyoto, Japan); theta flow optical contact angle measuring instrument (Bolin, Stockholm, Sweden); Instron 5300 universal testing machine (Instron, Inc., Shanghai, China); DSC1 differential scanning calorimeter (DSC) (Mettler Toledo International Ltd., Delaware, USA); TG 209 F3 thermogravimetric analyzer (TGA) (Netzsch, Selb, Germany); electronic balance ME202 (Mettler Toledo International Ltd, Delaware, USA); Nikon microscope (Nikon, Tokyo, Japan); SpectraMax ABS microtitration plate photometer (Molecular Devices, Sunnyvale, Cal-ifornia, USA); fluorescence microscope (Nikon, Tokyo, Japan); CytoFlex S Flow cytometer (Beckman, S. Kraemer Boulevard Brea, California, USA); FlowJo software (Becton, Dickinson & Company, Franklin, USA).
2.2. Preparation and characterization of PBS-SA composites
Before the operation of melt blending, PBS and SA were dried at 80 °C for 4 h and 40 °C for 12 h, respectively, in a vacuum drying oven to prevent excessive water in the composites. The two materials were pre-mixed and then placed into the internal mixer module of the torque rheometer preheated at 140 °C for 2 h, and the parameters of the module were set to 140 °C, 30 rpm, and 10 min. The composites were named PBS-SA5, PBS-SA10, PBS-SA15, and PBS-SA20 according to the mass ratio of SA increasing by 5% gradually. After cooling to room temperature, the composites were placed in sealed bags for the follow-up experiments.
The FTIR analysis range of 400–4000 cm−1 was determined for PBS, SA, and PBS-SA composites. SEM-EDS was used to study the microstructure and element distribution of PBS-SA composites treated with liquid nitrogen brittle fracture and platinum spraying. In addition, AFM was utilized to analyse the microscopic distribution described in the Supplementary material section.
The water contact angle of PBS and PBS-SA composites was measured using an optical contact angle analyser at 20 °C. Before the mechanical property test, the PBS and PBS-SA composites were formed into test samples with a size of 50 × 5 × 2 mm3 at 140 °C. Subsequently, a universal testing machine was used to conduct tensile tests at a constant tensile speed of 10 mm min−1 till breaking.
The thermal properties of the PBS and PBS-SA composites were determined using DSC and TGA. At a temperature change rate of 10 °C min−1, thermal history elimination (from room temperature to 200 °C), cooling curve (from 200 °C to 50 °C), secondary heating curve (from −50 °C to 300 °C), and thermogravimetric analysis (from room temperature to 800 °C) were conducted in nitrogen. During the measurement of thermal properties, such as glass transition temperature (Tg), melting point (Tm), crystallization temperature (Tc), enthalpy of cold crystallization (ΔHc), 5% initial decomposition temperature (Td), and the peak temperature of thermal decomposition in the derivative thermogravimetry (DTG) curve (Tmax), the following relationship was utilized to calculate the crystallinity of PBS in the composites:
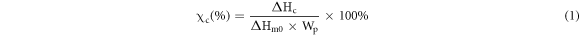
In this relationship, χc denotes the crystallinity of PBS in the composites, ΔHc denotes the enthalpy of cold crystallization we measured, ΔHm0 denotes the 100% theoretical crystallization enthalpy of PBS, taken to be 110.3 J g−1, and Wp denotes the weight percentage of PBS in the composites [28].
2.3. In vitro degradation
The samples for in vitro degradation were formed into thin discs with a diameter of 1 mm and a thickness of 0.3 mm at 140 °C. Each sample was placed into a glass bottle containing the Sorensen buffer solution, respectively. All sample bottles were sealed and placed in an incubator at 37 °C. Residual mass was examined and measured at 1, 2, 4, 8, and 12 weeks, and the Sorensen buffer solution in the bottles was replaced once a week. After drying each sample to constant weight at 40 °C in a vacuum oven, the sample mass was measured, and the mass loss percentage was determined using the following relationship:
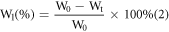
In this relationship, Wl denotes the mass loss percentage, W0 denotes the initial weight, and Wt denotes the weight at the pre-set time point.
2.4. In vitro cell assays
2.4.1. Cell culture and the samples preparation
The cells were incubated in DMEM high-glucose media at 37 °C with 5% CO2. After digestion with 0.25% trypsin and centrifugation at 1200 rpm, the cells were cultured in dishes at a density of 1 × 105 cells. The cells were used for cell experiments at a density of 60%–70%.
In compliance with ISO 10993.5–2009, the extract of the optimized composite was employed as the experimental sample, while the medium served as the control sample [29]. The medium was a DMEM high glucose medium with 10% bovine serum volume fraction, 100 IU ml−1 of penicillin, and 100 g ml−1 of streptomycin. The extract was prepared by immersing the composite in the medium at 37 °C for 24 h.
2.4.2. NRU cytotoxicity assay
After the samples co-cultured with BALB/c 3T3 cells at 37 °C in 5% CO2 for 24 h were removed by phosphate buffer solution, the cells were stained with neutral red staining solution for 0.5 h. Then, the optical density (OD) values were determined using a microtitration plate photometer with a 550 nm wavelength, and microscopy photographs were taken.
2.4.3. CCK-8 assay
NIH/3T3 cells with a density of 5000 cells/well were co-cultured with the 100 μl sample at 37 °C in 5% CO2 for 24 h, followed by the addition of 10 μl CCK-8 solution and incubation for 2 h. The OD values were measured at 450 nm with a microplate photometer.
2.4.4. Cytoskeleton Staining
After removing samples co-cultured with BALB/c 3T3 cells at 37 °C in 5% CO2 for 24 h with phosphate buffer solution, the cells were successively treated with 4% paraformaldehyde for 10 min, 0.1% Triton X-100 for 5 min, and 1 × Phalloidin-iFluor 555 for 1 h. The phosphate buffer solution was used to rinse the reagent thoroughly after each step of treatment. Finally, the cells were sealed with mounting medium and antifading (with DAPI) and observed using a fluorescence microscope equipped with an appropriate filter set at Ex/Em = 556/574 nm (Phalloidin-iFluor 555) and Ex/Em = 360/460 nm (DAPI).
2.4.5. Cell apoptosis
After co-culturing with the sample at 37 °C in 5 % CO2 for 24 h, the BALB/c 3T3 cells were digested with 0.25% trypsin (without EDTA) and rinsed with pre-cooled phosphate buffer solution 3 times and collected by centrifuging at 1200 rpm at 4 °C for 5 min. The phosphate buffer solution added in the previous step was discarded, and the cells were treated with 100 μl of 1 × binding buffer and mixed with 5 μl of Annexin V-FITC, followed by incubation at room temperature for 15 min without light. At last, cells were successively added with 10 μl of PI holding solution and 400 μl of 1 × binding buffer and detected by flow cytometry in 5 min.
2.4.6. ROS assay
After co-culturing with the sample at 37 °C in 5% CO2 for 24 h, the BALB/c 3T3 cells were coated with 2',7'-Dichlorofluorescin diacetate (DCFH-DA) (10 μmol l−1) diluted in serum-free medium and incubated at 37 °C for 20 min, followed by rinsing thoroughly with serum-free cell culture medium to remove extracellular DCFH-DA. Then, the cells were treated with 0.25% trypsin at 37 °C and collected after centrifugation at 1200 rpm for 5 min. Finally, the ROS-positive signal of the cells was analysed by flow cytometry in CytoFlex S using the FITC channel.
2.4.7. Statistical analysis
The statistical significance of the difference between the blank control group and the material group was determined by the Student's t-test in GraphPad Prism. In each instance, p < 0.05 was declared statistically significant.
3. Results and discussion
3.1. Composites characterization
FTIR, SEM-EDS, and AFM were utilized to analyse the composition and dispersion of PBS-SA composites. Figure 1 shows the FTIR spectra of SA, PBS, and PBS-SA composites with different proportions of SA. As demonstrated in figure 1(a), the band at 3260 cm−1 is assigned to hydrogen bonded O–H stretching vibration, the band from 1650 cm−1 to 1550 cm−1 is assigned to COO−, and the peak at 1420 cm−1 is assigned to C–OH deformation vibration with the contribution of O–C–O symmetric stretching vibration of the carboxylate group. The peaks at 1080 cm−1 and around 1000 cm−1 are assigned to C–O and C–C stretching vibrations of the pyranose ring, respectively [18, 30, 31]. As demonstrated in figure 1(b), the peaks around 2950 cm−1 are assigned to CH3 and CH2 stretching vibration, and the peak at 1710 cm−1 is assigned to the stretching vibration of the carbonyl group. The peaks around 1330 cm−1 are the out-of-plane oscillations of CH2. The peak at 1155 cm−1 is assigned to the C–O–C antisymmetric stretching vibration, and the peak at 1045cm−1 corresponds to the C–O–C symmetric stretching vibration, and the peaks at 955 cm−1, 806 cm−1, 654 cm−1 can be used to discriminate PBS from other polyesters [32, 33]. In figures 1(c)–(f), the spectra are mainly characteristic of PBS, while the bands and peaks of SA can be observed in them (black dotted lines), suggesting that SA was successfully incorporated into the PBS matrix.

Figure 1. FTIR spectra of SA (a), PBS (b), and PBS-SA composites with different proportions of SA (c)–(f).
Download figure:
Standard image High-resolution imageFigure 2 shows the SEM images of PBS-SA composites with different proportions of SA, as well as the EDS mapping of the PBS-SA10 composite. The particle size of SA in PBS-SA5 and PBS-SA10 is mainly between 100–200 nm and 200–300 nm, respectively. The dispersed phase can be relatively evenly distributed in the PBS matrix with a proportion of SA of 5% or 10%. As the proportion of SA increased to 15% or 20%, the particle size of the dispersed phase increased gradually, and agglomeration and uneven dispersion were more likely to be observed. The SA particle size determined by AFM (Figure S1 (available online at stacks.iop.org/MRX/9/085403/mmedia), Supplementary material) was basically consistent with the SEM findings shown in figure 2. For the composites, Na is an element found only in SA. In figures 2(e)–(f), the distribution of Na is essentially identical to the uniformity of C and O, demonstrating that SA in PBS-SA10 is uniformly spread in the PBS matrix.

Figure 2. SEM images of PBS-SA5 (a), PBS-SA10 (b) PBS-SA15 (c) PBS-SA20 (d);EDS mapping images of PBS-SA10 on elements N, O, and Na.
Download figure:
Standard image High-resolution imageSA is a hydrophilic polysaccharide, while PBS is a hydrophobic polyester. The composites presented the typical 'sea-island' structure of a heterogeneous phase, which was caused by thermodynamic incompatibility [34]. With an increase in SA loading, the spatial fraction and the spacing of dispersed phase particles increased and decreased, respectively, resulting in a steady increase in particle size and agglomeration and a reduction in dispersion [35].
3.2. Water contact angle
Since human body fluids are composed mostly of water, it is important for implantation applications to quantify the hydrophilicity of materials. By studying the water contact angle of PBS and PBS-SA composites, the hydrophilicity of the composites with different proportions of SA can be analysed. Figure 3 shows the water contact angle measurement photos of PBS and PBS-SA composites. As can be seen, the average water contact angle of PBS is 96.68°, and the average water contact angle from PBS-SA5 to PBS-SA20 is 91.75°, 88.40°, 84.00°, and 82.22°, respectively. SA is a hydrophilic polysaccharide with hydroxyl (−OH) and carboxyl (COO−) groups, while PBS is a hydrophobic polyester with a C–C bond and an ester group (−COOC−). The hydrophilicity of the composite gradually increased as the proportion of SA in the composite increased. When the SA loading was 10%, the water contact angle was less than 90%, indicating that the composite can be wetted by water and belongs to a hydrophilic material [36]. Good hydrophilicity promotes implant cell proliferation and adhesion after implantation, which is beneficial to the replacement of biodegradable implants by tissues in vivo [37].
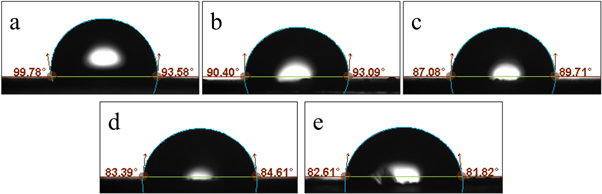
Figure 3. Water contact angle of PBS (a), PBS-SA5 (b), PBS-SA10 (c), PBS-SA15 (d), PBS-SA20 (e).
Download figure:
Standard image High-resolution image3.3. Mechanical properties
Mechanical properties of materials are one of the important parameters for the development of implantable medical devices. The mechanical characteristics of PBS and PBS-SA composites were measured to investigate the impact of SA loading on tensile properties. Figure 4 shows the tensile properties of PBS and PBS-SA composites with different proportions of SA. Figure 4(a) illustrates the stress-strain curve of tensile properties and figures 4(b)–(d) show the changes in Young's modulus, tensile strength, and elongation at break at various SA loading percentages.
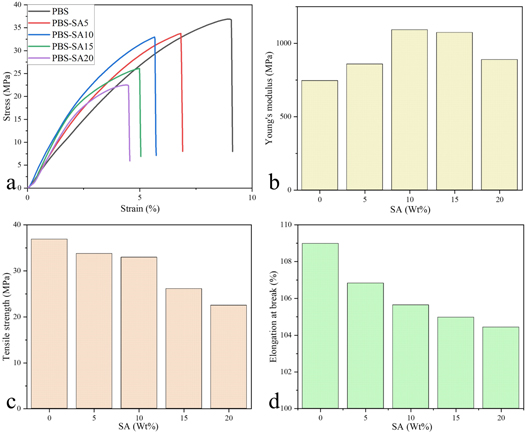
Figure 4. The mechanical properties of PBS, PBS-SA5, PBS-SA10, PBS-SA15, and PBS-SA20. Stress-strain curve (a), Young's modulus (b), tensile strength (c), elongation at break (d).
Download figure:
Standard image High-resolution imageWhen the loading amount of SA was 5% and 10%, the Young's modulus of the composite increased, which was 858.10 MPa and 1091.21 MPa, respectively, and was 115.13% and 146.40% of that of PBS at 745.35 MPa, respectively. With loadings of 15% and 20% SA, the Young's modulus showed a declining tendency. The Young's modulus of PBS-SA15 and PBS-SA20 was 1073.03 MPa and 888.01 MPa, or 143.96% and 119.14% of that of PBS, respectively. The tensile strengths of PBS and the composites with SA loadings ranging from 5% to 20% were 36.89 MPa, 33.75 MPa, 32.95 MPa, 26.11 MPa, and 22.50 MPa in sequence, indicating a declining trend with increasing SA addition. Similarly, as SA increased, elongation at break decreased gradually, with values of 108.98% for PBS, 106.83% for PBS-SA5, 105.64% for PBS-SA10, 104.97% for PBS-SA15, and 104.43% for PBS-SA20, respectively.
Many studies have shown that filler filling has a significant impact on the mechanical properties of composites. Mechanical property changes are primarily linked to filler dispersion and geometric dimensions, as well as interfacial adhesion between filler and matrix [9]. SA has good dispersion and smaller particle size in the PBS matrix when loaded at 5% or 10% (figure 2). The Young's modulus of composites was raised due to the increased effective interface contact area caused by good dispersion [38]. As a result of the further increase in SA loading, the agglomeration and microstructure discontinuity increased, resulting in a gradual decrease in Young's modulus [39, 40]. Due to the thermodynamic incompatibility and phase separation between SA and PBS, which limited the transfer of interface effective stress, the tensile strength and the elongation at break of the composites declined progressively with increasing SA loading [9].
The safety strain range of most human tendons is about 4% [41]. It is beneficial to reduce the strain of tendons and fix them onto bones more effectively with higher Young's modulus and tensile strength. Therefore, the mechanical properties of the PBS-SA10 were the most appropriate, and the PBS-SA10 composite was used for testing in vitro biocompatibility.
3.4. Thermal properties
The analysis of thermal properties is helpful in evaluating the applicable temperature and thermal processing temperature of composites. The DSC, TGA, and DTG curves of PBS and PBS-SA composites with different proportions of SA are shown in figure 5, and their thermal parameters are listed in table 1.

Figure 5. Thermal properties of PBS, PBS-SA5, PBS-SA10, PBS-SA15, and PBS-SA20: DSC secondary heating curves (a), DSC cooling curves (b), TGA curves (c), and DTG curves (d).
Download figure:
Standard image High-resolution imageTable 1. Thermal property parameters of PBS, PBS-SA5, PBS-SA10, PBS-SA15, and PBS-SA20.
| Tg | Tm1 | Tm2 | Tc | ΔHc (J g)−1 | χc (%) | Td | ΔT | Tmax1 | Tmax2 | |
|---|---|---|---|---|---|---|---|---|---|---|
| PBS | −34.01 | 96.59 | 109.79 | 77.49 | 69.55 | 63.06 | 322.33 | 212.54 | — | 396.93 |
| PBS-SA5 | −34.01 | 96.59 | 109.65 | 77.86 | 67.43 | 64.35 | 317.18 | 207.53 | 247.73 | 389.21 |
| PBS-SA10 | −34.19 | 96.59 | 109.99 | 78.16 | 65.13 | 65.61 | 309.47 | 199.48 | 242.58 | 386.63 |
| PBS-SA15 | −34.01 | 96.59 | 109.67 | 77.82 | 59.99 | 63.99 | 258.02 | 148.35 | 242.58 | 384.06 |
| PBS-SA20 | −33.27 | 96.59 | 109.84 | 78.32 | 53.93 | 61.12 | 237.44 | 127.60 | 237.44 | 384.06 |
Figure 5(a) shows the secondary heating curve of PBS and PBS-SA composites with different proportions of SA after the elimination of the thermal history. The Tg (black arrows in figure 5(a)) of PBS and PBS-SA composites was almost the same, indicating that the addition of SA had no significant effect on Tg, which may be due to the lack of strong interaction between the interface of the hydrophilic filler and the hydrophobic matrix [9]. The Tm of PBS and the composites also had no significant difference. Two melting peaks (Tm1 and Tm2 in table 1) could be observed in each secondary heating curve, and the morphology of melting peaks between different curves was basically the same. Due to the two melting peaks being attributed to two types of crystallization with different thermal stability in PBS and melt-recrystallization on heating, it could be inferred that SA loading has no discernible effect on PBS crystallisation [42].
Figure 5(b) shows the cooling curves of PBS and the composites after the elimination of the thermal history. The Tc of all the composites was slightly higher than that of PBS. Meanwhile, according to the crystallinity calculation method of PBS mentioned above and ΔHc we measured in DSC, the crystallinity of PBS and the composites with different percentages of SA was calculated as shown in table 1. The Tc and crystallinity of PBS in the composites were slightly increased with the SA loading, which may be attributed to the heterogeneous crystallization of SA dispersed in PBS. However, when the loading of SA was 15% and 20%, the crystallinity decreased, which may be attributed to the blocking effect of the higher filler loadings of SA, preventing the PBS polymer chain from folding into the crystal structure [43].
As can be seen from figures 5(c) and (d), when the temperature was between 50 °C and 150 °C, a band of increasing decomposition speed rate appeared in each curve of the composites, which represents the process of SA losing the binding water [44]. When the temperature was between 200 °C and 300 °C, the mass of the PBS-SA composites started to decrease obviously. The first peak temperature of thermal decomposition in the DTG curve (Tmax1) emerged at 247.73 °C, 242.58 °C, 242.58 °C, and 237.44 °C, respectively, which was the thermal decomposition of SA [43, 44]. The thermal decomposition temperature of PBS was the second peak temperature of thermal decomposition in the DTG curve (Tmax2) for PBS and PBS-SA composites [45]. As the SA loading increased, the thermal decomposition temperature of PBS decreased by 7.72°C–12.87°C. The decrease in thermostability of the composites may be attributed to the higher mobility of polymer chains caused by insufficient adhesion interaction between SA and PBS [9].
The decrease in thermostability of composites affects the thermal processing temperature, which is an important parameter for thermoplastic materials. The theoretical range of the thermal processing temperature (ΔT) was from Tm to Td (table 1). With the increase of SA loading, the thermostability of the composites was lower than PBS, the Td and ΔT gradually decreased. In order to ensure that SA did not decompose significantly during thermal processing, it is recommended that the actual thermal processing temperature range of PBS-SA composites be between 110 °C and 200 °C, which provides an adequate temperature range for the composites' thermal processing.
3.5. In vitro degradation
The mass loss curves of PBS, PBS-SA10, and PBS-SA20 degradation in vitro in Sorensen buffer solution at 37 °C are shown in figure 6. Because the swelling degree of hydrophilic SA was greater than that of PBS, the mass at 1 W and 2 W did not decrease but increased when the addition amount of SA was increased. The weight of the sample with higher SA content decreased more quickly as time passed. PBS, PBS-SA10, and PBS-SA20 lost 6.71 %, 8.00 %, and 12.09 % of their mass at 12 W, respectively. This indicates that SA can promote the biodegradability of the PBS-SA composites.

Figure 6. Mass loss percentages of PBS, PBS-SA10, and PBS-SA20 in in vitro degradation.
Download figure:
Standard image High-resolution image3.6. In vitro biocompatibility
Various in vitro cell experiments were carried out to determine the in vitro biocompatibility of the PBS-SA10 composites. Figure 7(a) shows the comparison of NRU levels in BALB/C 3T3 cells after 24 h of co-culture in the extract or the medium. The morphology and size of the material group were unchanged, and numeral cells ingested neutral red dye, while the total number of cells was not substantially different from that of the blank control group. Figure 7(b) illustrates a statistical comparison of the OD values between the two groups, indicating that there was no statistical difference (p > 0.05). The neutral red dye readily permeates the cell and lysosomal membranes. When the cell and lysosomal membranes are damaged, the cellular uptake of neutral red is lowered. In addition, when cell proliferation is harmed, the overall uptake of neutral red may be reduced owing to a reduction in the total number of cells [46]. As a result, we employed this approach to determine whether the material extract produced the aforementioned damage and its influence on cell growth. To the best of our knowledge, there is no study reported about NRU tests of SA, besides research on SA and polyglycolic acid composites that yielded non-cytotoxic results [47]. And there is no research reported to determine whether PBS affects the cell and lysosomal membranes of mammalian cells. It can be inferred that PBS-SA10 caused no significant damage to the cell membrane or lysosomal membrane and had no obvious influence on cell proliferation.

Figure 7. Comparison of NRU in BALB/c 3T3 cells of the material group and the blank control group (p > 0.05) (a, b);
Download figure:
Standard image High-resolution imageComparison of CCK-8 cell proliferation in NIH 3T3 cells of the material group and the blank control group (p > 0.05) (c).
Figure 7(c) illustrates the statistical comparison of the OD values in the CCK-8 cell proliferation test between the material group and the blank control group. The results indicated no statistically significant difference between the two groups (p > 0.05). The cell proliferation assay using CCK-8 was similar to the MTT cytotoxicity assay. It is a technique for determining cell proliferation by measuring the quantity of yellow formazan generated by cells metabolizing WST-8 in the mitochondria. Some studies have shown that SA composites with strontium-hydroxyapatite microspheres or chitosan can promote cell proliferation using CCK-8 to evaluate cell proliferation [48, 49]. Additionally, PBS has been shown to have no discernible effect on cell proliferation [5]. This finding is consistent with the results of our study.
The maintenance of cell morphology, which is dependent on the integrity of the cytoskeleton, is a critical aspect of cell health. The cytoskeleton and nucleus were stained with phalloidin-ifluor and DAPI, respectively (figure 8). The cytoskeletons of the two groups were almost identical and the nuclei were complete, with no notable variation between the two groups. These findings suggest that the PBS-SA10 composite had no discernible detrimental effects on the cytoskeleton.
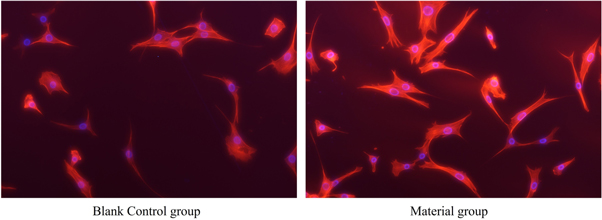
Figure 8. Comparison of cytoskeleton fluorescence staining between the material group and the blank control group (p > 0.05).
Download figure:
Standard image High-resolution imageFigure 9 illustrates the effects of the two groups on cell apoptosis. There was no statistically significant difference in the morphological range of the flow cytometry scatter diagram between the two groups, nor was there a statistically significant difference in the number of apoptotic cells (p > 0.05), indicating that the PBS-SA10 composite did not significantly promote apoptosis.

Figure 9. Comparison of cell apoptosis between the material group and the blank control group (p > 0.05).
Download figure:
Standard image High-resolution imageFigure 10 shows a comparison of the intracellular ROS levels between the material group and the blank control group. The number of ROS-positive cells was compared and examined by flow cytometry, and there was no statistically significant difference in the number of ROS-positive cells between the two groups (p > 0.05), indicating that the PBS-SA10 composite had no significant effect on the ROS level of cells. SA and its oligomers have been shown to scavenge oxygen free radicals, inhibit the production of caspase-9 and caspase-3, and protect against ROS-induced apoptosis [50].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 10. Comparison of ROS positive cells between the material group and the blank control group.
Download figure:
Standard image High-resolution image{kind=link}
PBS and PLA composites can stimulate the proliferation of epithelial fibroblasts [51]. Poly (butylene succinate-co-adipate) had no cytoskeleton toxicity [52] and had no effect on the apoptosis of human epithelial fibroblasts [53]. There are no investigations into the impact of PBS and its composites and copolymers on ROS levels in mammalian cells that we are aware of.
The good biocompatibility is also related to the monomers of polyesters. The body interacts with biodegradable polyesters primarily via the interaction of their degradation products with cells and tissues [54]. The soluble components of the PBS that has been dissolved in the medium is mostly composed of the monomers or oligomers of succinic acid and butanediol. They can be metabolized in vivo through the tricarboxylic acid cycle to generate CO2 and H2O, then eliminated from the body [55]. They are non-accumulative and quickly metabolize in the body [56].
4. Conclusion
In this study, PBS-SA composites were successfully prepared using the melt blending technique with an SA mass proportion of 5%, 10%, 15%, and 20%. By the determinations of FTIR and SEM-EDS, it was shown that SA was effectively blended into the PBS matrix. When the addition proportion of SA was 5% and 10%, it was uniformly distributed in the PBS matrix, and the Young's modulus increased by 15.13% and 46.40%, respectively. However, at higher SA loading, the Young's modulus was significantly dropped due to the filler agglomeration. The addition of SA had no significant influence on the Tg and Tm of the composites, as well as the crystallinity of the PBS matrix. As the SA loading increased, the PBS-SA composites improved the hydrophilicity and biodegradability and kept an adequate thermal processing temperature range.
Based on the results of the characterization, the PBS-SA10 composite has the best comprehensive mechanical properties, mild hydrophilicity, and good biodegradability. According to the results of the NRU cytotoxicity assay, the CCK-8 cell proliferation assay, the cell apoptosis assay, the cytoskeleton fluorescence staining assay, and the intracellular ROS assay, PBS-SA10 has no significant deleterious effects on the cell membrane, lysosomal membrane, cell proliferation, cell apoptosis, the cytoskeleton, or intracellular ROS levels and has good biocompatibility. The PBS-SA10 composite can be used as a substitute material for biodegradable medical devices such as suture anchors.
Acknowledgments
The authors are grateful for the research funding provided below.
Data availability statement
All data that support the findings of this study are included within the article (and any supplementary files).
Author contribution
Quansheng Xing: Conceptualization, methodology, software, formal analysis, resources, writing—review and editing, supervision, project administration, and funding acquisition.
Shanshan Wang: methodology, software, validation, formal analysis, investigation, resources, data curation, writing-original draft, visualization.
Research funding
This study was supported by the National Natural Science Foundation of China (NSFC) [Grant Nos.81770315 and 81570287] and the Taishan Scholars Program (2019).
Conflicts of interest
The authors declare no competing financial interests.